

Abstract
Background. Malaria and AIDS represent 2 leading causes of death from infectious diseases worldwide, and their high geographic overlap means coinfection is prevalent. It remains unknown whether distinct immune responses during coinfection with malaria and human immunodeficiency virus (HIV) affect clinical outcomes.
Methods. We tested this hypothesis by employing macaque models of coinfection with malaria and simian-human immunodeficiency virus (SHIV).
Results. Plasmodium fragile malaria coinfection of acutely SHIV-infected macaques induced hyperimmune activation and remarkable expansion of CD4+ and CD8+ T effector cells de novo producing interferon γ or tumor necrosis factor α. Malaria-driven cellular hyperactivation/expansion and high-level Th1-cytokines enhanced SHIV disease characterized by increasing CD4+ T-cell depletion, profound lymphoid depletion or destruction, and even necrosis in lymph nodes and spleens. Importantly, malaria/SHIV-mediated depletion, destruction, and necrosis in lymphoid tissues led to bursting parasite replication and fatal virus-associated malaria. Surprisingly, chronically SHIV-infected macaques without AIDS employed different defense mechanisms during malaria coinfection, and mounted unique ∼200-fold expansion of interleukin 17+/interleukin 22+ T effectors with profound Th1 suppression. Such remarkable expansion of Th17/Th22 cells and inhibition of Th1 response coincided with development of immunity against fatal virus-associated malaria without accelerating SHIV disease.
Conclusions. These novel findings suggest that virus infection status and selected Th1 or Th17/Th22 responses after malaria/AIDS-virus coinfection correlate with distinct outcomes of virus infection and malaria.
Approximately 300–500 million malaria infections occur annually, causing 1–3 million deaths (http://apps.who.int/malaria), mostly due to Plasmodium falciparum [1]. In addition, there are > 30 million people infected with human immunodeficiency virus (HIV) (http://www.who.int/hiv/data/en). Because both diseases are highly prevalent in overlapping geographic areas, coinfection is common [2], and mathematical modeling has demonstrated coinfection increases the spread of both pathogens in sub-Saharan Africa [3].
Although human studies of malaria/HIV coinfection have shown mutual impact on both diseases [4–12], no detailed study has been undertaken to elucidate potential immune mechanisms underlying this impact. Immune regulation against coinfection remains largely unknown, and studies cannot be performed in small lab animals because of resistance to HIV/AIDS. Using nonhuman primate models of malaria/AIDS-virus coinfection is therefore of central importance for circumventing these limitations and for enhancing our understanding of malaria-HIV interaction and disease pathogenesis.
In our current studies, we used our decades-long expertise in simian immunodeficiency virus (SIV)/mycobacterium coinfection and used Plasmodium fragile and simian-human immunodeficiency virus 89.6P (SHIV89.6P) to develop models of malaria/acute-SHIV coinfection and malaria/chronic-SHIV coinfection in Chinese-origin rhesus macaques. P. fragile causes malaria pathologically similar to that due to P. falciparum in macaques [13, 14], whereas SHIV89.6P resembles HIV by infecting CD4+ T cells or macrophages and inducing simian AIDS as HIV does in humans [15]. These features suggest that coinfection with P. fragile and SHIV is relevant to coinfection with P. falciparum and HIV. In these proof-of-concept studies, SHIV89.6P was used to optimally determine clinical outcome during highly pathogenic AIDS-virus coinfection, as Chinese rhesus macaques are somewhat resistant to simian immunodeficiency virus macaque (SIVmac). The nonhuman primate models of malaria and AIDS-virus coinfection allowed us to illustrate the reciprocal effects of malaria and AIDS-virus infections and potential immune mechanisms underlying coinfection-enhanced viral pathogenicity or malaria disease and immunity. We made novel observations that virus infection status and distinct Th1 or Th17/IL-22+ immune responses after malaria-coinfection of acutely or chronically SHIV-infected macaques correlate with different clinical outcomes of virus infection and virus-associated malaria.
METHODS
Animals
We used 19 Chinese rhesus macaques (4–8 years old), free of simian retrovirus, simian T-lymphotrophic virus type 1, and SIV infection. Macaques’ ages corresponded to young adult or adult humans. Of the 18, 4 macaques were simultaneously coinfected with SHIV/malaria; 5 chronically SHIV-infected macaques were coinfected with malaria; 4 were infected with malaria; 6 were infected with SHIV. All animals were maintained and used in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC). Chloroquine (West-Ward Pharmaceuticals) was given (37.5mg/kg by stomach gavage) to control life-threatening malaria, or to cure malaria (t2 doses given 2 days apart) at the end point. Animals infected with SHIV were humanely killed at the end of the study, or earlier if deemed necessary by veterinary staff.
Infections
Animals were inoculated intravenously with 1000AID50 SHIV89.6P or 104 P. fragile–parasitized red blood cells (pRBC). We propagated pRBC in a donor rhesus, stored them in liquid nitrogen until use, and thawed them as previously described [16]. SHIV89.6P was stored at −20°C, washed, and resuspended in RPMI-1040 + 10% heat-inactivated fetal bovine serum (FBS) [15]. For chronic SHIV/malaria coinfection, animals had been infected with SHIV89.6P for 296 days before infection with malaria. For SHIV-only infection, macaques had been infected with the same lot, dose, and route ∼1 year prior for use in another study [15]. Some of the data from these animals were previously published [15]. For chronic SHIV-only controls, data were collected from 16 weeks through 24 weeks after SHIV infection.
Estimation of Parasitemia
Thin blood smears were made by expressing a drop of blood from a tail prick of malaria-infected monkeys. Smears were air dried, then stained using DipQuick stain kit (Jorgenson Laboratories) according to manufacturer’s instructions. Parasitemia was estimated by comparing parasitized and total erythrocytes in blood film. Data were expressed as percentage of pRBC in total red blood cells.
Quantitative Measurement of SHIV RNA
Plasma SHIV RNA was assessed using real-time reverse transcription–polymerase chain reaction (RT-PCR) [15, 17, 18].
Histology
Tissue samples obtained from necropsy were formalin fixed (Protocol Formalin), embedded in paraffin, and sectioned at 5μm for routine staining with hematoxylin and eosin [19].
Isolation of Lymphocytes From Peripheral Blood
Peripheral blood lymphocytes (PBL) were isolated from freshly collected ethylenediaminetetraacetic acid (EDTA) blood by Ficoll-Paque Plus (Amersham,) density gradient centrifugation before analysis.
Immunofluorescent Staining and Flow Cytometric Analysis
For cell-surface staining, PBL were stained with ≤ 5 Abs (conjugated to fluorescein isothiocyanate, phycoerythrin (PE), allophycocyanin, pacific blue, and PE-Cy7) for 15 minutes. After staining, cells were fixed with 2% formaldehyde–phosphate-buffered saline (PBS) prior to analysis on a CyAn ADP flow cytometer (DakoCytomation). Lymphocytes were gated based on forward- and side-scatters, and pulse-width and ∼40000 gated events were generally analyzed using Summit Data Acquisition and Analysis Software (DakoCytomation).
The following mouse Abs were used: IL-17 (eBio64CAP17, eBioscience); IL-22-biotin (antihuman, R&D Systems); Streptavidin-Pacific Blue (Invitrogen); CD3 (SP34-2), CD4 (L200), CD8 (RPA-T8), IFNγ (4S.B3), TNFα (mAb11, BD Pharmingen).
Intracellular Cytokine Staining
Modified intracellular cytokine staining (ICS) without in vitro Ag stimulation was adopted as recently described [20]. Briefly, 0.5 × 106 PBL plus mAbs CD28 (1 μg/mL) and CD49d (1 μg/mL) were incubated in 100 μL final volume for 1 hour at 37°C, 5% CO2 followed by an additional 5-hour incubation with brefeldin A (GolgiPlug, BD). After staining for cell-surface markers (CD3, CD4, CD8) for 15 minutes, cells were permeabilized for 45 minutes (Cytofix/cytoperm, BD) and stained 45 minutes for IFNγ, TNFα, IL-17, or IL-22 before fixation in formalin.
As in vivo control experiments, longitudinal analyses during acute and chronic SHIV infection of 4 macaques did not have measurable increases in IFNγ-producing T cells in PBL [15]. In addition, we did not detect increased levels of IL-17– or IL-22–producing T cells during acute and chronic SHIV infection. Control data were obtained using CD28 and CD49d mAbs (without antigen restimulation) as described above.
Determination of Plasma Cytokine Levels
Cytokine levels were determined using Monkey 5-plex Cytokine Kit (Invitrogen), read on a Bio-Rad Bio-Plex machine (Bio-Rad), and analyzed using Bio-Plex Manager 5.0 software (Bio-Rad) according to manufacturers’ instructions.
Enzyme-Linked Immunosorbent Assay
Enzyme-linked immunosorbent assay (ELISA) was performed as previously described [15]. Plasma was heat inactivated for 30 minutes at 56°C before analysis. MSP-1 (19kDa fragment) antigen was obtained through Malaria Research and Reference Reagent Resource center (MRA-47), deposited by DC Kaslow. Antibody was detected using antimonkey immunoglobulin G and immunoglobulin M antibodies (KPL), and optical density at 405nm (OD405) was determined on an ELISA plate reader (Bio-rad model 550).
Statistical Analysis
Statistical analysis was performed using paired or unpaired 2-tailed t test [21] using Graphpad software (Prism). Similar trends were seen using nonparametric analysis. Data compared were based on percentage, unless otherwise stated.
RESULTS
Malaria Coinfection of Acutely SHIV-Infected Macaques Led to Fatal Virus-Associated Malaria
We presumed that mutual enhancement of malaria and HIV/AIDS could be best demonstrated in naive individuals who have not developed antimalaria or anti-HIV immune responses. Thus, a group of naive macaques was coinfected simultaneously with malaria and SHIV89.6P (malaria/acute SHIV). Additionally, chronically SHIV-infected macaques were similarly coinfected with malaria (malaria/chronic SHIV). As controls, a group of macaques was infected with malaria only, and another with SHIV only. The malaria-only group developed uncomplicated malaria, manifesting transient low hematocrits, and low (< 5.26%) but measurable parasitemia starting from day 19 and fluctuating through day 54 (Figures 1A and 1B). In contrast, malaria/acute SHIV–coinfected macaques developed fatal malaria, exhibiting sharp increases in parasitemia to > 70% at day 20 (Figure 1A; P = .0001, D19, between groups). All acutely coinfected macaques manifested severe anemia (pale mucosae; Figure 1B), shivering, and lethargy or comalike signs 2–3 weeks postcoinfection, and received chloroquine treatment. Despite treatment, 3 (7282, 7289, 7344) of these coinfected macaques became moribund and were euthanized by day 22 postcoinfection (Figure 1C). The 4th (7284) recovered from life-threatening malaria after chloroquine treatment. These results demonstrated profound impact of SHIV on malaria, as malaria/acute-SHIV-coinfection led to high-level parasitemia and rapidly progressed to fatal virus-associated malaria. The data also indicate that an active, progressive malaria/AIDS-virus coinfection model can be developed by simultaneous SHIV and P. fragile coinfection of macaques.
Figure 1.

Malaria coinfection of acutely simian-human immunodeficiency virus (SHIV)–infected macaques led to fatal virus-associated malaria; chronically SHIV-infected macaques developed moderately enhanced parasitemia without fatal outcome after malaria coinfection. A, Malaria/acute SHIV–coinfected macaques (left) had significantly higher parasite levels than malaria-only controls (center, P = .0004 at day 20). All acutely SHIV-infected macaques developed life-threatening malaria, and all but 1 (subject 7284) became moribund despite receiving chloroquine treatment. Malaria/chronicSHIV–coinfected macaques without AIDS (right) had higher parasitemia than that of malaria-only controls (P = .0245 at day 20), but much lower parasitemia than that of malaria/acute SHIV–coinfected macaques (P = .0034).* indicates chloroquine treatment. B, Hematocrit data for macaques as indicated on the top of the panels. Note that the macaques with fatal malaria had extremely low levels of hematocrit despite chloroquine treatment. All moribund macaques had clinical signs of severe anemia, although some were not tested for hematocrit at endpoints. C, Survival curves for the malaria/acute SHIV, malaria-only, and malaria/chronic SHIV groups. Only 1 macaque (subject 7284) in the malaria/acute SHIV group survived life-threatening malaria after chloroquine treatment. All macaques without AIDS in the malaria/chronic-SHIV group survived, with 2 (subjects 7393 and 7418) treated with chloroquine at day 20. The single macaque with prior AIDS (subject 7409) in the malaria/chronic SHIV group did not survive despite chloroquine treatment at day 19, and was euthanized on day 22. Abbreviation: pRBC, parasitized red blood cells.
Malaria Coinfection of Acutely SHIV-Infected Macaques Appeared to Accelerate SHIV Disease
We then sought to determine whether malaria coinfection could reciprocally enhance pathogenicity and accelerate SHIV disease. Acutely coinfected macaques appeared to have faster progressing SHIV disease than SHIV-only controls (Figures 2 and 3). Remarkable lymphoid destruction/depletion was found in both lymph nodes and spleen tissues collected at necropsy from malaria/acute SHIV–coinfected macaques 21 days post coinfection (Figures 2A and 2B). No or very few lymphocytes were detectable in T-cell zones of lymph nodes, with apparent disappearance of germinal centers and T-cell zones. Lymphoid structures were destroyed with notable necrosis in the lymph nodes (Figure 2A). Similarly, lymphoid destruction/depletion and necrosis were seen in spleen tissues (Figure 2B). In contrast, such profound destruction/depletion and necrosis in lymphoid tissues were not seen in lymph nodes and spleens from SHIV-only control macaques in acute-SHIV infection or 1.5 years after SHIV infection (Figures 2A and 2B). More apparent lymphoid depletion in rectal mucosa was detected 21 days post–malaria/acute SHIV coinfection compared with SHIV-only controls (Figure 2C). Blood CD4+ T-cell counts sharply declined to 54.9 ± 66.2/μl 19 days postcoinfection, whereas the SHIV-only group had CD4+ T-cell counts 362.67 ± 217.78/μL at day 21–22 (Figure 3; P = .0154). The acutely coinfected macaques exhibited high-level plasma SHIV RNA (Figure 3). Because of early chloroquine treatment and deaths of the macaques, a full comparison of peak viremia and set points between the acute-coinfected and SHIV-only groups could not be made. Despite this, it was surprising that profound destruction/depletion and necrosis in lymphoid tissues with sharp decline of blood CD4+ T-cell counts occurred in acutely coinfected macaques, as such severe pathogenic events would normally not be detected until end-stage AIDS in humans and nonhuman primates [22, 23]. These results suggested that malaria coinfection remarkably enhanced acute-SHIV disease.
Figure 2.

Malaria coinfection of acutely simian-human immunodeficiency virus (SHIV)–infected animals resulted in severe and rapid lymphoid depletion in the lymph nodes, spleen, and rectal mucosa. A, B, Histopathology images indicating lymphoid depletion/destruction in lymph nodes (top, 50× magnification) and spleens (bottom, 50× magnification) of 2 representative macaques (subjects 7282, 7289) in the malaria/acutely SHIV–coinfected group. Note the destruction of lymphoid structures, the disappearance of both germinal centers and T-cell zones, and apparent necrosis in lymph nodes and spleens collected at necropsy at day 21 after the coinfection. Shown on the left are 2 sets of control images (25× magnification). The first set from a representative uninfected macaque indicated normal structures of T-cell zones/germinal centers in the lymph node and white/red pulps in the spleen. The other set from SHIV-only macaque (subject 7351, necropsied on day 627 post-SHIV infection) showed enlarged germinal centers, but no lymphoid destruction or necrosis. A lack of necrosis and marked lymphoid depletion was also seen in acute viral infection phase of SHIV-only controls. Of note, acute SHIV infection of macaques did not cause lymphoid destruction, necrosis, or disappearances of germinal centers despite the minor decreases in cell density in T-cell zones and subtle paracortical hyperplasia were seen in lymph nodes collected 1 month after SHIV infection (data not shown). All samples were formalin-fixed sections stained by hematoxylin and eosin. Red arrows indicate germinal centers; green arrows indicate necrotic regions without germinal centers/T-cell zones or normal structures; and yellow arrows indicate depleted germinal centers and necrosis in spleen. Ci, Histopathology images of rectal mucosa collected at day 21 after SHIV from representative SHIVinfected control (left 2 panels) and malaria/acute SHIV–coinfected macaques (right 2 panels). Malaria/acute SHIV–coinfected macaques shows decreased densities of lymphoid cells in gut mucosa overall, with fewer lymphocytes seen (400× magnification).
Figure 3.

Malaria coinfection of acutely simian-human immunodeficiency virus (SHIV)–infected macaques enhanced SHIV disease; malaria coinfection of chronically SHIV-infected macaques induced only a transient increase in viral loads with stable CD4+ T-cell counts. Top: Plasma SHIV RNA levels detected in individual malaria/acute SHIV–coinfected macaques (left, n = 4), individual malaria/chronic SHIV–coinfected macaques (n = 5), and the SHIV-only group (means ± SD, n = 6). Bottom: Numbers of CD4+ T cells in malaria/acute SHIV and malaria/chronic SHIV macaques, and the SHIV-only group (means ± standard deviation [SD], n = 6). Data for the chronic SHIV-only group were derived from a 6-week period of chronic infection. The transient ∼1 log increase in SHIV level during the chronic SHIV coinfection was not statistically significant when compared with baseline. Absolute CD4+ T cell counts in malaria/acute SHIV coinfected macaques appeared to reach a lower level than those in SHIV-only controls. P = .0154 day 19 malaria/acute-SHIV vs day 22 SHIV-only (a progressive decline of CD4 T cell counts usually is most striking at weeks 3–5 after SHIV89.6P). No significant declines of CD4+ T cell counts in malaria/chronic SHIV-coinfected macaques were noted.
Malaria Coinfection During Chronic-SHIV Infection Caused Moderately Enhanced Malaria, With Stable CD4 Counts and Transiently Increased Viral Loads
Next, we sought to determine whether malaria coinfection of chronically SHIV-infected macaques led to similar or different clinical outcomes of SHIV disease and malaria compared with malaria/acute-SHIV coinfection. Chronically SHIV-infected macaques without AIDS did not develop clinical signs of fatal malaria or accelerated SHIV disease after malaria coinfection. Whereas macaque 7409 with AIDS (CD4+ count < 200/μL) exhibited rapidly increased parasitemia, and deteriorated to moribund status despite chloroquine treatment (Figure 1A–C), other coinfected macaques remained clinically stable, despite exhibiting moderately increased malaria parasitemia (up to 15% pRBC) compared with that of malaria-only controls (Figure 1A and 1C).
The malaria impact on chronic SHIV coinfection was much less dramatic than on acute SHIV coinfection. Malaria coinfection of chronically SHIV-infected macaques led to transient ∼1 log increase in plasma viral RNA (Figure 3). Malaria/chronic SHIV coinfected monkeys did not have significant changes in CD4+ T-cell counts (Figure 3). These results demonstrated that malaria coinfection in chronically SHIV-infected macaques without AIDS led to uncomplicated, moderately enhanced malaria disease, with stable CD4 counts and transient/subtle increases in viral loads.
Hyperactivation or Expansion of Th1 Effector Cells and High-Level Proinflammatory Cytokines During Malaria/Acute SHIV Coinfection Coincided With Accelerated SHIV Disease and Fatal AIDS-Associated Malaria
We then undertook mechanistic studies to determine if different host factors or immune responses after malaria/SHIV coinfection correlated with different outcomes of disease. This compelling question has not been addressed despite decades-long studies of malaria/HIV coinfection. We first explored whether AIDS-related fatal malaria resulted from SHIV-mediated inhibition of antimalaria T-cell responses or parasite-mediated hyperproinflammatory responses in malaria/acute-SHIV-coinfection. We employed our novel methods for detecting activated/differentiated T-cells capable of de novo production of antimicrobial cytokines without in vitro antigen restimulation (Figure 4A; [20]). We focused on CD4+ and CD8+ T effector cells producing IFNγ, TNFα, or IL-17/IL-22, as IFNγ or Th1/Th17 cells may have a role in both malaria protection and pathology [24, 25], and TNFα is likely involved in proinflammatory responses during malaria [26]. Malaria-only controls showed remarkable increases in IFNγ-producing CD4+ and CD8+ T effectors, which emerged at week 2, peaked at week 3, and were sustained at least through week 7 after infection (Figure 4A–E, see P values in legend). TNFα-producing CD4+PBL were also increased (Figure 4D).
Figure 4.

Hyperactivation/expansion of Th1 effector cells during malaria/acute simian-human immunodeficiency virus (SHIV) coinfection coincided with accelerated SHIV disease and fatal AIDS-associated malaria; suppressed Th1 T effector function associated with uncomplicated malaria and ”stabilized” SHIV infection in malaria/chronic SHIV–coinfection. A, Representative histograms showing increases in CD4+ and CD4-(CD8+) T effector cells producing interferon (IFN) γ at day 22 postinfection in malaria/acute SHIV–coinfected macaque (left) and malaria-only control (center), but not malaria/chronic SHIV–coinfected macaque (right). Data were gated on CD3. Numbers in upper right and brackets were percentages of CD3+ T cells and of CD4+ T cells, respectively. B, Hyperactivation/expansion of IFNγ+CD4+ T-effector cells in malaria/acute SHIV–coinfected macaques (left, coinfected, P = .0096, baseline vs day 15, n = 4; P = .0665, baseline vs day 22, n = 2) and malaria-only controls (center, n = 4, P < .0001, baseline vs day 15; P = .0018, baseline vs day 22). Almost no CD4+ T effectors were detected in malaria/chronic SHIV–coinfected macaques (right, P = .0008 compared with malaria/acute SHIV group, and P = .0004 compared with malaria-only group at day 22, n = 5). SHIV-only controls had few or no IFNγ+ CD4+ T effector cells measured by intracellular cytokine staining (ICS) without Ag stimulation (n = 6, P = .0001 at day 22 compared with malaria/acute SHIV coinfected group). Data are presented as fold increase % IFNγ+ cells among CD4+ T cells. C, Hyperactivation/expansion of IFNγ+CD8+ T effectors (CD8 Ab used for ICS) in malaria/acute SHIV coinfected and malaria-only infected macaques (P = .0099 for malaria/acute SHIV-coinfected macaques, baseline vs day 15, n = 4; P < .0001 for malaria-only infected macaques, baseline vs day 15, n = 2). Much lower increases of CD8+ T effectors were detected in malaria/chronic SHIV-coinfected macaques (right, P = .0002 compared with malaria/acute-SHIV group, and P = .0015 compared with malaria-only group at day 22). SHIV-only controls (n = 6) showed very few or no IFNγ+ CD8+ T effector cells (P = .0004 at day 22, compared with malaria/acute SHIV coinfected group). D, Hyperactivation/expansion of tumor necrosis factor (TNF) α+CD4+ T effectors in malaria/acute SHIV coinfected and malaria-only macaques (P = .0100, baseline vs day 22; malaria/acute SHIV n = 2 at day 22, malaria-only n = 4). However, few TNFα+CD4+ T effectors were detected in malaria/chronic-SHIV (n = 5 at day 22) group (right, P = .005 compared with malaria/acute-SHIV group, and P = .0030 compared with malaria-only group at day 22). E, The absolute number of IFNγ+CD4+ and IFNγ+CD8+ T effectors increased 50- to 100-fold and 100- to 300-fold, respectively, in malaria/acute SHIV-coinfected macaques and malaria-only infected macaques by day 21. The fold increase in malaria/chronic SHIV-coinfected macaques was much less significant.
Interestingly, malaria/acute SHIV-coinfected macaques exhibited hyperactivation and expansion of Th1 cytokine–producing T effectors, rather than suppression. Malaria/acute SHIV–coinfected macaques with fatal malaria and profound AIDS showed 50- to 70-fold increases in CD4+ and CD8+ T effectors producing IFNγ de novo (Figure 4A–E, see P values in legend), and up to 300-fold increases in TNFα-producing T effectors (Figure 4D). It was noteworthy that despite a marked loss of CD4+ T cells (Figure 3), increased IFNγ- and TNFα-producing CD4+ T effectors were detected in malaria/acute SHIV-coinfected macaques (Figure 4A–E). In contrast, SHIV-only controls did not show any detectable increases in the number of IFNγ-producing CD4+ or CD8+ T effector cells during acute and chronic stages of SHIV infection, as no or very few IFNγ-producing CD4+ or CD8+ T cells were detected by direct ICS without in vitro Ag restimulation before and after SHIV infection (Figure 4B–D; [15]). This suggested that marked increases in CD4+ and CD8+ T effectors were driven by malaria.
The remarkable increases in Th1 cells were consistent with high plasma levels of Th1 cytokine IFNγ but not Th2 cytokine IL-4 in malaria/acute SHIV–coinfected macaques (Figure 5). Interestingly, IFNγ levels in plasma of malaria/acute SHIV–coinfected macaques were higher than those of the malaria-only controls (Figure 5A), although the frequencies of increased Th1 cells were comparable (Figure 4). These results suggested that hyperactivation/expansion of Th1 cytokine–producing CD4+ and CD8+ T effectors and high-level plasma Th1 proinflammatory cytokines during malaria/acute-SHIV coinfection correlated with accelerated SHIV disease and fatal AIDS-associated malaria. We also noted small increases of IL-17–producing CD4+ T effectors, but not IL-22–producing CD4+ T effectors (Figure 6). Of note, no or very few IL-22+ and IL-17+ T effectors were detected in SHIV-only controls during acute and chronic phase infections (Figure 6A; [20]). The marked increases in IFNγ+CD4+ and CD8+ T effectors, but small changes in IL-17+ and IL-22+ T effectors, were associated with enhanced CD4+ T cell depletion and lymphoid destruction/necrosis (Figure 7), suggesting malaria-activated CD4+ T cells served as productive SHIV reservoirs leading to accelerated SHIV/AIDS. Reciprocally, increased Th1 effector cells also coincided with SHIV-associated increases in parasitemia in malaria/acute SHIV–coinfected macaques (Figure 7), implicating a role of these effector cells in development of fatal virus-associated malaria.
Figure 5.

Malaria induced high-level Th1 proinflammatory cytokines in acute simian-human immunodeficiency virus (SHIV) coinfection, whereas Th1 cytokine responses were suppressed in chronic SHIV coinfection. A, Plasma interferon (IFN) γ increased in malaria/acute SHIV-coinfected macaques (left), but not malaria-only controls (middle) or malaria/chronic SHIV-coinfected macaques (right). IFNγ was not detectable in acute SHIV-only controls. B, There was no increase in plasma interleukin 4 (IL-4) in malaria/acute SHIV-coinfected macaques except the chloroquine-treated survivor (left) or malaria-only controls (middle) or malaria/chronic SHIV-coinfected macaques (right). C, Overall, there was no significant increase in plasma IL-2 in malaria/acute-SHIV (left), malaria-only (middle), or malaria/chronic-SHIV groups of macaques (right) detected. D, There was no significant difference in the development of malaria–specific Ab responses between malaria/acute-SHIV (left), malaria-only (middle), and malaria/chronic-SHIV groups of macaques (right). Shown were the data of MSP-1-specific immunoglobulin G (IgG) and immunoglobulin M (IgM) Ab levels (optical density [OD] values) derived from ELISA using antimonkey IgG and IgM as second Ab. A lack of differences in total Abs between groups suggests that humoral immune responses did not appear to correlate with clinical outcomes of malaria coinfection.
Figure 6.
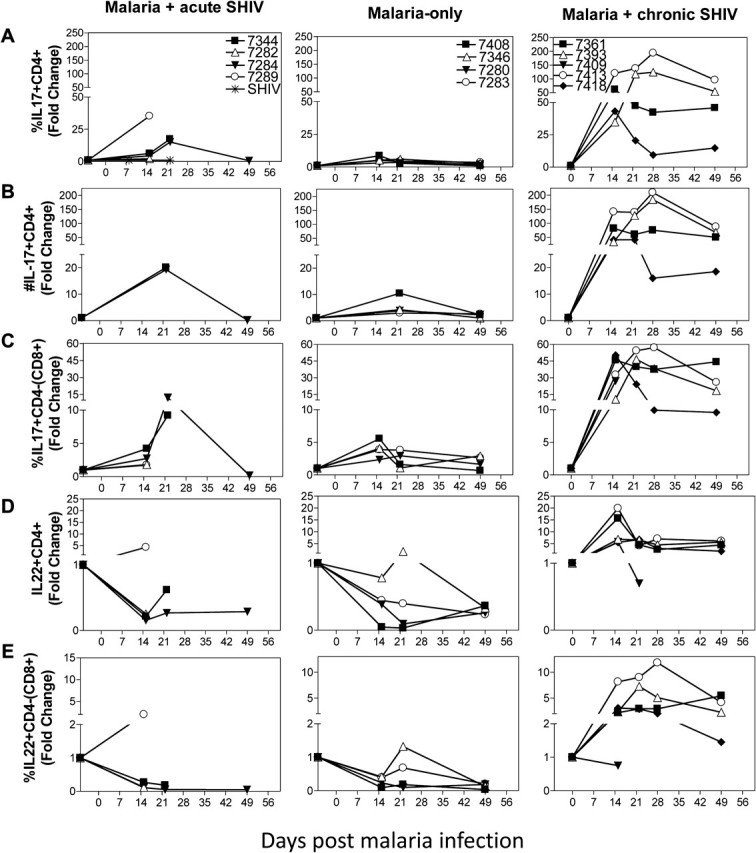
Remarkable expansion of Th17 cells and unique increases in interleukin (IL) 22+ T effector cells coincided with the absence of fatal malaria and enhanced simian-human immunodeficiency virus (SHIV) disease in malaria/chronic SHIV–coinfection. A, IL-17–producing CD4+ T effector cells increased remarkably up to 200-fold during malaria/chronic SHIV–coinfection (right; P = .0167 at day 15, n = 6; P = .0021 at day 22, n = 5), and the magnitude was much greater than that in malaria-only group (P = .0181 at day 15; P = .0173 at day 22, using fold-change data) or malaria/acute SHIV–coinfected group (P = .0294 at day 15; P = .0757 at day 22, using fold-change data). SHIV-only infected animals did not show any detectable IL-17– or IL-22–producing T cells during the acute and chronic SHIV infection ([20], and data not shown). B, The absolute number of IL-17+CD4+ T cells increased < 25-fold in malaria/acute SHIV-coinfected macaques and malaria-only–infected macaques. In contrast, the absolute number increased up to 200-fold in malaria/chronic SHIV-coinfected macaques. C, Malaria/chronic SHIV–coinfected macaques also developed marked increases in IL-17–producing CD3+CD4-(CD8+) T effectors (P = .0181, baseline vs day 15, n = 5; P = .0081 baseline vs day 22, n = 5), and the expansion magnitude was significantly greater than that of the malaria-only group (P = .0218 at day 15, P = .0338 at day 22, using fold change data) and malaria/acute SHIV coinfection (P = .0440 at day 15; P = .1990 at day 22, using fold-change data). D, IL-22–producing CD4+ T cells are increased significantly, ≤ 22-fold from baseline, after malaria infection of chronically SHIV-infected macaques (P = .0038 at day 15, n = 5; P = .035 at day 22, n = 5). In contrast, the frequency of IL-22–producing CD4+ T effector cells actually decreased after malaria infection of acutely SHIV-infected or SHIV-naive macaques. Baseline IL-22–producing T effector cells were low in chronic SHIV-infected macaques, a finding similarly seen in simian immunodeficiency virus (SIV)–infected macaques [26]. No detectable IL-22–producing T cells during the chronic SHIV-only infection without malaria coinfection ([19], and data not shown). E, IL-22–producing CD3+CD4-(CD8+) T cells increased during malaria infection of chronically SHIV-infected macaques (P = .0260 at day 15, n = 5; P = .0097 at day 22, n = 5). Conversely, IL-22–producing CD3+CD4-(CD8+) T effector cells did not significantly increase during malaria/acute SHIV–coinfection and actually decreased during malaria-only infection (P = .0323 at day 15, n = 4). Similar trends of fold changes and P values were seen by comparative analyses of absolute numbers of T effectors.
Figure 7.

Overreactive Th1 responses coincided with progressive simian-human immunodeficiency virus (SHIV) disease (lymphoid depletion/destruction/necrosis) and fatal AIDS-associated malaria, whereas marked expansion of Th17 cells and unique increases in interleukin (IL) 22+ T effector cells with Th1 suppression correlated with uncomplicated malaria and stable SHIV infection. A, Spiking interferon γ–producing CD4+ T-cell levels representing hyperactivation of T cells correlated with CD4+ T cell depletion (top), bursting SHIV replication (middle), and high parasitemia (bottom) in malaria/acute SHIV–coinfected macaques. Increases in SHIV levels were seen earlier than those in detectable expansion of CD4+ T effector cells. This might be because virus replication was enhanced dramatically on cellular activation and that CD4+ T cells underwent activation events before they proliferated to a detectable expansion. Note that at week 3 after malaria coinfection of acutely SHIV-infected macaques, lymphoid depletion and destruction/necrosis occurred (Figure 2). B, High-frequency Th17 cells and IL-22–producing CD4+ T effector cells correlated with stable CD4+ T cell counts (top), subtle change in plasma SHIV RNA (middle), and moderate parasitemia in malaria/chronic SHIV–coinfected macaques. No or few Th1 cells also coincided with the uncomplicated SHIV/malaria diseases (Figures 3). Abbreviation: pRBC, parasitized red blood cells.
Remarkable Expansion of IL-17+ T Effectors, Unique Increases in IL-22+ T Effectors, and Suppression of Overreacting Th1 Effectors Coincided with Stable SHIV Infection and Absence of Fatal Malaria During Malaria/Chronic SHIV Coinfection
Because malaria coinfection of chronic SHIV-infected macaques without AIDS did not lead to fatal malaria or rapid progression of SHIV disease, we examined whether different immune responses correlated with the favorable clinical outcome. Despite moderately enhanced parasitemia, these coinfected survivors developed few CD4+ and CD8+ T effectors producing IFNγ or TNFα de novo, indicating profound suppression of Th1 effectors compared with malaria/acute SHIV-coinfected macaques and malaria-only controls (Figure 4; P < .0001 malaria/chronic-SHIV vs malaria/acute-SHIV; P = .0007 malaria/chronic-SHIV vs malaria-only at day 22). Consistently, plasma IFNγ levels were much lower in malaria/chronic SHIV–coinfected macaques than in malaria/acute SHIV–coinfected macaques (Figure 5). Interestingly, plasma IL-4 and antimalaria Abs were not significantly different between malaria/chronic SHIV coinfected, malaria/acute SHIV coinfected, and malaria-only groups (Figure 5B and 5D).
Surprisingly, IL-17–producing CD4+ T effectors were remarkably increased in malaria/chronic SHIV–coinfected macaques, undergoing ∼200-fold expansion after coinfection (Figure 6A and 6B; P = .0021 baseline vs D22). IL-17–producing CD4- (presumed CD8+) T cells were also increased during malaria/chronic SHIV coinfection (Figure 6C). In addition, malaria/chronic SHIV-coinfected macaques developed unique 10- to 20-fold expansion of IL-22–producing CD4+ T effectors after malaria coinfection (Figure 6D). We also detected increases in IL-22–producing CD3+CD4-(CD8+) T effectors in malaria/chronic SHIV-coinfected macaques (Figure 6E). Importantly, IL-17+ and IL-22+ T effectors were detected without additional in vitro Ag restimulation, implicating previous in vivo activation and maturation. In contrast, malaria/acute SHIV–coinfected macaques exhibited decreases in IL-22+CD4+ T effectors (Figure 6), a down-regulation as described in SIV-infected macaques [27]. In SHIV-only controls, no or very few IL-17+ or IL22+ T cells were detected in acute or chronic SHIV infection [20]. As reported by us and others, IL-17–producing and IL-22–producing T effectors were distinct populations (data not shown and [20, 28]). Notably, the remarkable expansion of IL-17+ and IL-22+ T effectors coincided with controllable parasitemia and absence of fatal malaria, with stable CD4+ T cell counts and transient minor increase in SHIV viremia (Figure 7B). Thus, remarkable expansion of IL-17+ T effectors, unique increases in IL-22+ T effectors, and suppression of overreacting Th1 effectors correlated with absence of fatal malaria in malaria/chronic SHIV–coinfected macaques without AIDS.
DISCUSSION
The proof-of-concept studies in macaque models of malaria/SHIV coinfection allowed us to make the novel observations that AIDS-virus infection stages dictate coinfection-induced immune responses and that distinct Th1 or Th17/IL-22+ immune responses correlate with different outcomes of SHIV disease and virus-associated malaria. Our studies extend human HIV/malaria coinfection studies [4–12, 29, 30] and provide new information regarding pathogenesis of AIDS-virus and malaria coinfection. Our findings also suggest that macaque models of SHIV and P. fragile can be useful for studying pathologic and immunologic events in human patients coinfected with HIV and P. falciparum.
The macaque model of P. fragile/acute SHIV coinfection suggests that malaria coinfection during acute AIDS-virus infection can induce fatal virus-associated malaria, characterized by bursting parasitemia and severe anemia. We temporarily use the term “fatal virus-associated malaria” because SHIV89.6P infection usually does not induce acutely fatal disease within a month after infection. Moreover, earlier reports and this study demonstrated that P. fragile infection of naive macaques often does not induce acutely fatal malaria, although some individuals may develop complications due to unusually high-level parasitemia and associated anemia [13]. The extraordinary effects of acute HIV or SIVmac infection on malaria coinfection in naive individuals have not been reported in humans or nonhuman primates, perhaps because of difficulty to recruit and study newly coinfected patients or because of previous exposures to malaria in the acutely AIDS virus–infected individual [4–12, 31, 32].
Fatal virus-associated malaria and rapidly accelerated AIDS go hand-in-hand during P. fragile/acute SHIV coinfection. CD4+ T-cell depletion appeared more profound in acute coinfected macaques than in SHIV-only controls. More importantly, dramatic lymphoid destruction/depletion and necrosis in lymph nodes and spleens were seen in malaria/acute SHIV coinfected monkeys as early as 21 days postinfection. Significant lymphoid depletion was also seen in gut mucosae. The rapid and profound lymphoid destruction/depletion and necrosis (temporarily defined as AIDS in the study) are attributed to malaria coinfection of acutely SHIV-infected macaques, because such fulminating changes can rarely be seen even in very advanced or end stages of AIDS induced by HIV, SIVmac, or SHIV. Such lymphoid destruction/necrosis might potentially occur in acute malaria/HIV coinfection, as P. fragile causes P. falciparum–like malaria [13, 14], and as SHIV induces simian AIDS–like HIV [15].
The hyperactivation or expansion of IFNγ– and TNFα–producing CD4+ and CD8+ T effectors after malaria coinfection of acutely SHIV-infected macaques appears to be the mechanism underlying accelerated AIDS. This hyperactivation appeared to be driven mainly by malaria, because similar magnitudes of T effectors are detected in malaria-only controls, and SHIV-only control macaques did not develop detectable IFNγ–, IL-17–, or IL-22–producing CD4+ or CD8+ T effectors in either acute (3–4 week) or chronic SHIV infection (Figure 3 and [15]). Notably, because of SHIV coinfection, peak IFNγ plasma levels in malaria/acute SHIV–coinfected macaques were almost 100-fold higher than those in malaria-only controls. Hyperactivation of the immune system can readily transactivate the HIV 5′ LTR [33, 34] for massive viral replication, provide more CD4+ T cell or macrophage sources for productive virus infection, and make infected CD4+ T cells more susceptible to virus-mediated destruction [35, 36]. We did not detect much higher SHIV viral loads 2–3 weeks after acute SHIV/malaria coinfection than those on SHIV-only controls, perhaps because of extremely high turnover of virions in plasma during acute coinfection. The malaria/SHIV-driven hyperimmune activation may contribute to the profound lymphoid destruction/depletion and necrosis.
Because of acutely bursting SHIV replication, malaria-driven hyperactivation/expansion of Th1 effectors and overproduction of proinflammatory cytokines IFNγ and TNFα in malaria/acute SHIV-coinfected macaques might compromise antimalaria immune responses and lead to high-level parasitemia and fatal malaria. The extremely high-level Th1 cytokines in AIDS-virus coinfection may not be protective against malaria, as IFNγ has been shown to have a role in both protection and inflammation/pathology in malaria [24]. Conversely, the suppression or disruption of CD4+ T cell-mediated antimalaria immunity may have taken place as a result of profound lymphoid destruction/depletion and necrosis. Because cell-mediated immunity is believed to occur primarily in the spleen for blood stage malaria [24], profound lymphoid depletion in the spleen would lead to a loss of antimalaria effector function. Furthermore, the disappearance of germinal centers and lymphoid depletion of CD4+ T cells would block early development of antimalaria Ab responses.
One of the novel findings in the current study is that chronically SHIV-infected macaques employed a different defense mechanism to respond to malaria coinfection. In sharp contrast to acutely SHIV-infected macaques, chronically SHIV-infected macaques without AIDS showed marked suppression of Th1 responses after malaria coinfection. Rather, they mounted 50- to 200-fold expansion of IL-17–producing CD4+ T effectors, as well as unique, potent IL-22 responses. Unique expansion of IL-22–producing T effectors with Th1 suppression in malaria/chronic SHIV coinfection is consistent with our recent finding that IFNγ networking pathways can down-regulate IL-22+ T effector cells in tuberculosis [20]. By mounting IL-17+ and IL-22+ T-cell responses and suppressing overreacting Th1 responses to malaria coinfection, chronically SHIV-infected macaques exhibit only transient, low-magnitude increases in viremia, maintain stable CD4+ T cell counts, and avoid rapid progression to AIDS. In addition, marked expansion of Th17 cells and unique increase in IL-22+ T effectors might act in concert with Ab and other immune components to attenuate moderately enhanced malaria in a timely manner, therefore avoiding fatal virus-associated malaria. IL-17 and IL-22 primarily act by recruitment of neutrophils and induction of antimicrobial peptide production by responder cells, and have been shown to have protective roles in infections of various pathogens, including toxoplasmosis [37], candidiasis [38], Listeria [39], Klebsiella pneumonia [40, 41], and others (reviewed in [42]). It would be interesting to see if these responses play a role in resistance to multiple malaria exposures in future studies.
Thus, our proof-of-concept study demonstrates for the first time to our knowledge that virus infection status and distinct Th1 or Th17/IL-22 responses after malaria coinfection of AIDS virus–infected individuals correlate with different clinical outcomes. As far as we know, this is also the first illustration of the dichotomy of Th1 and Th17/IL-22 responses during infection/coinfection of higher primates. The findings may be potentially useful in the development of AIDS and malaria vaccines and immunotherapeutics.
Notes
Acknowledgments.
We would like to thank B. Paige, J. Graves, J. Chen, and Dr. K. Hagen for technical assistance with flow cytometry. We also thank F. Villinger for technical assistance with qRT-PCR analysis of SHIV levels. We thank BRL staff for animal care and assistance with obtaining samples.
Author Contributions.
B. R.-P. designed and performed experiments, analyzed data, and wrote the paper; Z. A. designed and performed experiments; D. H., L. Y., R. W., and Y. W. performed experiments; C. Y. C. wrote the paper; W. E. C. provided reagents; Z. W. C. designed experiments and wrote the paper.
Financial support.
This work was supported by the National Institutes of Health (R01 HL64560 and R01 RR13601, both to Z. W. C).
Potential conflicts of interest.
All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Miller LH, Good MF, Milon G. Malaria pathogenesis. Science. 1994;264:1878–83. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- 2.Slutsker L, Marston BJ. HIV and malaria: Interactions and implications. Curr Opin Infect Dis. 2007;20:3–10. doi: 10.1097/QCO.0b013e328012c5cd. [DOI] [PubMed] [Google Scholar]
- 3.Abu-Raddad LJ, Patnaik P, Kublin JG. Dual infection with HIV and malaria fuels the spread of both diseases in sub-Saharan Africa. Science. 2006;314:1603–6. doi: 10.1126/science.1132338. [DOI] [PubMed] [Google Scholar]
- 4.Butcher GA. T-cell depletion and immunity to malaria in HIV-infections. Parasitology. 2005;130:141–50. doi: 10.1017/s003118200400650x. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman IF, Jere CS, Taylor TE, et al. The effect of Plasmodium falciparum malaria on HIV-1 RNA blood plasma concentration. AIDS. 1999;13:487–94. doi: 10.1097/00002030-199903110-00007. [DOI] [PubMed] [Google Scholar]
- 6.Kublin JG, Patnaik P, Jere CS, et al. Effect of Plasmodium falciparum malaria on concentration of HIV-1-RNA in the blood of adults in rural Malawi: A prospective cohort study. Lancet. 2005;365:233–40. doi: 10.1016/S0140-6736(05)17743-5. [DOI] [PubMed] [Google Scholar]
- 7.Grimwade K, French N, Mbatha DD, Zungu DD, Dedicoat M, Gilks CF. HIV infection as a cofactor for severe falciparum malaria in adults living in a region of unstable malaria transmission in South Africa. AIDS. 2004;18:547–54. doi: 10.1097/00002030-200402200-00023. [DOI] [PubMed] [Google Scholar]
- 8.Patnaik P, Jere CS, Miller WC, et al. Effects of HIV-1 serostatus, HIV-1 RNA concentration, and CD4 cell count on the incidence of malaria infection in a cohort of adults in rural Malawi. J Infect Dis. 2005;192:984–91. doi: 10.1086/432730. [DOI] [PubMed] [Google Scholar]
- 9.Cohen C, Karstaedt A, Frean J, et al. Increased prevalence of severe malaria in HIV-infected adults in South Africa. Clin Infect Dis. 2005;41:1631–7. doi: 10.1086/498023. [DOI] [PubMed] [Google Scholar]
- 10.French N, Nakiyingi J, Lugada E, Watera C, Whitworth JA, Gilks CF. Increasing rates of malarial fever with deteriorating immune status in HIV-1-infected Ugandan adults. AIDS. 2001;15:899–906. doi: 10.1097/00002030-200105040-00010. [DOI] [PubMed] [Google Scholar]
- 11.Whitworth J, Morgan D, Quigley M, et al. Effect of HIV-1 and increasing immunosuppression on malaria parasitaemia and clinical episodes in adults in rural Uganda: A cohort study. Lancet. 2000;356:1051–6. doi: 10.1016/S0140-6736(00)02727-6. [DOI] [PubMed] [Google Scholar]
- 12.Laufer MK, van Oosterhout JJ, Thesing PC, et al. Impact of HIV-associated immunosuppression on malaria infection and disease in Malawi. J Infect Dis. 2006;193:872–8. doi: 10.1086/500245. [DOI] [PubMed] [Google Scholar]
- 13.Collins WE, Warren M, Sullivan JS, et al. Studies on sporozoite-induced and chronic infections with Plasmodium fragile in Macaca mulatta and New World monkeys. J Parasitol. 2006;92:1019–26. doi: 10.1645/GE-848R.1. [DOI] [PubMed] [Google Scholar]
- 14.Fujioka H, Millet P, Maeno Y, et al. A nonhuman primate model for human cerebral malaria: Rhesus monkeys experimentally infected with Plasmodium fragile. Exp Parasitol. 1994;78:371–6. doi: 10.1006/expr.1994.1040. [DOI] [PubMed] [Google Scholar]
- 15.Ali Z, Yan L, Plagman N, et al. Gammadelta T cell immune manipulation during chronic phase of simian-human immunodeficiency virus infection [corrected] confers immunological benefits. J Immunol. 2009;183:5407–17. doi: 10.4049/jimmunol.0901760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moll K, Ljungtrom I, Perlmann H, Scherf A, Wahlgren M, editors. Methods in malaria research Malaria Research and Reference Reagent Resource Center/ American Type Culture Collection. 5th ed. Manassas, VA: 2004. pp. 141–3. [Google Scholar]
- 17.Amara RR, Villinger F, Altmen JD, et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science. 2001;292:69–74. doi: 10.1126/science.1058915. [DOI] [PubMed] [Google Scholar]
- 18.Cline AN, Bess JW, Piatak M, Jr., Lifson JD. Highly sensitive SIV plasma viral load assay: practical considerations, realistic performance expectations, and application to reverse engineering of vaccines for AIDS. J Med Primatol. 2005;34:303–12. doi: 10.1111/j.1600-0684.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 19.Huang D, Chen CY, Ali Z, et al. Antigen-specific VgammacVdeltad T effector cells confer homeostatic protection against pneumonic plaque lesions. Proc Natl Acad Sci U S A. 2009;106:7553–8. doi: 10.1073/pnas.0811250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yao S, Huang D, Chen CY, et al. Differentiation, distribution and gammadelta T cell-driven regulation of IL-22-producing T cells in tuberculosis. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1000789. e1000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Y, Zhou D, Qiu L, et al. Adaptive immune response of Vgamma2Vdelta2 T cells during mycobacterial infections. Science. 2002;295:2255–8. doi: 10.1126/science.1068819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimada T, Suzuki H, Motohara M, et al. Comparative histopathological studies in the early stages of acute pathogenic and nonpathogenic SHIV-infected lymphoid organs. Virology. 2003;306:334–46. doi: 10.1016/s0042-6822(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 23.van Grevenynghe J, Halwani R, Chomont N, et al. Lymph node architecture collapse and consequent modulation of FOXO3a pathway on memory T- and B- cells during HIV infection. Semin Immunol. 2008;20:196–203. doi: 10.1016/j.smim.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Stephens R, ALbano FR, Quin S, et al. Malaria-specific transgenic CD4(+) T cells protect immunodeficient mice from lethal infection and demonstrate requirement for a protective threshold of antibody production for parasite clearance. Blood. 2005;106:1676–84. doi: 10.1182/blood-2004-10-4047. [DOI] [PubMed] [Google Scholar]
- 25.Good MF, Xu H, Wykes M, Engwerda CR. Development and regulation of cell-mediated immune responses to the blood stages of malaria: implications for vaccine research. Annu Rev Immunol. 2005;23:69–99. doi: 10.1146/annurev.immunol.23.021704.115638. [DOI] [PubMed] [Google Scholar]
- 26.Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci U S A. 1992;89:3200–3. doi: 10.1073/pnas.89.8.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raffatellu M, Santos RL, Verhoeven DE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–8. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10:864–71. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 29.Renia L, Potter SM. Co-infection of malaria with HIV: an immunological perspective. Parasite Immunol. 2006;28:589–95. doi: 10.1111/j.1365-3024.2006.00903.x. [DOI] [PubMed] [Google Scholar]
- 30.Troye-Blomberg M, Berzins K. Immune interactions in malaria co-infections with other endemic infectious diseases: implications for the development of improved disease interventions. Microbes Infect. 2008;10:948–52. doi: 10.1016/j.micinf.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 31.Hisaeda H, Tetsutani K, Imai T, et al. Malaria parasites require TLR9 signaling for immune evasion by activating regulatory T cells. J Immunol. 2008;180:2496–503. doi: 10.4049/jimmunol.180.4.2496. [DOI] [PubMed] [Google Scholar]
- 32.Koehler JW, Bolton M, Rollins A, et al. Altered immune responses in rhesus macaques co-infected with SIV and Plasmodium cynomolgi: an animal model for coincident AIDS and relapsing malaria. PLoS One. 2009;4 doi: 10.1371/journal.pone.0007139. e7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao L, Owen SM, Rudolph DL, Lal RB, Lal AA. Plasmodium falciparum antigen-induced human immunodeficiency virus type 1 replication is mediated through induction of tumor necrosis factor-alpha. J Infect Dis. 1998;177:437–45. doi: 10.1086/514212. [DOI] [PubMed] [Google Scholar]
- 34.Nti BK, Slingluff JL, Keller CC, et al. Stage-specific effects of Plasmodium falciparum-derived hemozoin on blood mononuclear cell TNF-alpha regulation and viral replication. AIDS. 2005;19:1771–80. doi: 10.1097/01.aids.0000189862.44311.36. [DOI] [PubMed] [Google Scholar]
- 35.Margolick JB, Volkman DJ, Folks TM, Fauci AS. Amplification of HTLV-III/LAV infection by antigen-induced activation of T cells and direct suppression by virus of lymphocyte blastogenic responses. J Immunol. 1987;138:1719–23. [PubMed] [Google Scholar]
- 36.Douek DC, Roederer M, Koup RA. Emerging concepts in the immunopathogenesis of AIDS. Annu Rev Med. 2009;60:471–84. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly MN, Kolls JK, Happel K, et al. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infect Immun. 2005;73:617–21. doi: 10.1128/IAI.73.1.617-621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conti HR, Shen F, Nayyar N, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamada S, Umemura M, Shiono T, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. 2008;181:3456–63. doi: 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–9. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 41.Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Connor W, Jr., Zenewicz LA, Flavell RA. The dual nature of T(H)17 cells: shifting the focus to function. Nat Immunol. 2010;11:471–6. doi: 10.1038/ni.1882. [DOI] [PubMed] [Google Scholar]