

Abstract
Lactic acid, an attractive, renewable chemical for production of biobased plastics (polylactic acid, PLA), is currently commercially produced from food-based sources of sugar. Pure optical isomers of lactate needed for PLA are typically produced by microbial fermentation of sugars at temperatures below 40 °C. Bacillus coagulans produces L(+)-lactate as a primary fermentation product and grows optimally at 50 °C and pH 5, conditions that are optimal for activity of commercial fungal cellulases. This strain was engineered to produce D(−)-lactate by deleting the native ldh (L-lactate dehydrogenase) and alsS (acetolactate synthase) genes to impede anaerobic growth, followed by growth-based selection to isolate suppressor mutants that restored growth. One of these, strain QZ19, produced about 90 g L-1 of optically pure D(−)-lactic acid from glucose in < 48 h. The new source of D-lactate dehydrogenase (D-LDH) activity was identified as a mutated form of glycerol dehydrogenase (GlyDH; D121N and F245S) that was produced at high levels as a result of a third mutation (insertion sequence). Although the native GlyDH had no detectable activity with pyruvate, the mutated GlyDH had a D-LDH specific activity of 0.8 μmoles min-1 (mg protein)-1. By using QZ19 for simultaneous saccharification and fermentation of cellulose to D-lactate (50 °C and pH 5.0), the cellulase usage could be reduced to 1/3 that required for equivalent fermentations by mesophilic lactic acid bacteria. Together, the native B. coagulans and the QZ19 derivative can be used to produce either L(+) or D(−) optical isomers of lactic acid (respectively) at high titers and yields from nonfood carbohydrates.
Keywords: bioplastics, biomass conversion
Petroleum serves as the primary source of automotive fuel and as the dominating feedstock for organic chemicals and plastics. The finite nature of the petroleum reserves and the negative environmental impact of petroleum use have shifted attention towards alternatives from renewable feedstocks (1–4). Fermentation of carbohydrates has been shown to produce short chain hydroxy acids as well as other chemicals that can be polymerized into plastics and replace petroleum (5). Commercial production of lactic acid using pure bacterial cultures started as early as 1895 (6) and current annual production is > 300,000 metric tons. Although lactic acid is primarily used by food and pharmaceutical industries, its application in production of polylactic acid biopolymers (PLA) is expected to exceed other uses provided the cost of PLA production can be lowered (7, 8).
Lactic acid is condensed into lactide dimers, purified, and polymerized into a thermoplastic (7, 9). Blending D(−)- and L(+)- lactic acid polymers provides substantial control on the physical and thermochemical properties, and the rate of biodegradation (9). Although lactic acid can be synthesized from petroleum, chemical synthesis produces a mixture of isomers that are not suitable for PLA. Optically pure lactic acid required for PLA synthesis can be readily produced by microbial fermentation (8). L(+)-lactic acid is produced commercially by lactic acid bacteria such as Lactobacillus, Lactococcus, etc., at high yield and titers from glucose and sucrose at temperatures between 30 °C and 40 °C (8, 10). Derivatives of Escherichia coli are the only known commercial D(−)-lactic acid producers (11, 12) and these also operate optimally at 40 °C or lower.
Alternative sources of fermentable sugars such as lignocellulosic biomass and improved microbial biocatalysts are needed to eliminate the use of food carbohydrates (glucose, sucrose) for lactic acid production (13). With cellulose as a feedstock, however, commercial fungal cellulases represent a significant process cost. This cost could be reduced by the development of thermotolerant biocatalysts that effectively ferment under conditions that are optimal (pH 5.0, 50 °C) for fungal cellulases (14, 15).
Lactic acid biocatalysts used by industry also metabolize pentose sugars (from hemicellulose) by the phosphoketolase pathway, preventing efficient conversion of these sugars to lactic acid. Lactic acid produced from pentoses using this pathway is contaminated with an equimolar amount of acetic acid (Fig. 1) (8, 16–18). Attempts to improve the xylose fermentation properties of these lactic acid bacteria have met with limited success (18, 19). Although all the pentoses in hemicellulose are efficiently fermented by the E. coli derivatives to D(−)- or L(+)- lactic acid through the pentose-phosphate pathway, the temperature and pH tolerances of this microbial biocatalyst are insufficient to permit cellulosic fermentations at the conditions that are optimal (50 °C and pH 5.0) for commercial cellulases (20).
Fig. 1.

Anaerobic metabolic pathways of glucose and xylose in B. coagulans strain P4-102B. Thickness of the arrow indicates glucose or xylose flux to L-lactate (broken line) as the preferred route during anaerobic growth of wild type and to D-lactate in strain QZ19 constructed in this study. Broken arrows (L-LDH and ALS) represent mutations in the two pathways introduced in this study. Xylose fermentation pathway in lactic acid bacteria, such as L. lactis utilizing phosphoketolase pathway resulting in an equimolar lactate and acetate is also presented for comparison. See Patel, et al. for details on pentose fermentation by B. coagulans and L. lactis (17).
Bacillus coagulans has many desirable properties for the fermentation of lignocellulosic sugars into lactic acid. This bacterium ferments both hexoses and pentoses to L(+)-lactic acid using the highly efficient pentose-phosphate pathway (Fig. 1) at 50–55 °C and pH 5.0, conditions that are optimal for commercial fungal cellulases (15, 17, 21). Native strains produce optically pure L(+)-lactic acid at concentrations as high as 180 g L-1 in fed-batch fermentations from either glucose or xylose, and perform well during simultaneous saccharification and fermentation (SSF) of cellulose using fungal cellulases (21, 22). This match in optima both for the fermentation of B. coagulans and for fungal cellulase activity allowed a fourfold reduction in cellulase usage in comparison to SSF with mesophilic bacterial biocatalysts (21). B. coagulans also grows and ferments sugars in mineral salts medium with inexpensive corn steep liquor (0.25%, wt/vol) supplementation in contrast to lactic acid bacteria that require complex nutrients (8, 15, 18). Although B. coagulans has excellent potential as a biocatalyst for the conversion of cellulose to optically pure L(+)-lactic acid, an equivalent microbe for production of D(−)-isomer of lactic acid has not been previously described.
In this communication, a general method for genetic engineering of B. coagulans is described because genetic recalcitrance is a major limiting factor in the use of this bacterium at industrial level. A derivative of B. coagulans strain P4-102B obtained after mutagenesis and adaptive evolution was found to use a mutated form of glycerol dehydrogenase to produce D(−)-lactic acid. With this strain (QZ19), over 1 M D(−)-lactic acid can be produced in less than 48 h at 50 °C and pH 5.0.
Results and Discussion
Lactate Dehydrogenase Genes in B. coagulans.
B. coagulans strain P4-102B typically produces L-lactate with low (15, 17) or undetectable level of the D-isomer (Table 1). Using the annotated genome sequence of a related B. coagulans strain 36D1 (GenBank Accession number, CP003056) as a guide, homologous genes encoding both L-lactate dehydrogenase (ldh) and D-lactate dehydrogenase (ldhA) activities in strain P4-102B were amplified by PCR and confirmed by sequencing. The gene encoding D-lactate dehydrogenase in strain P4-102B was also identified by its ability to suppress anaerobic growth defect of an E. coli mutant (ldhA, pflB) followed by sequencing the corresponding cloned ldhA gene. With appropriate deletions to block competing pathways (Fig. 1), this organism has the potential to produce either D(−)-lactate or L(+)-lactate using only native genes.
Table 1.
Fermentation profiles of B. coagulans derivatives on the path to D(−)-lactic acid production at 50 °C
| Strain | Genotype | Culture | Cell | Glucose | Qs* | Product (mM) | Yield † | |||||||
| pH | Yield | Consumed | Lactate | Pyruvate | Acetate | Succ | Formate | Ethanol | Lactate | Total | ||||
| (mM) | L(+)- | D(−) | ||||||||||||
| P4-102B | wild type | 5.0 | 3.0 | 144.3 | 5.7 | 255.6 | UD | UD | 5.7 | 0.6 | UD | 10.5 | 0.89 | 1.00 |
| 7.0 | 7.1 | 188.6 | 18.4 | 336.4 | UD | UD | 15.9 | 0.4 | UD | 4.6 | 0.89 | 0.96 | ||
| QZ4 ‡ | Δldh | 5.0 | 1.8 | 53.8 | 0.8 | UD | 1.0 | UD | UD | 1.0 | 6.5 | 20.3 | 0.01 | 1.03 |
| 7.0 | 8.1 | 226.0 | 4.3 | UD | 9.7 | UD | 42.3 | 6.7 | 90.6 | 224.5 | 0.02 | 1.17 | ||
| QZ5 | Δldh Δ alsS | 5.0 | 2.5 | 32.8 | 0.5 | UD | 9.8 | 4.5 | 4.6 | 0.1 | 25.3 | 25.6 | 0.15 | 0.72 |
| 7.0 | 5.8 | 152.5 | 6.8 | UD | 12.8 | 10.1 | 95.4 | 2.1 | 154.6 | 164.5 | 0.04 | 0.94 | ||
| QZ13 | evolved § | 5.0 | 4.3 | 128.8 | 2.4 | UD | 212.4 | 6.1 | 2.7 | 0.6 | 24.3 | 23.4 | 0.82 | 0.95 |
| 7.0 | 6.0 | 162.6 | 4.0 | UD | 219.8 | 9.3 | 14.8 | 0.4 | 66.9 | 58.1 | 0.68 | 0.93 | ||
| QZ14 | evolved § | 7.0 | ND ¶ | 265.7 | 5.4 | UD | 468.4 | 1.6 | UD | 1.3 | 21.5 | 28.4 | 0.88 | 0.93 |
| QZ15 | evolved § | 7.0 | ND ¶ | 562.6 | 17.0 | UD | 928.2 | UD | 22.6 | 8.1 | UD | 97.5 | 0.83 | 0.94 |
| QZ19 | evolved § | 5.0 ∥ | ND | 580 | 12.5 | UD | 1,108.1 | UD | 10.2 | UD | UD | 40.9 | 0.96 | 1.00 |
| 7.0 | ND ¶ | 590.0 | 19.4 | UD | 993.0 | UD | 45.3 | 5.7 | UD | 84.2 | 0.84 | 0.96 | ||
All fermentations were in LB medium with glucose and the reported values were after 72 h, unless indicated otherwise. Cell yield is expressed in OD420 nm.
*Qs, rate of glucose consumption—mmoles glucose consumed L-1 h-1
†Product yield is presented as a fraction of theoretical yield from glucose (for lactate, the theoretical yield is two per glucose).
‡Strain QZ4 also produced acetoin and 2,3-butanediol; pH 5.0 culture, 44.5 mM 2,3-butanediol; pH 7.0 culture, 31.6 mM acetoin, and 93.1 mM 2,3-butanediol. These two products were not detected in the broths from other cultures.
§Evolved, various stages in evolution (Fig. S2).
¶Due to the presence of CaCO3 in the medium, the cell density of these cultures was not determined.
∥Fermentation pH was maintained by addition of Ca(OH)2 and cell density was not determined due to the presence of Ca-salts. UD—undetectable, less than 0.5 mM; Succ, succinate.
Deletion of Native L-(+)-Lactate Dehydrogenase (ldh) and Acetolactate Synthase (alsS) Genes.
B. coagulans strain P4-102B produced L-lactic acid as the primary fermentation product at pH 5.0 (Table 1; Fig. S1) together with small amounts of acetate and ethanol. Deletion of the ldh gene encoding L(+)-lactate dehydrogenase (L-LDH) (strain QZ4) using methods developed for B. coagulans (SI Text) eliminated the primary route for NADH oxidation, slowing growth and sugar metabolism. However, production of 2,3-butanediol was increased dramatically with only a small increase in D(−)-lactate (Table 1; Fig. S1). At pH 7.0, the primary product of fermentation was ethanol as seen previously with an ldh mutant of a related strain 36D1 (23).
Strain QZ4 was further modified by deleting acetolactate synthase (alsS), essential for 2,3-butanediol production (Fig. 1). The resulting strain (QZ5) metabolized glucose rapidly at pH 7.0 with ethanol, acetate, and formate as primary products (Table 1). In this strain and at this pH, pyruvate flux to D-lactate was only about 5% of the total. However, strain QZ5 did not grow anaerobically when cultured at pH 5.0 in the same medium. Fermentation profiles of strain QZ5 at pH 5.0 were determined by starting cultures with air in the gas phase to support growth. Depletion of O2 in the medium, due to increasing cell density, initiated and maintained anaerobic metabolism of the bacterium. The specific rate of glucose consumption by strain QZ5 at pH 5.0 during the O2-limited phase (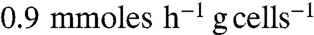 ) was about 15% of the pH 7.0 culture (
) was about 15% of the pH 7.0 culture (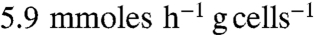 ) under similar condition. The sixfold lower specific glucose consumption rate and formate titer (Table 1) suggest that pyruvate formate-lyase (PFL) activity is limiting anaerobic growth of strain QZ5 at pH 5.0. Strain QZ5 also had higher D-LDH activity and a corresponding increase in ldhA mRNA as compared to strain QZ4 (Table 2). However, the D(−)-lactate titer was only about 10 mM in the QZ5 broth irrespective of the culture pH with yields of < 0.15 lactate per glucose metabolized (theoretical yield of 2 per glucose).
) under similar condition. The sixfold lower specific glucose consumption rate and formate titer (Table 1) suggest that pyruvate formate-lyase (PFL) activity is limiting anaerobic growth of strain QZ5 at pH 5.0. Strain QZ5 also had higher D-LDH activity and a corresponding increase in ldhA mRNA as compared to strain QZ4 (Table 2). However, the D(−)-lactate titer was only about 10 mM in the QZ5 broth irrespective of the culture pH with yields of < 0.15 lactate per glucose metabolized (theoretical yield of 2 per glucose).
Table 2.
Enzyme and mRNA levels of B. coagulans strains during evolution for high level of D-lactate production
| Strain | Genotype | mRNA level | Enzyme activity | Ratio | |||
| ldh | gldA | ldhA | GlyDH | D-LDH | D-LDH/GlyDH | ||
| P4-102B | wild type | 61.90 | 1.13 | 0.12 | 0.19 | < 0.001 | < 0.005 |
| QZ4 | Δldh | 0.02 | 1.29 | 1.08 | 0.13 | 0.002 | 0.015 |
| QZ5 | Δldh, ΔalsS | UD | 1.51 | 4.87 | 0.13 | 0.007 | 0.050 |
| QZ13 | evolution | UD | 148.50 | 3.62 | 7.06 | 0.028 | 0.003 |
| QZ14 | evolution | UD | 175.20 | 3.77 | 7.12 | 0.021 | 0.003 |
| QZ15 | evolution | UD | 176.70 | 5.12 | 5.60 | 0.112 | 0.020 |
| QZ19 | evolution | UD | 243.50 | 6.12 | 1.01 | 0.221 | 0.219 |
All cultures were grown in pH-controlled fermentations in LB + glucose (0.167 M) at pH 5.0. mRNA levels are ng mL-1 of total RNA. ldh and ldhA represent the mRNA encoding L-LDH and D-LDH, respectively. gldA, glycerol dehydrogenase mRNA.
Enzyme activities were determined in cell extracts and expressed as μmoles min-1 mg protein-1. UD—Undetectable, < 0.01 ng mL-1 of total RNA.
Because the double mutant produced significant amounts of formate and other products of PFL activity, a triple mutant lacking PFL was constructed. However, this triple mutant lacking L-LDH, ALS, and PFL activities failed to grow anaerobically under all conditions tested and was not investigated further.
Growth-Based Selection Increased D(−)-Lactate Production.
Sugar metabolism, ATP production, and anaerobic growth are apparently constrained in strain QZ5, especially at pH 5.0, by inadequate routes for NADH oxidation (Table 1; Fig. 1). Increasing the level of D(−)-lactate produced by strain QZ5 is expected to restore anaerobic growth of the double mutant to a level similar to that of the parent during L(+)-lactate production. Growth-based metabolic evolution is a powerful tool for selection of advantageous mutations (24) but this requires design of appropriate selection. Because strain QZ5 produced low level of PFL activity at pH 5.0, as determined by the relative concentration of formate in the broth (Table 1), metabolic evolution of this strain for anaerobic growth at this pH offers a route to coselect increased production of D-lactate.
Metabolic evolution of strain QZ5 at pH 5.0 for higher cell yield in LB + glucose fermentation yielded a derivative with improved growth after 113 d of selection. Cell yield of this strain (QZ13) was higher than the wild type, with a 20-fold increase in D-(-)-lactate production (Table 1; Fig. S2). To further increase the D-lactate yield and titer, strain QZ13 was further evolved at pH 7.0 in LB + glucose fermentations with CaCO3. Addition of CaCO3 to the medium was previously shown to overcome lactate inhibition of B. coagulans fermentation and permit titers approaching 2 M in fed-batch fermentations (22). Because solid CaCO3 buffered the medium better at pH 7.0 than at pH 5.0, further metabolic evolutions were at this higher pH. After 42 d of selection, strain QZ14 was isolated and tested. With strain QZ14, D(−)-lactate represented over 90% of the total fermentation products with a yield of 88% of the fermented glucose (Table 1).
Strain QZ15 was isolated after an additional 60 d of serial transfers at pH 7.0 with increasing glucose concentrations (Fig. S2 and S3). The D(−)-lactic acid titer of strain QZ15 at pH 7.0 was close to 1 M in 72 h (Table 1). Continued metabolic evolution of strain QZ15 using 100 g L-1 (0.56 M) glucose led to further increase in glucose flux to lactic acid. Strain QZ19 was isolated after 63 d of transfer (Fig. S3) and this strain produced about 1.0 M D(−)-lactate in less than 48 h at pH 7.0 (Table 1). Fermentation of glucose by strain QZ19 at pH 5.0 started after a short lag with a titer of 1.1 M lactate in 72 h (Fig. 2). Formate was not detected in the fermentation broth of strains QZ15 and QZ19 producing high levels of lactate. Formate is an indicator of PFL activity because these strains lack formate hydrogen-lyase activity. The small amount of ethanol and acetate (< 5% of glucose carbon) produced by strain QZ19 is probably derived from acetyl-CoA produced by pyruvate dehydrogenase complex (25).
Fig. 2.

Fermentation profile of D-lactate producing B. coagulans strain QZ19. Glucose concentration for the fermentation at 50 °C was 110 g L-1 (0.6 M) and the medium pH at 5.0 was maintained by addition of Ca(OH)2.
These results show that deletion of the ldh and alsS genes combined with metabolic evolution led to alteration of the primary fermentation product of B. coagulans from L(+)-lactate to D(−)-lactate. Although the pfl genes are still present and transcribed in strain QZ19 at levels that are comparable to that of strain QZ4 (about 1.2 ng mL-1 of total RNA), glucose flux through PFL was minimal as seen by the absence of formate in the fermentation broth. This redirection of glucose carbon is apparently a consequence of increasing metabolic flux to D-lactate that supplanted any pyruvate flux through PFL to acetyl-CoA during growth-based selection (Table 1).
Fermentation Characteristics of D-Lactate Producing Strain QZ19.
Strain QZ19 produced close to 2M D-lactate in a fed-batch fermentation of glucose at pH 5.0 (Fig. S4). In such a fed-batch fermentation, the first 100 g L-1 (0.56 M) of glucose was fermented in about 50 h and fermentation of the second 100 g L-1 (0.56 M) of glucose required an additional 72 h, although the rate of lactate production was linear during this phase. Strain QZ19 also fermented xylose to D-lactic acid after a short lag. Approximately 80 g L-1 (0.53 M) of xylose was converted to D(−)-lactate in 72 h at 90% yield by weight (Fig. S5).
Simultaneous Saccharification and Fermentation (SSF) of Cellulose to D-Lactic Acid by Strain QZ19.
One of the advantages of using B. coagulans as a microbial biocatalyst for lactic acid production is to lower the cellulase enzyme loading in SSF of cellulose to lactic acid due to its higher operating temperature and lower pH that match the optimum for commercial cellulase activity (50 °C and pH 5.0) (14, 15). Strain QZ19 reached the highest volumetric productivity for D-lactate with crystalline cellulose as feedstock at about 7.5 FPU (g cellulose)-1 (Fig. S6) With this enzyme loading, the productivity of a typical lactic acid bacterium Lactococcus lactis at 40 °C was only 1/3 that of strain QZ19. These results are in agreement with previous studies on lower cellulase requirements for optimal L-lactate production by unmodified B. coagulans compared to other lactic acid bacteria (21). Strain QZ19 is better suited for SSF of cellulose to D-lactate and requires significantly lower level of cellulase for cost-effective metabolism of this nonfood carbohydrate.
Identification of Glycerol Dehydrogenase as the Source of D(−)-LDH Activity in Strain QZ19.
Cell-free extracts of strain QZ19 had D(−)-lactate dehydrogenase activity (0.2 unit per mg protein) while the wild type B. coagulans had no detectable D-LDH activity (Table 2). An observed 30-fold increase in D-LDH activity in strain QZ19 compared to strain QZ5 did not correlate with the 1.3-fold increase in the mRNA of ldhA gene identified as encoding D-LDH. To identify potential mutation(s) in the D-LDH protein that could be responsible for the unusually high D-lactate dehydrogenase activity, the ldhA and flanking DNA from strains P4-102B and QZ19 were sequenced. However, both sequences were identical indicating that ldhA encoded D-LDH is not responsible for the large increase in D(−)-lactate production in strain QZ19. Only a single copy of ldhA was also found in the chromosome of strains QZ19 and P4-102B eliminating gene duplication as a potential basis for D(−)-lactate production. This conclusion was further supported by comparable ldhA mRNA levels of strain QZ19 and its preevolution parent, strain QZ5 (Table 2). Higher level of ldhA mRNA in these strains compared to the wild type could be responsible for the small amount of D(−)-lactate produced by strain QZ5 and other evolved strains, but does not explain the high D-lactate titer and yield observed with strain QZ19. The highest level of ldhA mRNA (D-LDH) in strain QZ19 was only about 10% of the level of ldh mRNA encoding L(+)-LDH, the primary fermentation route in the parent (Fig. 1; Table 2). These results suggest that another enzyme with D-LDH activity is present in strain QZ19 contributing to high D-lactate titer.
Cell-free extracts from strain QZ13 through QZ19 contained an abundant protein (apparent molecular mass of 37 KDa) that was absent in the wild type strain P4-102B and in the preevolution strain QZ5 (Fig. 3). This protein was calculated to represent 11% of the total proteins in the extract of these strains. A protein with D-LDH activity was purified from strain QZ19. The size of this purified protein, 37 KDa, was comparable to the additional protein found in the evolved strains. After trypsin digestion of the protein with D-LDH activity, the fragments were identified after separation by liquid chromatography and analysis by tandem mass spectrometry. Of the 22 peptides generated, 9 were identical to predicted tryptic peptides from a protein in B. coagulans strain 36D1 genome (Bcoa_1919; Accession number AEP01106.1) that was identified as glycerol dehydrogenase (gldA) by comparative sequence analysis (26) (Fig. S7). Based on the gldA gene sequence of B. coagulans strain 36D1, the gldA gene of strains P4-102B and QZ19 was amplified, cloned, and sequenced. The gldA gene from strain QZ19 had two mutations compared to the native gene: G361A and T734C (“A” in ATG as +1). These two mutations led to amino acid changes, D121N and F245S and the allele is designated as gldA101 and the protein as GlyDH*. In addition to these two mutations in the coding region, a 1,821 bp DNA fragment was found to be inserted upstream of −62 (“A” in ATG as +1) of the gldA gene in QZ19 (Fig. 4). Two ORFs were identified within this insert (103 and 232 amino acids in length), both with similarity to a putative transposase (ISLhe15) from Lactobacillus helveticus strain H10 (25% identity and 48% similarity) (27).
Fig. 3.

SDS-PAGE of proteins from the crude extracts of various B. coagulans strains on the path to D-lactic acid producing strain QZ19. Cultures grown in LB + glucose (0.17M) in pH-controlled (5.0) fermentations were harvested during mid- to late-exponential phase of growth and cell extracts were prepared and analyzed. Left lane, molecular weight standards. Numbers on the left represent the corresponding molecular mass of the proteins in kilodaltons. Right lane, pure GlyDH* from strain QZ19. Only the SDS-PAGE gel in the region of 35–50 KDa is presented.
Fig. 4.

Insertion of DNA in the upstream region of gldA gene of B. coagulans strain QZ13 that enhanced gldA expression. An insert DNA (1,821 bp) indicated in italics starts after “C” at −62 of the gldA gene (“A” in ATG as +1) and continues upstream. An 11 base direct repeat (enclosed within the oval) is present at the ends of the insertion, one at the 5-end of the insert and the second at the 5-end of the gldA gene immediately after the 3-end of the insert. ORF1, a putative DeoR family transcriptional regulator; ORF2, and ORF3 in the insert DNA, putative transposase; gldA, glycerol dehydrogenase. Putative −10, −35 and Shine-Dalgarno sequences (SD) are indicated.
The purified protein from strain QZ19 corresponding to the 37 KDa protein in the SDS-PAGE gel had both glycerol oxidation and pyruvate reduction activities. The GDH activity (glycerol oxidation to dihydroxyacetone) was 1.2 units (μmole min-1) per mg protein and the pyruvate reduction to D-lactate activity was 0.8 unit per mg protein. The native enzyme purified from recombinant E. coli had a GlyDH activity of 7.1 units and an LDH activity of 0.01 unit per mg protein. These results suggest that a GlyDH in the B. coagulans genome acquired D-LDH activity through two mutations during the metabolic evolution of strain QZ19. High level of expression of this protein apparently resulted from the upstream transposon insertion.
GlyDH* from Strain QZ19 Produced D-LDH Activity in E. coli.
To confirm that the gldA101 allele in strain QZ19 encodes D-LDH activity, the gene was cloned from strain QZ19 (plasmid pQZ115) and introduced into E. coli strain AH242. E. coli strain AH242 is anaerobic minus due to mutations in ldhA and pflB that abolished the ability to oxidize NADH produced during glycolysis (25). Anaerobic growth of strain AH242 was restored by plasmid pQZ115 with D(−)-lactate as the fermentation product. Strain AH242 carrying plasmid pQZ109 that lacks the C-terminal 10 amino acids of the GlyDH* did not grow anaerobically indicating that the truncated form of this protein lacks D-LDH activity. These results establish that GlyDH* from B. coagulans strain QZ19 has D(−)-LDH activity.
To further confirm that the gldA101 from strain QZ19 encodes a protein with D-LDH activity, the gene was cloned from strain QZ19 (plasmid pQZ113) and expressed in recombinant E. coli with N-terminal His-tag for protein purification. The recombinant GlyDH* also had both GlyDH and D-LDH activities. The specific activity of the enzyme with glycerol and NAD+ was 0.68 unit per mg protein. With pyruvate and NADH as substrates (LDH reaction), the specific activity of the protein was an unexpectedly high 6.9 units per mg protein and the reaction product was identified as D-lactic acid by HPLC. In the reverse reaction, the enzyme was active only with D(−)-lactate and NAD+ as substrates (specific activity of 0.17 unit per mg protein). Activity with L(+)-lactate as substrate was undetectable.
These results show that during metabolic evolution of strain QZ5, the native GlyDH acquired D-LDH activity. Native GlyDH from B. stearothermophilus has been reported to lack LDH activity (28) and in agreement with this, the LDH activity of B. coagulans native GlyDH purified from recombinant E. coli had less than 0.15% of the GlyDH activity.
Boosting gldA Transcription by Insertion of DNA in the D-Lactate Producing Strains.
In addition to evolution of the enzyme as D-LDH, the level of expression of gldA101 in strain QZ19 was the highest of the genes analyzed in this study (Table 2). This increase in transcription is apparently due to the 1,821 bp DNA insertion upstream of the gldA101 gene contributing a strong promoter for gldA101 expression in the evolved strain (Fig. 4).
Although consensus sequences corresponding to Shine-Dalgarno (GGAG) and −10 region (TATGAT) can be identified in the upstream DNA of gldA gene of the wild type, a corresponding −35 region could not be discerned. An inverted repeat with a 6 base pairs stem and 11 bases loop was found at the projected −35 region suggesting that expression of this gene is only moderate, at best, in the native bacterium. Insertion of the 1,821 bp sequence in the upstream region of gldA gene in strain QZ19 introduced a consensus −35 sequence to the gldA (Fig. 4). The high level of expression of the gldA gene and GDH* protein in strain QZ19 (Table 2) is apparently due to this reconstruction of the gldA upstream region in strain QZ13 and its derivatives yielding a strong promoter.
Evolutionary Path to D-Lactate Production in Strain QZ19.
There are apparently three seminal events that occurred in the evolutionary path of B. coagulans strain QZ5 to strain QZ19 that produced over 90 g of D-lactate in about 48 h; two mutations in the GlyDH (D121N and F245S) and change in the promoter structure of gldA101 by insertion. DNA sequence analysis of the various intermediate strains in the evolutionary path revealed that the transposon DNA insertion occurred first in strain QZ13. This proposed promoter alteration due to transposition is in agreement with the elevated level of gldA mRNA and GlyDH activity in strain QZ13 (about 100-fold and 50-fold, respectively, over the levels of strain QZ5) and other derivatives of this strain (Table 2). The higher GlyDH activity in strain QZ13 led to a slight increase in D-LDH activity also. However, the ratio of D-LDH to GlyDH activity in this strain was only 0.003 suggesting that the GlyDH still lacks significant D-LDH activity. The first GlyDH mutation (F245S) was detected in strain QZ15 after the increase in transcription of gldA and this mutation apparently increased D-LDH/GlyDH ratio by about sevenfold to 0.02 (Table 2). The second mutation (D121N) occurred during the evolution of strain QZ19 from strain QZ15 and this second mutation increased the D-LDH/GlyDH ratio by about 10-fold to 0.22. It is interesting to note that with the increase in D-LDH activity, the enzyme lost part of its GlyDH activity. A 10-fold increase in D-LDH activity of the enzyme due to the two mutations (QZ19 vs QZ14) in strain QZ19 lowered the GlyDH activity by about sevenfold from that of strain QZ14 (Table 2). This loss in GlyDH activity in the extracts of strain QZ19 is apparently not due to lower protein level in the cell since this protein band still accounted for about 11% of the total proteins (Fig. 3).
The mutations in the GlyDH of strain QZ19 occurred in two of nine critical amino acids forming a deep pocket where the nicotinamide ring of NAD+ binds in the enzyme from B. stearothermophilus (29). A model of the GlyDH of B. coagulans also shows the deep cleft between the two domains of the enzyme with the mutated amino acids in the edge (Fig. S8). Glycerol C1 and C3 are reported to be stabilized in the B. stearothermophilus GlyDH by van der Walls interactions with the benzyl ring of Phe247. Changing the corresponding Phe245 of B. coagulans GlyDH to serine is expected to abolish this interaction resulting in the observed reduction in glycerol oxidation activity of the enzyme from strain QZ19. The Asp121 and Ser245 probably stabilize pyruvate at the active site through their interactions with C1 and C2 of pyruvate (Fig. S8) resulting in the observed LDH activity.
These results show that an increase in the expression level of gldA and acquisition of D-LDH activity by the GlyDH through two mutations are responsible for the increase in D-lactate titer of B. coagulans strain QZ19 fermentations (Table 1).
Conclusion
Using a method developed for the genetically recalcitrant B. coagulans, the ldh and alsS genes were deleted. These two mutations reduced anaerobic growth and sugar metabolism by B. coagulans at pH 5.0. Although a gene encoding D-LDH (ldhA) is present in the chromosome, metabolic evolution of the ldh, alsS double mutant recruited glycerol dehydrogenase (GlyDH; gldA) to restore sugar fermentation with the production of D(−)-lactate as the fermentation product at a yield and titer equivalent to L(+)-lactate with the parent. This shift in lactic acid isomer produced by the mutant from either glucose or xylose is due to mutational evolution of glycerol dehydrogenase to catalyze reduction of pyruvate to D(−)-lactate plus a strong promoter from an upstream insertion sequence. SSF of crystalline cellulose to D-lactate required only about 1/3 of the commercial cellulases needed to achieve highest volumetric productivity compared to mesophilic lactic acid bacteria. This change in the primary fermentation product from L-lactic acid to D-lactic acid in the thermotolerant bacterium was accomplished using only the native genes. The availability of thermotolerant B. coagulans strains for production of optically pure D(−)- or L(+)- lactic acid at 50–55 °C and pH 5.0 using cellulose as a feedstock can be expected to reduce the cost of lactic acid production for biopolymers.
Materials and Methods
Bacterial Strains and Plasmids.
Bacterial strains, plasmids, and primers used in this study are listed in Table S1. B. coagulans strain P4-102B used as the wild type strain was described previously (17). Plasmid pGK12 replicates in several Gram-positive bacteria and E. coli (30, 31) and its replication is naturally restricted to temperatures ≤ 42 °C. This temperature sensitive nature of plasmid pGK12 replication at 50 °C provides an opportunity to select for chromosomal DNA integrants of B. coagulans that can grow at 50–55 °C.
Medium and Growth Condition.
Growth and fermentation conditions have been described previously (15, 21–23). Cultures were grown in L-broth (LB) at pH 5.0 or 7.0, as needed. Sterile glucose was added before inoculation.
Gene Deletions in B. coagulans.
Construction of deletion mutants of B. coagulans was based on previously described methods (32) and are described in detail in SI Text. Plasmid pGK12 was used as the primary vehicle for transfer of DNA to B. coagulans for deletion construction. Presence of plasmid in each cell in a population (109 CFU mL-1) at 37 °C helps to overcome the low transformation efficiency of plasmid DNA into this bacterium and chromosomal integrants in the population were readily identified when the plasmid was eliminated by growth at 50–55 °C.
Metabolic Evolution.
Metabolic evolution was carried out by sequentially subculturing in small pH-controlled fermenters under indicated conditions. Sequential transfers (2% inoculum; vol/vol) were made every 2–3 d, as growth permitted.
Enzyme Assays.
Cultures were grown (LB-glucose medium in pH-controlled fermenters) to midexponential phase, harvested, and used for enzyme assays as previously described (23). Glycerol dehydrogenase from strain QZ19 was purified using differential ammonium sulfate precipitation followed by anion-exchange and hydrophobic interaction chromatography (SI Text). GlyDH and GlyDH* were also purified as N-terminal His-tagged enzymes (33). GlyDH and LDH were assayed using standard methods (34, 35).
Analytical Methods.
Glucose and fermentation products were determined by HPLC as previously described (36). Optical isomers of D-(−)- and L-(+)- lactic acids were determined by HPLC [Chirex 3126(D)-penicillamine column;150 × 4.6 mm, 5 μm; Phenomenex] using 2 mM CuSO4 as mobile phase, and enzymatically using D-lactate dehydrogenase (Sigma Chemical Co.).
Supplementary Material
Acknowledgments.
We thank J. Maupin-Furlow for the use of Bioflo fermenter, G. Lorca for plasmid pGK12 and L. Govindasamy for the GlyDH active site model. This study was supported in part by grants from the Department of Energy (DE-FG36-04GO14019 and DE-FG36-08GO88142), Department of Agriculture, National Institute of Food and Agriculture (2011-10006-30358), the State of Florida, University of Florida Agricultural Experiment Station, the Florida Energy Systems Consortium, and Myriant Technologies.
Footnotes
Conflict of interest statement: Myriant Corp has licensed technology from University of Florida. L.O.I. is a member of their advisory board, and is a minor share-holder.
This article is a PNAS Direct Submission.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database, http://www.ncbi.nlm.nih.gov/genbank (accession nos. HQ699676, HQ699678, and HQ699677).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1111085108/-/DCSupplemental.
References
- 1.Demain AL. Biosolutions to the energy problem. J Ind Microbiol Biot. 2009;36:319–332. doi: 10.1007/s10295-008-0521-8. [DOI] [PubMed] [Google Scholar]
- 2.Lynd LR, et al. How biotech can transform biofuels. Nat Biotechnol. 2008;26:169–172. doi: 10.1038/nbt0208-169. [DOI] [PubMed] [Google Scholar]
- 3.Mecking S. Nature or petrochemistry?-biologically degradable materials. Angew Chem Int Ed. 2004;43:1078–1085. doi: 10.1002/anie.200301655. [DOI] [PubMed] [Google Scholar]
- 4.Ragauskas AJ, et al. The path forward for biofuels and biomaterials. Science. 2006;311:484–489. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]
- 5.Chen G-Q. A microbial polyhydroxyalkanoates (PHA) based bio- and materials industry. Chem Soc Rev. 2009;38:2434–2446. doi: 10.1039/b812677c. [DOI] [PubMed] [Google Scholar]
- 6.Benninga H. A history of lactic acid making. Dordrecht, The Netherlands: Kluyver Academic Publishers; 1990. [Google Scholar]
- 7.Datta R, Henry M. Lactic acid: recent advances in products, processes and technologies—a review. J Chem Technol Biot. 2006;81:1119–1129. [Google Scholar]
- 8.Hofvendahl K, Hans-Hagerdal B. Factors affecting the fermentative lactic acid production from renewable resources. Enzyme Microb Tech. 2000;26:87–107. doi: 10.1016/s0141-0229(99)00155-6. [DOI] [PubMed] [Google Scholar]
- 9.Tsuji H. Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol Biosci. 2005;5:569–597. doi: 10.1002/mabi.200500062. [DOI] [PubMed] [Google Scholar]
- 10.Teusink B, Smid EJ. Modelling strategies for the industrial exploitation of lactic acid bacteria. Nat Rev Microbiol. 2006;4:46–56. doi: 10.1038/nrmicro1319. [DOI] [PubMed] [Google Scholar]
- 11.Grabar TB, Zhou S, Shanmugam KT, Yomano LP, Ingram LO. Methylglyoxal bypass identified as source of chiral contamination in L(+) and D(−)-lactate fermentations by recombinant Escherichia coli. Biotechnol Lett. 2006;28:1527–1535. doi: 10.1007/s10529-006-9122-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhou S, Shanmugam KT, Yomano LP, Grabar TB, Ingram LO. Fermentation of 12% (w/v) glucose to 1.2 M lactate by Escherichia coli strain SZ194 using mineral salts medium. Biotechnol Lett. 2006;28:663–670. doi: 10.1007/s10529-006-0032-5. [DOI] [PubMed] [Google Scholar]
- 13.Tenenbaum DJ. Food vs.fuel: diversion of crops could cause more hunger. Environ Health Perspect. 2008;116:A254–A257. doi: 10.1289/ehp.116-a254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer Pv, Lee YY. Product inhibition in simultaneous saccharification and fermentation of cellulose into lactic acid. Biotechnol Lett. 1999;21:371–373. [Google Scholar]
- 15.Patel MA, Ou M, Ingram LO, Shanmugam KT. Simultaneous saccharification and co-fermentation of crystalline cellulose and sugar cane bagasse hemicellulose hydrolysate to lactate by a thermotolerant acidophilic Bacillus sp. Biotechnol Progr. 2005;21:1453–1460. doi: 10.1021/bp0400339. [DOI] [PubMed] [Google Scholar]
- 16.Garde A, Jonsson G, Schmidt AS, Ahring BK. Lactic acid production from wheat straw hemicellulose hydrolysate by Lactobacillus pentosus and Lactobacillus brevis. Bioresource Technol. 2002;81:217–223. doi: 10.1016/s0960-8524(01)00135-3. [DOI] [PubMed] [Google Scholar]
- 17.Patel MA, et al. Isolation and characterization of acid-tolerant, thermophilic bacteria for effective fermentation of biomass-derived sugars to lactic acid. Appl Environ Microbiol. 2006;72:3228–3235. doi: 10.1128/AEM.72.5.3228-3235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka K, et al. Two different pathways for D-xylose metabolism and the effect of xylose concentration on the yield coefficient of L-lactate in mixed-acid fermentation by the lactic acid bacterium Lactococcus lactis IO-1. Appl Microbiol Biotechnol. 2002;60:160–167. doi: 10.1007/s00253-002-1078-5. [DOI] [PubMed] [Google Scholar]
- 19.Okano K, et al. Improved production of homo-D-lactic acid via xylose fermentation by introduction of xylose assimilation genes and redirection of the phosphoketolase pathway to the pentose phosphate pathway in L-Lactate dehydrogenase gene-deficient Lactobacillus plantarum. Appl Environ Microbiol. 2009;75:7858–7861. doi: 10.1128/AEM.01692-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarboe LR, et al. Metabolic engineering for production of biorenewable fuels and chemicals: contributions of synthetic biology. J Biomed Biotechnol. 2010;2010:1–18. doi: 10.1155/2010/761042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ou MS, Mohammed N, Ingram LO, Shanmugam KT. Thermophilic Bacillus coagulans requires less cellulases for simultaneous saccharification and fermentation of cellulose to products than mesophilic microbial biocatalysts. Appl Biochem Biotechnol. 2009;155:379–385. doi: 10.1007/s12010-008-8509-4. [DOI] [PubMed] [Google Scholar]
- 22.Ou MS, Ingram LO, Shanmugam KT. L: (+)-Lactic acid production from non-food carbohydrates by thermotolerant Bacillus coagulans. J Ind Microbiol Biot. 2011;38:599–605. doi: 10.1007/s10295-010-0796-4. [DOI] [PubMed] [Google Scholar]
- 23.Su Y, Rhee MS, Ingram LO, Shanmugam KT. Physiological and fermentation properties of Bacillus coagulans and a mutant lacking fermentative lactate dehydrogenase activity. J Ind Microbiol Biot. 2010;38:441–450. doi: 10.1007/s10295-010-0788-4. [DOI] [PubMed] [Google Scholar]
- 24.Turner NJ. Directed evolution drives the next generation of biocatalysts. Nat Chem Biol. 2009;5:567–573. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 25.Kim Y, Ingram LO, Shanmugam KT. Construction of an Escherichia coli K-12 mutant for homoethanologenic fermentation of glucose or xylose without foreign genes. Appl Environ Microbiol. 2007;73:1766–1771. doi: 10.1128/AEM.02456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang H, et al. Complete genome sequence of Lactobacillus helveticus strain H10. J Bacteriol. (2666) 2011;193 doi: 10.1128/JB.00166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer P, Bown KJ, Scawen MD, Atkinson T, Gore MG. Isolation and characterization of the glycerol dehydrogenase from Bacillus stearothermophilus. Biochim Biophys Acta. 1989;994:270–279. doi: 10.1016/0167-4838(89)90304-x. [DOI] [PubMed] [Google Scholar]
- 29.Ruzheinikov SN, et al. Glycerol dehydrogenase. structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure. 2001;9:789–802. doi: 10.1016/s0969-2126(01)00645-1. [DOI] [PubMed] [Google Scholar]
- 30.Kok J, van der Vossen JM, Venema G. Construction of plasmid cloning vectors for lactic streptococci which also replicate in Bacillus subtilis and Escherichia coli. Appl Environ Microbiol. 1984;48:726–731. doi: 10.1128/aem.48.4.726-731.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luchansky JB, Muriana PM, Klaenhammer TR. Application of electroporation for transfer of plasmid DNA to Lactobacillus, Lactococcus, Leuconostoc, Listeria, Pediococcus, Bacillus, Staphylococcus, Enterococcus and Propionibacterium. Mol Microbiol. 1988;2:637–646. doi: 10.1111/j.1365-2958.1988.tb00072.x. [DOI] [PubMed] [Google Scholar]
- 32.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989;171:4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim Y, Ingram LO, Shanmugam KT. Dihydrolipoamide dehydrogenase mutation alters the NADH sensitivity of pyruvate dehydrogenase complex of Escherichia coli K-12. J Bacteriol. 2008;190:3851–3858. doi: 10.1128/JB.00104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin EC, Magasanik B. The activation of glycerol dehydrogenase from Aerobacter aerogenes by monovalent cations. J Biol Chem. 1960;235:1820–1823. [PubMed] [Google Scholar]
- 35.Yoshida A, Freese E. Lactate dehydrogenase from Bacillus subtilis. Method Enzymol. 1975;41:304–309. doi: 10.1016/s0076-6879(75)41069-2. [DOI] [PubMed] [Google Scholar]
- 36.Underwood SA, et al. Genetic changes to optimize carbon partitioning between ethanol and biosynthesis in ethanologenic Escherichia coli. Appl Environ Microbiol. 2002;68:6263–6272. doi: 10.1128/AEM.68.12.6263-6272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.