

Summary
Motoneuron-derived agrin clusters nicotinic acetylcholine receptors (AChRs) in mammalian muscle cells. We used two-hybrid screens to identify a protein, tumorous imaginal discs (Tid1), that binds to the cytoplasmic domain of muscle-specific kinase (MuSK), a major component of the agrin receptor. Like MuSK, Tid1 colocalizes with AChRs at developing, adult and denervated motor endplates. Knockdown of Tid1 by short hairpin RNA (shRNA) in skeletal muscle fibers dispersed synaptic AChR clusters and impaired neuromuscular transmission. In cultured myotubes, Tid1 knockdown inhibited AChR clustering, as well as agrin-induced activation of the Rac and Rho small GTPases and tyrosine phosphorylation of the AChR, without affecting MuSK activation. Tid1 knockdown also decreased Dok-7-induced clustering of AChRs. Overexpression of the N-terminal half of Tid1 induced agrin- and MuSK-independent phosphorylation and clustering of AChRs. These results demonstrate that Tid1 is an essential component of the agrin signaling pathway, crucial for synaptic development.
Introduction
During development of the neuromuscular junction (NMJ), acetylcholine receptors (AChRs) become clustered in the postsynaptic membrane, where their high density is critical for efficient synaptic transmission (Hall and Sanes, 1993; Burden, 2002). Clustering of AChRs and other muscle proteins, including the receptor-associated muscle protein rapsyn, can occur spontaneously in the absence of nerves (Yang et al., 2001; Lin et al., 2001), but it is facilitated by motoneuron-derived agrin, whose action localizes AChRs to the subsynaptic muscle membrane in developing muscle and sustains their localization at adult NMJs. Agrin acts through a membrane receptor complex that includes the muscle-specific kinase MuSK, a receptor tyrosine kinase (RTK) (Valenzuela et al., 1995; Glass et al., 1996). The signaling pathway by which activation of MuSK leads to clustering of AChRs remains poorly understood. Although other kinases, such as src and abl, are likely involved (Mittaud et al., 2001; Finn et al., 2003), MuSK does not appear to signal through the common MAP or PI3 kinase pathways (Herbst and Burden, 2000). Recent studies have uncovered roles for Dishevelled (Luo et al., 2002), the Rac1/RhoA and Cdc42 small GTPases (Weston et al., 2000, 2003), and the muscle protein Dok-7 (Okada et al., 2006) in mediating MuSK-dependent AChR clustering.
We report here experiments that identify a new MuSK-binding protein, the rat homologue (Tid1) of the Drosophila tumor suppressor, tumorous imaginal discs (Tid56), a heat shock protein (hsp) 40 homologue. Tid1 and other hsps have recently been implicated in a variety of signaling pathways (Gaestel, 2006), including those involving tyrosine phosphorylation. Tid1 has been shown to interact with, and to modulate the signaling of, the Jak family of protein tyrosine kinases (Sarkar et al., 2001), the Ras-GTPase activating protein (Ras-GAP) (Trentin et al., 2001), ErbB2 (Kim et al., 2004), and the Trk family of RTKs (Liu et al., 2005).
Our experiments demonstrate that Tid1 is closely associated with MuSK at the neuromuscular junction and that it is essential for spontaneous and agrin-induced clustering of AChRs, as well as for the maintenance of clustered AChRs at adult synapses. Whereas Tid1 is not required for agrin-induced MuSK activation, an N-terminal fragment of Tid1 can induce AChR phosphorylation and clustering independently of agrin and MuSK. Thus, Tid1 is a critical factor that acts downstream of MuSK activation in the neurally-mediated differentiation and maintenance of the neuromuscular synapse.
Results
Tid1 Binds MuSK
To identify proteins that interact with MuSK, we carried out two bacterial two-hybrid screens (Joung et al., 2000), first using the entire cytoplasmic domain (residues 515–869) of mouse MuSK as bait and then using the juxtamembrane region (residues 515–692), which contains a tyrosine phosphorylation site required for agrin-induced AChR clustering (Herbst and Burden, 2000; Watty et al., 2000); in both cases, a cDNA library of embryonic day 19 (E19) rat skeletal muscle was the target (Supplementary Fig. 1A). Tumorous imaginal discs (Tid1), a rat homologue of the Drosophila tumor suppressor Tid56 and the heat shock protein hsp40, appeared twice in the initial screen and six times in the second screen (Fig. 1A and Supplementary Fig. 1B). Tid1 has two alternatively spliced forms, “Tid1 short” (Tid1S) and “Tid1 long” (Tid1L), which differ in the last 33 amino acid residues of their C-termini (Syken et al., 1999; Supplementary Fig. 1C). Importantly, the bacterial two-hybrid screens only identified the short isoform of Tid1 as a MuSK-binding partner (Fig. 1A).
Figure 1. Bacterial Two-Hybrid Screening and Co-Immunoprecipitation Demonstrate that Tid1S Binds MuSK.
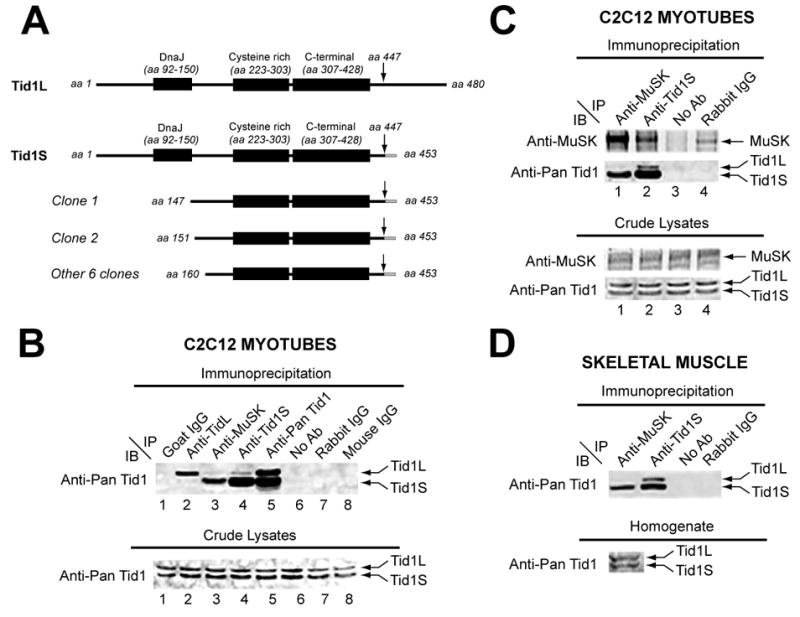
(A) Schematic representations of the full-length long (Tid1L) and short (Tid1S) isoforms of Tid1 and the eight overlapping clones of Tid1S identified in the bacterial two-hybrid screens. (B) Top panel: Lysates of C2C12 myotubes were immunoprecipitated (IP) with specific antibodies as indicated (lanes 2 – 5), Protein-A Sepharose beads without an antibody (lane 6), or control goat, rabbit, or mouse immunoglobulins (IgGs; lanes 1, 7–8). mAb RS-13 (anti-pan Tid1), a mouse antibody that recognizes both Tid1S and Tid1L, was used for immunoblotting (IB). Bottom panel: Corresponding crude lysates, immunoblotted with mAb RS-13. (C) Top panels: Lysates of C2C12 myotubes were immunoprecipitated (IP) with specific antibodies as indicated (lanes 1 and 2), Protein-A Sepharose beads without an antibody (lane 3), or purified rabbit IgGs (lane 4). A rabbit polyclonal antibody against MuSK (anti-MuSK) and mAb RS-13 were used for immunoblotting. Bottom panel: Corresponding crude lysates were immunoblotted similarly. (D) Top panel: Detergent extracts of adult rat tibialis anterior (TA) muscle were immunoprecipitated as in C and immunoblotted with mAbRS-13. Bottom panel: corresponding muscle homogenate.
To determine whether Tid1 binds to MuSK in skeletal muscle cells, we immunoprecipitated detergent extracts of C2C12 myotubes with a polyclonal antibody against MuSK (anti-MuSK; Fuhrer et al., 1997; Sugiyama et al., 1997) and immunoblotted with a monoclonal antibody that recognizes both Tid1S and Tid1L (anti-pan Tid1). A band of ~40 KDa, corresponding to Tid1S (453 residues), was detected on the immunoblots, but Tid1L, also present in the cell lysates, was not (Fig. 1B, lane 3). The co-immunoprecipitation of Tid1S with MuSK was highly specific, as the band did not appear in negative control lanes (Fig. 1B, lanes 1–2, 6–8). Similar results were obtained when a polyclonal antibody specific for Tid1S (anti-Tid1S, see Experimental Procedures) was used for immunoprecipitation and anti-MuSK was employed for immunoblotting (Fig. 1C, lane 2). “Pull-down” assays using anti-MuSK were also performed with detergent extracts of the tibialis anterior (TA) muscle from adult rats. Anti-MuSK specifically co-immunoprecipitated Tid1S (Fig. 1D, lane 1). Thus, both two-hybrid and biochemical studies indicate that MuSK is associated with Tid1S, but not Tid1L.
Tid1 Co-Clusters with AChRs and Associates with the Membrane in a MuSK-Dependent Manner
In cultured myotubes, AChRs are normally distributed diffusely along the muscle cell surface, except for occasional spontaneous, or aneural, AChR clusters. Co-culturing myotubes with motoneurons or treatment with recombinant neural agrin induces aggregation of AChRs into high-density clusters resembling those on the postsynaptic membranes of NMJs (Christian et al., 1978; Ferns et al., 1992). Using a pan-Tid1 polyclonal antibody (anti-Tid1; Lo et al., 2004), we examined the localization of Tid1 in C2C12 mouse myotubes by immunofluorescence. In the absence of agrin, Tid1 immunofluorescence was found in the cytoplasm and of the myotubes; it was also associated with spontaneous AChR clusters seen on the cell surface (Fig. 2A, a–c). Treatment with recombinant neural agrin (CAg12,4,8, Ferns et al., 1992) dramatically increased the number of AChR and Tid1 clusters, which in each case were precisely colocalized (Fig. 2A, g-i). Thus, like the postsynaptic proteins rapsyn and MuSK, Tid1 co-clusters with AChRs on the muscle cell membrane in both agrin-stimulated and unstimulated muscle cells. As with MuSK (Valenzuela et al., 1995), the overall level of Tid1 expression in C2C12 cells was low in undifferentiated myoblasts and high in myotubes and as with the AChR (Godfrey et al., 1984), it was not altered by agrin (Fig. 2B). The induction of Tid1 clusters by agrin thus does not represent increased expression of Tid1, but rather a redistribution of Tid1, either among subcellular fractions or within the cell membrane.
Figure 2. Colocalization of Tid1 and AChRs in C2C12 Myotubes and on the Postsynaptic Membrane of the NMJ.
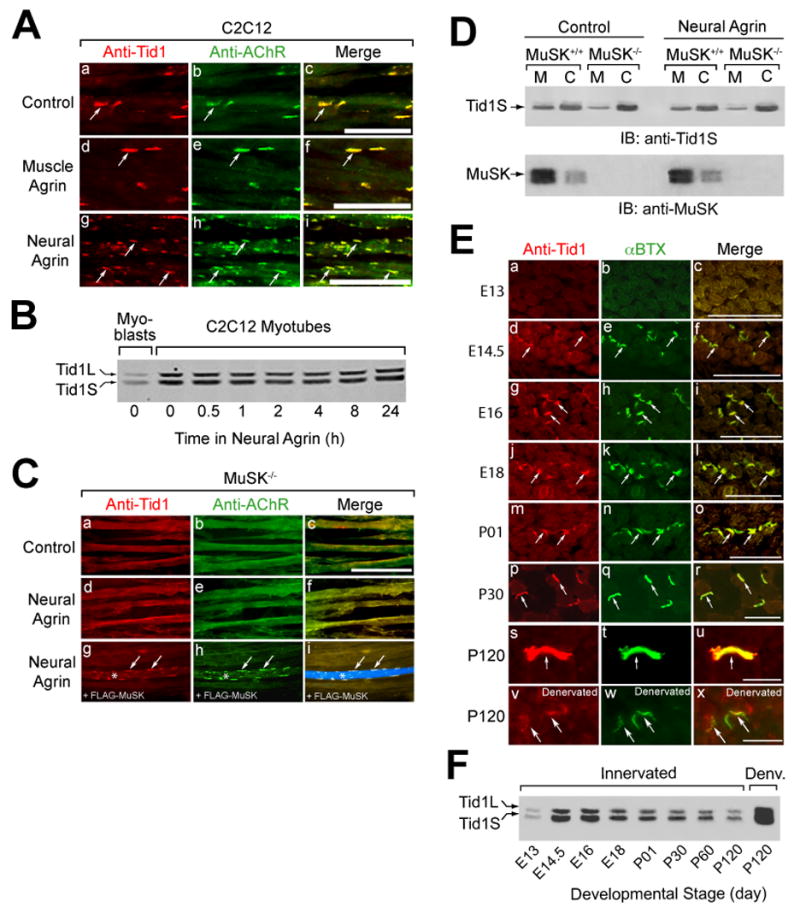
(A) Tid1 is co-clustered with AChRs (arrows) on the surface of C2C12 myotubes. C2C12 myotubes were incubated with control medium, muscle agrin (CAg12,0,0, an alternatively-spliced isoform with little AChR clustering activity), or neural agrin (CAg12,4,8) for 4 h, fixed, and double-stained with a pan-Tid1 rabbit polyclonal antibody (anti-Tid1) and mAb 35, a rat monoclonal antibody against the AChR. Cy3-conjugated goat anti-rabbit and Cy2-conjugated goat anti-rat IgGs were used as secondary antibodies. Scale bar = 25 μm in (A), (C), and (E). (B) The effects of differentiation and agrin on the expression of Tid1 in C12C12 cells. Myoblasts (lane 1) were maintained in growth medium; myotubes were grown in fusion medium for 48 hrs. Myotubes (lanes 2–8) were treated with agrin for the indicated time, lysed with extraction buffer, and immunoblotted with mAb RS-13. (C) Agrin does not cluster Tid1 or AChRs without MuSK. Myotubes derived from MuSK−/− mice were treated for 4 h with neural agrin and stained as in (A) for Tid1 and with Alexa Fluor 488-conjugated α-bungarotoxin (αBTX) to label AChRs (d–f). In a rescue experiment (g-i), MuSK−/− cells were transfected with a plasmid cDNA encoding FLAG-MuSK, treated with neural agrin, and stained with the anti-FLAG M2 antibody followed by an Alexa Fluor 633-conjugated rabbit anti-mouse secondary antibody. A FLAG-MuSK-positive myotube is clearly visible in i (*, arrows). (D) More Tid1S is found in the cytosolic versus the membrane fraction of MuSK-deficient muscle cells, as compared to wildtype myotubes. Myotubes derived from the skeletal muscles of MuSK+/+ or MuSK−/− mice were treated with or without neural agrin, homogenized, and fractionated by ultracentrifugation. The amount of Tid1S protein in the membrane (M) and cytosolic (C) fractions was determined by immunoblotting with a rabbit polyclonal anti-Tid1S antibody (anti-Tid1S). (E) Tid1 is concentrated at motor endplates (arrows) during development. Cross-sections of mouse hindlimb muscles at different ages were stained as in (C). 7.5 weeks after denervation, denervated (v-x) and contralateral innervated (s-u) soleus muscles were dissected, cryosectioned, fixed, and stained. (F) The expression of Tid1 in mouse skeletal muscle during development and following denervation. After homogenization of mouse hindlimb muscles, cleared lysates were immunoblotted with mAb RS-13. An equal amount of total protein was loaded into each of the lanes.
The strong association of MuSK with Tid1S, a protein without a transmembrane sequence, suggests that Tid1S may become bound to the membrane and clustered through its association with MuSK. To test this idea, we examined the distribution of Tid1 in myotubes derived from skeletal muscles of MuSK knockout (MuSK−/−) mice (Glass et al., 1996), cultured with and without agrin. Immunofluorescence studies demonstrated that neither AChRs nor Tid1 formed spontaneous or agrin-induced clusters in MuSK-deficient myotubes (Fig. 2C, a–f). Transfection of the mutant myotubes with FLAG-tagged MuSK rescued their ability to cluster AChRs and Tid1 on the cell surface (Fig. 2C, g–i).
Subcellular fractionation assays showed significant differences in the distribution of Tid1S in the membrane versus cytosolic fractions of wildtype (MuSK+/+) and MuSK−/− myotubes (Fig. 2D). In wildtype myotubes, 29.3% of Tid1S was found in the membrane fraction and 70.7% in the cytosol. In MuSK−/− cells, membrane-bound Tid1S was decreased to 8.6%, whereas the cytosolic form increased to 91.4% (p < 0.05, n=4). There was no significant difference in the total amount of Tid1S in the wildtype and MuSK−/− myotubes, and agrin did not change the proportion of Tid1S in the membrane versus the cytosolic fraction. Taken together (see also Fig. 8C), our data suggest that Tid1S is constitutively associated with MuSK on the myotube membrane. Binding to MuSK appears to be required for Tid1 membrane association and for both the spontaneous and agrin-induced clustering of Tid1. Moreover, as with the AChR (Anderson et al., 1977), agrin appears to cluster Tid1 by redistributing it within the membrane, rather than by recruiting additional Tid1S to the cell surface.
Figure 8. Tid1 Regulates Dok-7/MuSK Binding, but Binds MuSK Constitutively.
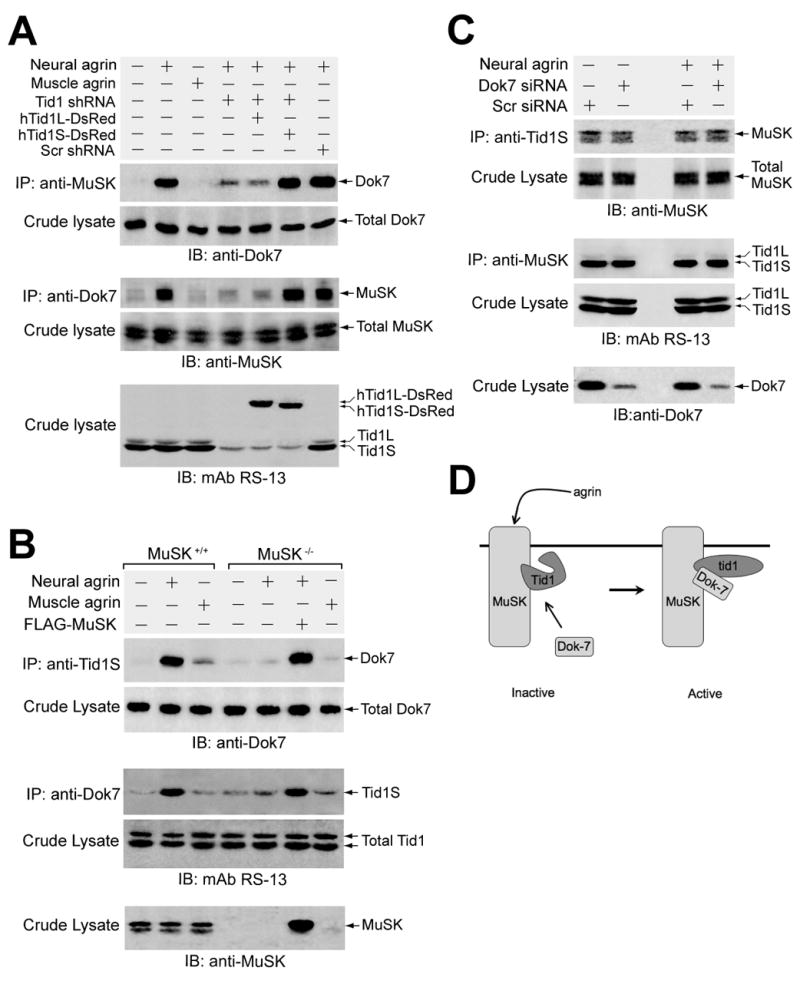
(A) Overexpression of Tid1 shRNA diminishes the agrin-induced association of MuSK and Dok-7. C2C12 myoblasts were transfected overnight with each of the indicated plasmid cDNAs and then differentiated for 48–72 h. Myotubes were incubated with recombinant agrins for 1 h, lysed, and then immunoprecipitated and immunoblotted using the indicated antibodies. Aliquots of crude lysates were immunoblotted with anti-Dok-7, anti-MuSK, and RS-13 antibodies. (B) Tid1S interaction with Dok-7 is dependent on MuSK. Myoblasts from wildtype or MuSK−/− mice were differentiated, incubated with recombinant agrin for 1 h, lysed, immunoprecipitated, and immunoblotted using antibodies as indicated. One dish of MuSK−/− myoblasts was transfected with a plasmid encoding FLAG-tagged MuSK approximately 24 hours before differentiation was initiated. (C) The interaction between MuSK and Tid1S is independent of neural agrin and Dok-7. C2C12 myotubes expressing Scr or Dok-7-specific siRNA were incubated with or without neural agrin for 1 hour. Detergent extracts of the cells were immunoprecipitated and immunoblotted using antibodies as indicated. (D) Schematic representation of the activation of Tid1 by the binding of Dok-7 to the agrin-stimulated MuSK/Tid1 complex.
Tid1 is Concentrated at Motor Endplates During Development and Dispersed After Chronic Denervation
To examine the distribution of Tid1 in developing and adult skeletal muscles, we double-stained frozen sections of hindlimb muscles from embryonic and postnatal mice for AChRs (α-bungarotoxin, αBTX) and Tid1 (Fig. 2E, a–u). At E13, before nerve contact, muscle staining for both AChRs and Tid1 was weak and diffusely distributed. At E14.5, shortly after innervation, faint but discernible Tid1 immunofluorescence appeared, coincident with small AChR clusters. Clusters containing AChR and Tid1 were more numerous and brighter by E16. Similar to MuSK (Bowen et al., 1998), colocalization of Tid1 and AChRs increased throughout embryogenesis, until the two proteins appeared precisely in register at neonatal and adult NMJs (Fig. 2E, a–u). Consistent with the immunostaining, and similar to the pattern of MuSK expression, Western blots revealed markedly enhanced levels of Tid1 protein in fetal muscles shortly after nerve-muscle contact (day E14.5). Expression remained high throughout the embryonic period, but gradually decreased after birth (Fig. 2F).
We then transected the sciatic nerves of adult mice and stained cryosections of denervated soleus muscles for Tid1, AChR (αBTX), and synaptophysin. 8 days after denervation, the precise colocalization of Tid1 and AChRs at endplates was maintained (Supplementary Fig 4., a–c) whereas synaptophysin immunoreactivity was abolished (j, m), confirming the postsynaptic localization of Tid1.
At 8 weeks after denervation (Supplementary Fig. 5), AChR clusters were fragmented, with significantly reduced AChR staining (Fig. 2E, w). Their mean size was 33% of those in innervated soleus muscles (Supplementary Fig. 5D, p < 0.001), consistent with earlier experiments (Frank et al., 1976). Tid1 remained colocalized with AChRs at the denervated endplates, but with reduced intensity of immunofluorescence (Fig. 2E, v). The mean size of Tid1 clusters was 18% of those in control muscles (Supplementary Fig. 5D, p < 0.001). The mean ratio of Tid1 cluster size to AChR cluster size was 98% in control muscles, but only 58% after denervation (Supplementary Fig. 5E, p < 0.001), suggesting that dispersal of endplate Tid1 after denervation precedes that of AChRs. In one denervated hindlimb, myofibers appeared to be reinnervated, as judged by synaptophysin staining. At these endplates, Tid1 and AChR staining were restored to near-normal levels (Supplementary Fig. 5C–E).
Western blotting showed a dramatic increase in the level of Tid1 protein after chronic denervation (Fig. 2F), similar to that seen with MuSK (Valenzuela et al., 1995) and the AChR (Merlie et al., 1984). The increase in all three proteins may reflect a general activation of synaptic protein gene transcription in extrasynaptic regions of muscle following denervation (Hall and Sanes, 1993). Thus, as with AChRs, both endplate and non-endplate Tid1 levels appear to be regulated by innervation.
Tid1 Knockdown by Short Hairpin RNA Inhibits AChR Clustering in Myotubes
To determine whether Tid1 is required for AChR clustering, we utilized RNA interference (RNAi) to inhibit mouse Tid1 expression. Short hairpin RNA (shRNA) targeted to the region encoding the N-terminal domain of Tid1 (see Experimental Procedures) was inserted into a vector also containing a gene for the modified red fluorescent protein marker DsRed-Express. Transfection of cultured myotubes with the shRNA resulted in a dramatic decrease in the level of Tid1 expression, whereas transfection with a vector containing a scrambled shRNA sequence showed no change (Supplementary Fig. 6A). About 50% of the cells in both cases expressed the fluorescent marker (Supplementary Fig. 6B). When shRNA-transfected cultures were incubated for 5 hours, either with or without neural agrin, and stained for AChRs, the number of clusters was significantly reduced both in the absence (Fig. 3A, d–f) and presence (Fig. 3A, j–l) of agrin. In contrast, expression of the scrambled control shRNA had no effect on the number of AChR clusters in either case (Fig. 3A, a–c, g–i). In individual myotubes, the observed reduction in receptor clusters correlated well with the degree of Tid1 immunofluorescence intensity (Supplementary Fig. 6C, e–l). When the time course of cluster formation was examined, Tid1 knockdown significantly reduced the number of AChR clusters at all time points (Fig. 3B, p < 0.01).
Figure 3. Inhibition of AChR Clustering by ShRNA-Mediated Tid1 Knockdown in Myotubes.
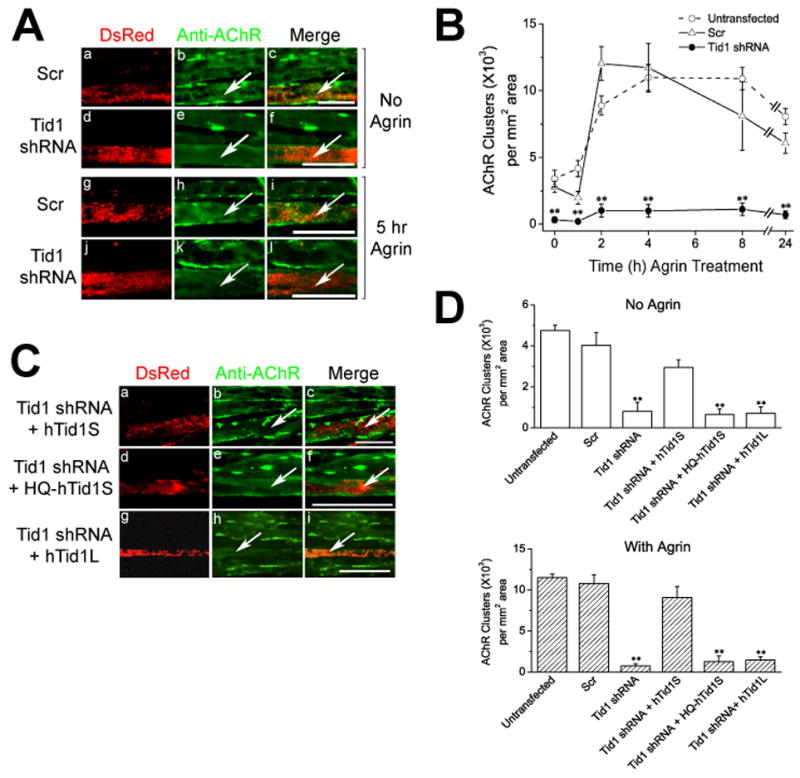
C2C12 myoblasts were transfected with a pSIREN vector encoding the scrambled control (Scr) or Tid1-targeted short hairpin RNA (shRNA) under the control of a U6 promoter. (A) Tid1 knockdown inhibits AChR clustering. At ~72 h post transfection and fusion, myotubes were incubated without (top panels), or with (bottom panels) neural agrin for 5 h. Transfected myotubes express the red fluorescent protein marker DsRed-Express under the control of a CMV promoter. The cells were fixed and stained with Alexa Fluor 488-conjugated αBTX for AChRs. Arrows highlight transfected myotubes and scale bar = 25 μm in (A) and (C). (B) Time course of AChR cluster formation following agrin stimulation. Myotubes transfected with Scr or Tid1 shRNA were incubated with neural agrin for the indicated time, fixed and stained with mAb 35 followed by Cy2-conjugated goat anti-rat IgG to label AChRs. Data are expressed as mean ± S.E.M. **, p < 0.01, Student’s t-tests comparing values between scrambled and Tid1-targeted shRNA-transfected myotubes. n = 10 for Scr and Tid1-shRNA transfected cells and n = 20 for untransfected myotubes for each time point. (C) Co-expression of human Tid1S (hTid1S: a–c), but not Tid1L (hTid1L: g–i) or the H121Q mutant (HQ-hTid1S: d–f), rescues AChR clustering from interference by mouse Tid1-targeted shRNA. Myotubes were treated with neural agrin and stained as in (A). (D) The effects of shRNA constructs on the formation of spontaneous (top) and agrin-induced (bottom) AChR clusters in myotubes. Data are expressed as mean ± S.E.M. **, p < 0.01. Student’s t-tests were performed comparing values between scrambled and the various Tid1-targeted shRNA-transfected myotubes. n = 120 for untransfected myotubes and n = 20 for every other group.
To rule out the possibility that the reduction in AChR clusters was caused by interference with the expression of genes unrelated to Tid1 (“off target” effects), cDNAs encoding human Tid1S or Tid1L were co-expressed along with the Tid1-targeted shRNA. To ensure that the mouse Tid1-specific shRNA would not affect their expression, selected nucleotides were silently mutated in the shRNA-targeted region of the human Tid1 cDNAs (see Experimental Procedures). Co-expression of the mouse Tid1-targeted shRNA with the full-length human Tid1S (hTid1S), but not human Tid1L (hTid1L), successfully rescued both spontaneous and agrin-induced AChR clustering activity in myotubes (Fig. 3C, a–c, g–i; Fig. 3D). Thus, the inhibitory effect of shRNA on AChR clustering in cultured myotubes is mediated specifically by interference with Tid1S expression.
Previous studies in other cell types have shown that mutation of a key residue in human Tid1 cDNA (H121Q) functionally inactivates Tid1 by allowing it to bind to, but not activate, its co-chaperone, hsp70 (Syken et al., 1999). We found that a silently-mutated human Tid1S with the H121Q mutation did not rescue shRNA-mediated inhibition of AChR clustering (Fig. 3C–D). Thus, the effect of Tid1 on AChR clustering likely depends upon its ability to activate hsp70.
An N-Terminal Domain of Tid1 Clusters AChRs Independently of Agrin and MuSK
The inability of the mutated (H121Q) human Tid1S cDNA to rescue clustering after shRNA inhibition led us to investigate the role of the different Tid1S domains in AChR clustering. Tid1 contains three well-characterized domains: an N-terminal DnaJ domain (residues 92–150), a cysteine-rich region (residues 223–303), and a C-terminal domain (residues 307–428). The DnaJ domain was of particular interest, since it contains the critical H121Q mutation. We constructed a set of truncated fragments of Tid1S lacking some or all of the domains (Fig. 4A). Cultured muscle cells were transfected with plasmid cDNAs encoding the myc-tagged fragments and 3–4 days later were incubated with or without neural agrin for 4 hours.
Figure 4. Induction of AChR Clustering by Tid1 in C2C12 Myotubes.

(A) Schematic diagram of full-length and four truncated fragments of Tid1S. (B) The N-terminal half of Tid1 induces AChR clustering. C2C12 myoblasts were transfected with the control plasmid pCS2+MT, which expresses 6 myc tags (Vector), or plasmid cDNAs encoding the various myc-tagged (fused in-frame to the N-terminus) Tid1S constructs shown in (A). At ~72 h after transfection and fusion, myotubes were incubated with or without neural agrin for 4 h, fixed, permeabilized, and double-stained with Rhodamine-conjugated αBTX and the anti-myc antibody mAb 9E10 followed by FITC-conjugated goat anti-mouse IgG. Arrows highlight transfected cells. Scale bar = 100 μm. (C) The effect of full-length and truncated fragments of Tid1S on the number of spontaneous and agrin-induced AChR clusters. n = 60 myotubes for each experimental group. *, 0.01 < p < 0.05; **, p < 0.01. (D) Overexpression of the N-terminal half of Tid1 does not induce tyrosine phosphorylation of MuSK. C2C12 myoblasts were transfected and fused as in (B), then incubated without (lanes 1 – 6), or with, neural agrin (lane 7) for 20 min, lysed, immunoprecipitated with anti-MuSK, and immunoblotted with the phosphotyrosine-specific antibodies mAb 4G10 and PY20. The blot was stripped and re-probed for MuSK. Crude lysates were immunoblotted with mAb 9E10. (E) MuSK is dispensable for the AChR clustering induced by overexpression of Tid1(1–222). Myoblasts from wildtype or MuSK−/−mice were transfected with plasmid cDNAs encoding DsRed alone (control) or DsRed-tagged Tid1(1–222), respectively, and incubated in fusion medium for 48–72 h. Myotubes were fixed and stained with Alexa Fluor 488-conjugated αBTX. The results represent mean ± SEM for the number of AChR clusters per DsRed-positive myotube from four independent experiments. **, p < 0.01. n = 60 myotubes for each of the experimental groups.
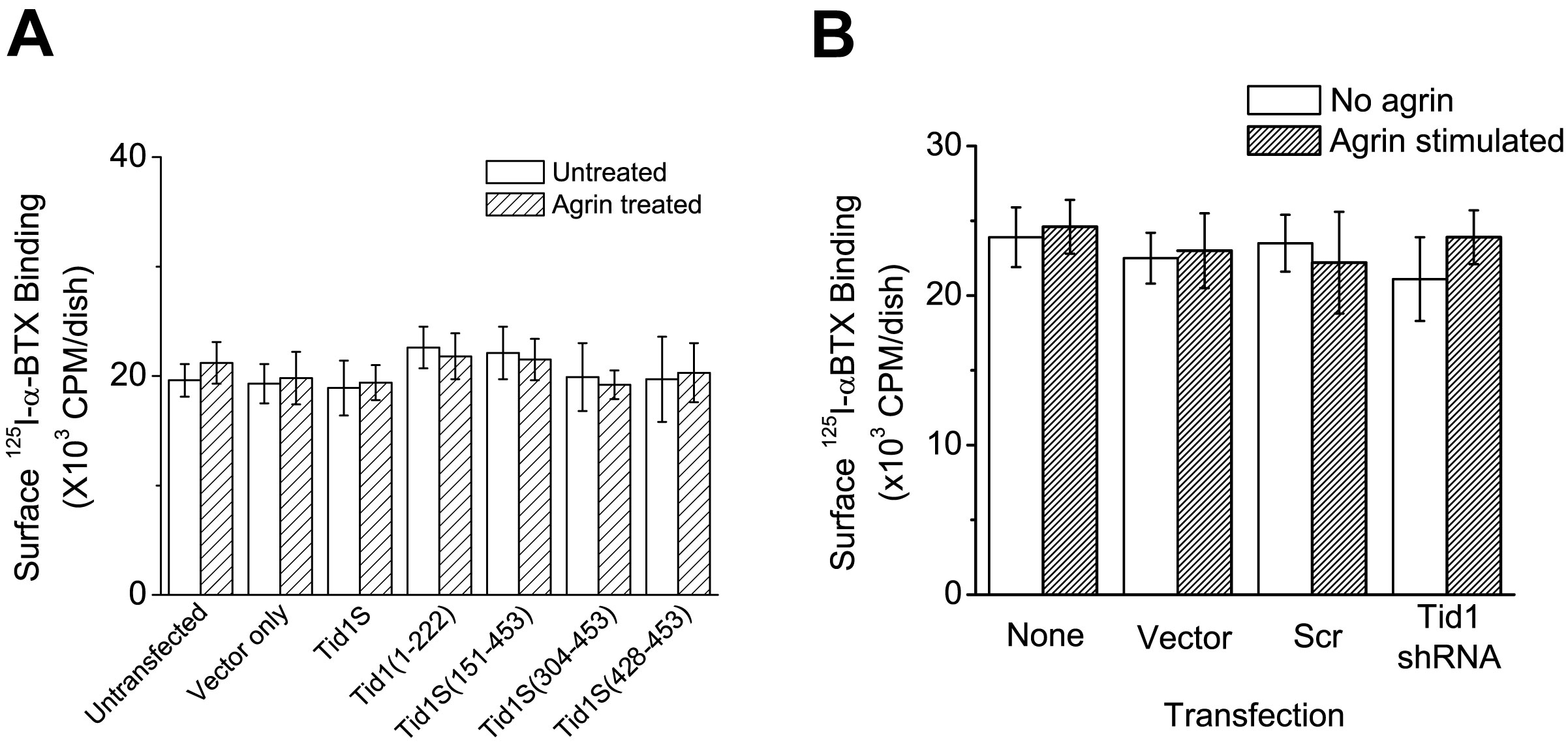
Whereas other constructs had no significant effect on the number and size of spontaneous AChR clusters (Fig. 4B, left panels), transfection of cDNA encoding the N-terminal half of Tid1 [Tid1(1–222)] induced robust clustering of AChRs (Fig. 4B, VII–IX; Fig. 4C). The clusters induced by Tid1(1–222) were smaller than those induced by agrin (Fig. 4B, i - iii), but were similar to receptor micro-clusters seen in the early stages of agrin-induced AChR clustering (Weston et al., 2003). In agrin-treated myotubes, transfection with Tid1(1–222) led to a slight, but statistically significant (0.01 < p < 0.05), increase in the number of clusters on the myotube surface (Fig. 4B, right panels; Fig. 4C) as compared to control myotubes. The physiologic activity of Tid1(1–222) was confirmed by showing that, like full-length human Tid1S, it is able to rescue the inhibition of AChR clustering caused by Tid1-targeted shRNA. The rescue was ineffective when the fragment contained the H121Q mutation (Supplementary Fig. 6D). Binding assays with 125I-αBTX revealed no increase in surface AChRs after transfection with Tid1(1–222) or other truncated fragments in either agrin-treated or untreated myotubes (Supplementary Fig. 7A). Interestingly, Tid1S was unable to induce AChR clustering in the absence of shRNA inhibition (Fig. 4, B, C).
As MuSK activation is the initial step in the agrin pathway, we examined whether Tid1(1–222) induces phosphorylation of MuSK by immunoprecipitating transfected cells with anti-MuSK and subsequently immunoblotting with antibodies to phosphotyrosine. None of the Tid1S fragments caused MuSK phosphorylation (Fig. 4D), suggesting that activation of MuSK is not required for Tid1(1–222)-induced AChR clustering. Transfecting muscle cultures from MuSK knockout mice with Tid1(1–222) revealed that even the total absence of MuSK had little or no effect on the fragment’s ability to cluster AChRs (Fig. 4E). Our experiments thus show that not only is Tid1 expression required for AChR clustering, but that an active fragment of Tid1 can induce AChR clustering in the absence of agrin or MuSK.
ShRNA–Mediated Tid1 Knockdown Disrupts the Structure and Function of the NMJ
To determine whether Tid1 is required for the maintenance of AChR clusters at mature endplates, we used electroporation to introduce either the Tid1-specific or scrambled shRNA into the TA muscles of adult mice. 5.5 weeks later, transfected and normal muscle fibers were stained for AChRs and Tid1. Untransfected muscle fibers and those expressing scrambled shRNA had a single large central AChR and Tid1-positive cluster that appeared as a brightly stained, “pretzel”-like structure with sharp boundaries, ~40 μm in diameter (Fig. 5A, a–d). In contrast, fibers expressing the Tid1-targeted shRNA showed mild to severe disruption of AChR clusters. In fibers with a slight decrease in Tid1 staining, individual receptor clusters were broken into several large fragments (Fig. 5A, e–h). In fibers in which Tid1 staining was severely reduced, the AChR clusters were fragmented into many (> 20) discontinuous structures, ~2–5 μm in diameter (Fig. 5A, i–p). In the most severe cases, Tid1 immunostaining was completely absent and the NMJ was massively disorganized, with numerous small, weakly-stained AChR clusters whose boundaries were poorly defined. These structures were scattered over a larger area than a normal AChR cluster, suggesting diffusion away from the endplate region (Fig. 5B, b, e). Ten weeks after electroporation, only a few fragments remained in prior sites of innervation (Fig. 5C, a–c). Tid1 is thus clearly required to maintain the normal structure of AChR clusters at adult motor endplates.
Figure 5. Tid1 Knockdown Disrupts the Structure and Function of the NMJ.
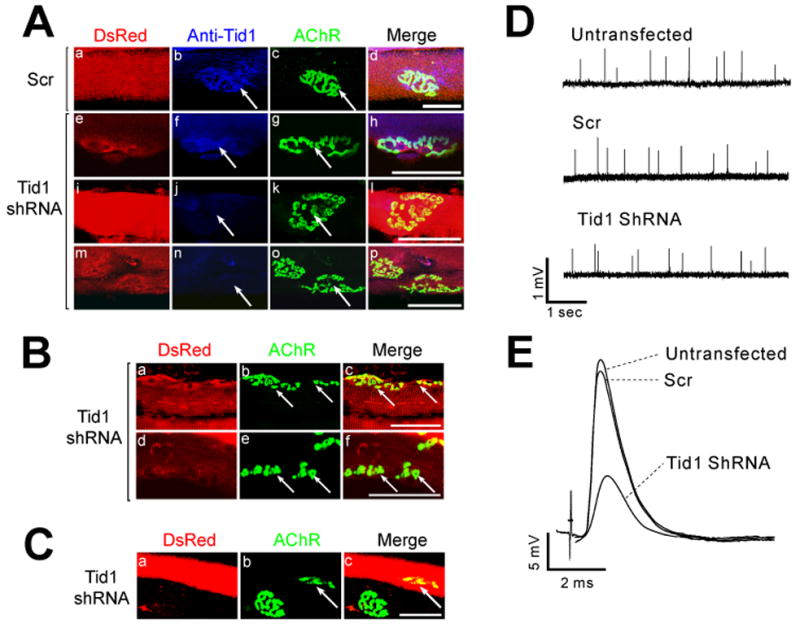
(A) TA muscles of adult mice were injected with 5 μg of plasmid pSIREN encoding either scrambled (Scr) or Tid1-targeted shRNA and electroporated in vivo. 5.5 weeks later the muscles were stained with Alexa Fluor 488-conjugated αBTX and anti-Tid1 followed by Alexa Fluor 633-conjugated goat anti-rabbit IgG. Transfected myotubes expressed DsRed-Express. Arrows highlight the NMJs. Scale bar = 50 μm for (A)–(C). (B) A higher dose (12 μg) of Tid1 shRNA causes fragmentation and diffusion of AChR clusters. (C) 10 weeks after electroporation, AChR clusters shrink (arrow) or disappear in transfected myofibers. (D, E) Tid1 knockdown disrupts the function of the NMJ. 5 weeks after electroporation, spontaneous miniature endplate potentials (mEPPs, D) and nerve stimulation-evoked endplate potentials (EPPs, E) were recorded as described in Experimental Procedures.
When myofibers expressing Tid1-shRNA were stained with a polyclonal antibody against synaptophysin, the pattern of presynaptic innervation was dramatically changed, closely matching the observed changes in AChR clusters (Supplementary Fig. 8). In fibers in which endplate Tid1 immunostaining was reduced or undetectable, AChRs were dispersed and appeared as small, isolated, bead-like aggregates. Motor nerve endings remained, forming numerous branches in register with the fragmented AChR clusters. These results are similar to findings that motor nerve termini rapidly withdraw and redistribute when AChRs at the NMJ are blocked by αBTX (Balice-Gordon and Lichtman, 1994).
We also found that shRNA-mediated Tid1 knockdown reduced synaptic transmission at the mouse NMJ. 5 weeks after electroporation, the amplitudes of miniature endplate potentials (mEPPs) and evoked endplate potentials (EPPs) in electroporated soleus muscle fibers were reduced by approximately 40% and 50%, respectively, when compared to control myofibers (Fig. 5D, E; Table 1). Tid1 knockdown had no effect on the mEPP frequency or the corrected quantal content, suggesting that Tid1 knockdown did not alter the release of ACh from presynaptic terminals. The reduced amplitudes of mEPPs and EPPs therefore reflect a reduction in postsynaptic AChR density at the NMJ, consistent with the morphological changes. Thus, Tid1 is essential for the maintenance of normal synaptic structure and function in adult muscles.
Table 1.
mEPP and EPP Properties at Control and Tid1 shRNA-Treated NMJs
| Untransfected | Scrambled shRNA | Tid1 shRNA | |
|---|---|---|---|
| RMP (mV) | −74.3 ± 0.9 | −73.9 ± 0. 6 | −74.1 ± 0.8 |
| mEPP rise time (ms) | 0.86 ± 0.11 | 0.86 ± 0.08 | 0.87 ± 0.12 |
| mEPP Amplitude (mV) | 0.80 ± 0.04 | 0.79 ± 0.04 | 0.45 ± 0.02 * |
| mEPP frequency (s−1) | 1.4 ± 0.09 | 1.5 ± 0.06 | 1.4 ± 0.07 |
| EPP rise time (ms) | 0.79 ± 0.03 | 0.77 ± 0.02 | 0.82 ± 0.05 |
| EPP amplitude (mV) | 25.3 ± 0.12 | 26.1 ± 0.09 | 13.7 ± 0.04 ** |
| QC (Corrected) | 64.8 ± 0.7 | 65.1 ± 0.6 | 62.19 ± 1.2 |
The values in the table are the means ± S.E.M. for 30 endplates from each group (three animals). The rise time was calculated as the time taken for the EPP or mEPP to rise from 10% to 90% of its full amplitude. QC, quantal content.
0.01 < P < 0.05 and
P < 0.01 by One-Way ANOVA.
Tid1S Is Required for, and Can Induce, Agrin-Induced Signaling Downstream of MuSK
Although the agrin signaling pathway remains poorly defined, several of its downstream steps have been identified, including phosphorylation and activation of MuSK, tyrosine phosphorylation of AChRs, and activation of the small GPTases Rac and Rho (reviewed in Sanes and Lichtman, 2001). To determine the position of Tid1 in relation to these events, we examined the effect(s) of shRNA-mediated knockdown of Tid1 on them.
Consistent with the results described above, knockdown of Tid1 did not change the overall expression or the activation of MuSK in response to stimulation by neural agrin (Fig. 6A). In contrast, Tid1 knockdown markedly reduced agrin-induced tyrosine phosphorylation of the AChR β subunit (Fig. 6B). The effect was specific, as it was counteracted by co-transfection with silently-mutated human Tid1S (hTid1S-DsRed). Tid1-targeted shRNA did not alter the overall expression level of the β subunit (Fig. 6B) or the entire AChR (Supplementary Fig. 7B) in muscle cells. The knockdown experiments suggest that Tid1’s effects on AChR phosphorylation occur downstream of MuSK. This conclusion was supported by experiments with Tid1(1–222). Transfection of myotubes with Tid1(1–222) induced tyrosine phosphorylation of the β subunit in the absence of agrin (Fig. 6C). Moreover, Tid1(1–222) also induced AChR phosphorylation in myotubes derived from MuSK−/− mice (Fig. 6D).
Figure 6. Tid1S Is Indispensable for Agrin-induced Signaling Events Downstream of MuSK.

(A) Tid1 shRNA has little effect on agrin-induced MuSK activation. C2C12 myoblasts were transfected overnight with plasmid cDNAs encoding human Tid1S, Tid1L, control (Scr) or Tid1-specific shRNA, differentiated in fusion medium for ~48 h, and incubated with recombinant neural or muscle agrin for 20 minutes. Detergent extracts of the cells were immunoprecipitated with anti-MuSK and immunoblotted with a mixture of mAb 4G10 and mAb PY20. The blot was stripped and re-probed for MuSK. (B) Tid1 shRNA diminishes agrin-induced tyrosine phosphorylation of the AChR β subunit. Transfected myotubes were incubated with recombinant agrins for 1 h and then lysed. AChRs were “pulled-down” using αBTX-Sepharose beads and immunoblotted with mAbs 4G10 and PY20. The blot was stripped and re-probed with mAb 124 for the AChR β subunit. (C) Overexpression of the N-terminal half of Tid1 induces agrin-independent phosphorylation of the AChR β subunit. C2C12 myoblasts were transfected as described for Fig. 4B and incubated without (lanes 1–6), or with, neural agrin for 1 h (lane 7). The cell lysates were immunoprecipitated and immunoblotted as described in (B). Crude lysates were probed with mAb 9E10. (D) MuSK is dispensable for β subunit phosphorylation induced by overexpression of the N-terminal half of Tid1. Myoblasts from wildtype or MuSK knockout mice were transfected with plasmid cDNA encoding myc-tagged Tid1(1–222) and differentiated in fusion medium. Detergent extracts were immunoprecipitated and immunoblotted as described in (B) and crude lysates were immunoblotted with anti-MuSK and mAb 9E10. (E) Tid1 shRNA inhibits the activation of small GTPases in response to agrin stimulation. C2C12 myoblasts were transfected overnight with plasmids encoding Scr shRNA, Tid1-specific shRNA, or Tid1 shRNA co-expressed with silently-mutated hTid1S fused to DsRed (hTid1S-DsRed). Myotubes grown in fusion medium for 48–72 h were incubated with recombinant neural agrin for different periods of time, as indicated. Detergent extracts were assayed with the “EZ-Detect” Rac or Rho activation kit. (F) Rac and Rho activation were quantified using densitometry. Rac-GTP and Rho-GTP levels were indexed against those from unstimulated myotubes (time 0). The results represent mean ± SEM of four independent experiments. *, 0.01 < p < 0.05; **, p < 0.01.
We next examined the effect of Tid1 knockdown on agrin-induced activation of the small GTPases Rac and Rho (Weston et al., 2000; 2003; Fig. 6E, F). In both cases, Tid1 knockdown diminished GTPase activation and the effects were reversed by co-transfection with silently-mutated human Tid1S. Taken together, our results show that Tid1S occupies an essential position in the agrin signaling pathway that is subsequent to MuSK activation, but prior to AChR phosphorylation and the activation of Rac and Rho.
Tid1 Acts Downstream of Dok-7 to Induce AChR Clustering
Like Tid1S, the cytoplasmic protein Dok-7 is an essential mediator of MuSK-dependent AChR clustering that can induce robust clustering in the absence of agrin (Okada et al., 2006). To determine whether Tid1 is required for this induction, we expressed DsRed (control), Dok-7-DsRed, or Dok-7-DsRed together with scrambled (Scr) or Tid1-specific shRNA, respectively, in C2C12 myotubes. ShRNA-mediated knockdown of Tid1 significantly reduced the AChR clustering induced by overexpression of Dok-7 (Fig. 7A). Silently-mutated hTid1S, but not hTid1L, rescued the clustering effect of Dok-7 (data not shown). Thus the effects of Dok-7 on AChR clustering are at least partially dependent on Tid1S.
Figure 7. Tid1 Acts Downstream of Dok-7.
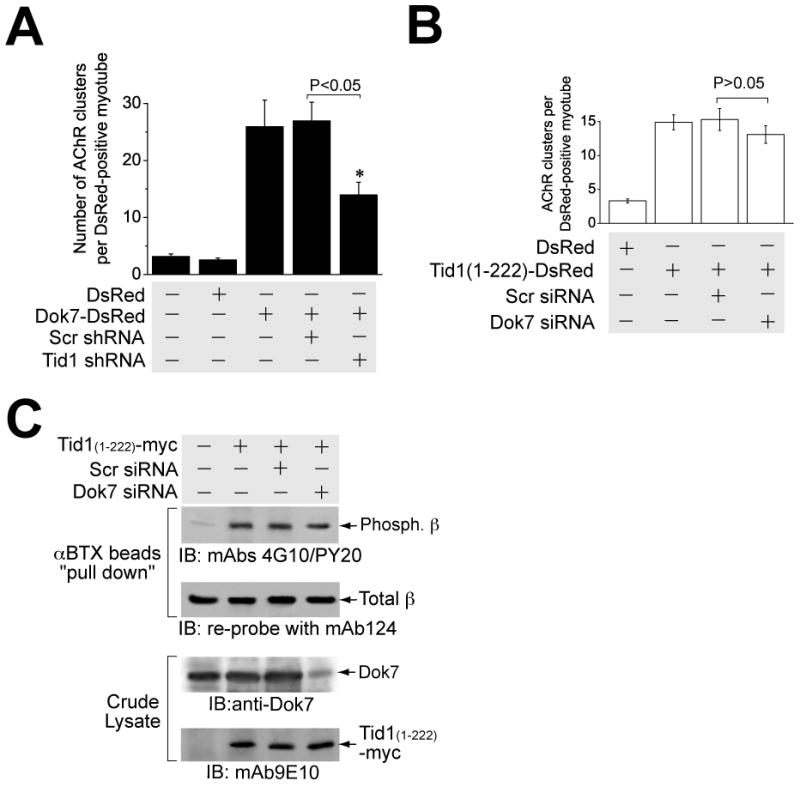
(A) Tid1 shRNA disrupts Dok-7-induced AChR clustering. C2C12 myoblasts were transfected with plasmids encoding DsRed (control), Dok-7-DsRed, or Dok-7-DsRed together with Scr or Tid1-specific shRNA, respectively. After differentiation, myotubes were fixed briefly and stained with Alexa Fluor 488-conjugated αBTX. The results represent mean ± SEM of four independent experiments. *, p < 0.05. n = 60 myotubes for each of the experimental groups. (B) Dok-7 siRNA has no significant effect on AChR clustering induced by the overexpression of Tid1(1–222). C2C12 myoblasts were transfected with plasmid cDNAs encoding DsRed (control), Tid1(1–222)-DsRed, or Tid1(1–222)-DsRed together with Scr siRNA or Dok-7 siRNA, respectively. After differentiation, myotubes were fixed and stained with Alexa Fluor 488-conjugated αBTX. The results represent mean ± SEM of four independent experiments. n = 80 myotubes for each of the experimental groups. (C) AChR β subunit phosphorylation induced by the N-terminal domain of Tid1 is not affected by Dok-7-targeted siRNA. C2C12 myoblasts were transfected with plasmid cDNAs encoding myc-tagged Tid1(1–222), along with either control (Scr) or Dok-7 siRNA, respectively. Myotubes were lysed with extraction buffer and assayed for β phosphorylation as described in Fig. 6B. Crude lysates were immunoblotted with rabbit anti-Dok-7 and mAb 9E10 antibodies.
To determine whether Dok-7 is required for the induction of AChR clustering by Tid1(1–222), we transfected myotubes with cDNA for Tid1(1–222) along with either a Dok-7-targeted or a scrambled siRNA (Okada et al., 2006). Although the siRNA reduced levels of Dok-7 by > 75% (Fig. 7C), we observed no significant difference in the induction of AChR clusters by Tid1(1–222) (Fig. 7B). Additionally, Dok-7 knockdown had no effect on the ability of Tid1(1–222) to induce phosphorylation of the AChR (Fig. 7C). Because Tid1(1–222) acts independently of Dok-7 to induce AChR clusters and Dok-7-induced clustering is diminished by Tid1 knockdown, we conclude that Tid1S acts downstream of Dok-7 in the AChR clustering signaling pathway.
Tid1 Regulates Dok-7 Binding to MuSK
Dok-7 binds to MuSK through a phosphotyrosine binding domain (Okada et al., 2006). As Tid1 constitutively associates with MuSK, we examined whether Dok-7 requires Tid1 to bind to MuSK. In co-immunoprecipitation experiments with C2C12 myotubes, shRNA-mediated Tid1 knockdown markedly reduced agrin-induced association of MuSK and Dok-7 without affecting the total levels of Dok-7 or MuSK (Fig. 8A). Similar results were found when lysates of myotubes expressing Tid1 shRNA were immunoprecipitated with anti-MuSK and then subjected to immunoblotting with an antibody against Dok-7 (anti-Dok-7) or when the lysates were immunoprecipitated with anti-Dok-7 and immunoblotted with anti-MuSK. The effect of Tid1 knockdown on the MuSK-Dok-7 interaction was reversed by co-transfection with silently-mutated hTid1S, but not hTid1L.
Dok-7 is Associated with Tid1S Through MuSK, but is Dispensable for Tid1S Binding to MuSK
To see whether Dok-7 is associated with Tid1S, we cultured myotubes from wildtype mice with neural agrin, immunoprecipitated the cell lysates either with anti-Tid1S (Fig. 8B, top panels) or with anti-Dok-7 (Fig. 8B, middle panels), and probed the resulting immunoblots with the complementary antibody. The results clearly demonstrate agrin-induced association between the two proteins. The interaction between Tid1S and Dok-7 is apparently mediated by MuSK, since, in lysates of MuSK-deficient myotubes, Dok-7 was barely detected in precipitates pulled-down by anti-Tid1S, and Tid1S was diminished in precipitates pulled-down by anti-Dok-7. Transfection of the MuSK−/− cells with FLAG-tagged MuSK cDNA completely restored the agrin-induced interaction between Tid1S and Dok-7 (Fig. 8B).
Since Tid1S regulates Dok-7 binding to MuSK, Dok-7 could also regulate the association of Tid1S with MuSK. C2C12 myotubes expressing control (Scr) or Dok-7-specific siRNA were incubated with or without neural agrin and their detergent extracts were subjected to co-immunoprecipitation with anti-MuSK or anti-Tid1S. Although Dok-7 siRNA significantly reduced the level of Dok-7, it did not change the amount of Tid1S co-immunoprecipitated by anti-MuSK, nor of MuSK co-immunoprecipitated by anti-Tid1S (Fig. 8C), either with or without agrin treatment. Thus, although Tid1S and agrin are required for Dok-7 to bind to MuSK, the association between Tid1S and MuSK appears to be independent of both Dok-7 and agrin.
Discussion
We used two-hybrid assays to identify a new binding protein for MuSK, a component of the agrin receptor responsible for nerve-induced AChR clustering at the neuromuscular junction. The short form of Tid1, Tid1S, a member of the hsp40 protein family, bound specifically to the juxtamembrane region of MuSK in two-hybrid experiments and immunoprecipitation showed that Tid1S is associated with MuSK in mouse myotubes and adult muscles (Fig. 1). As with MuSK (Bowen et al., 1998), Tid1 is precisely co-localized with spontaneous and agrin-induced AChR clusters in the membranes of cultured myotubes (Fig. 2). In muscle, Tid1 co-localizes with AChR clusters from their first appearance at developing endplates and remains co-distributed with AChRs throughout the morphological changes that occur in the postsynaptic muscle membrane during maturation, denervation and reinnervation (Fig. 2 and Supplementary Fig. 4, 5). Thus, Tid1S, along with rapsyn and MuSK, appears to be an important component of the complex of proteins associated with AChR clusters in both synaptic and non-synaptic muscle membrane.
Protein knockdown by shRNA interference demonstrated that Tid1 is required for both spontaneous and agrin-induced AChR clustering in myotubes (Fig. 3) and for the maintenance of clusters at synapses in adult muscle fibers (Fig. 5). In myotubes, Tid1S, but not Tid1L, can rescue the inhibition of AChR clustering induced by shRNA (Fig. 3). Thus, the short form of Tid1 appears to be essential for AChR cluster formation and maintenance in aneural cultures and at developing and mature synapses. In the absence of agrin, introduction of the N-terminal half of Tid1 into myotubes can induce AChR phosphorylation (Fig. 6) and robust AChR clustering, without MuSK activation (Fig. 4). Since Tid1S does not affect surface AChR levels, it presumably acts, as does the nerve (Anderson et al., 1977), by redistributing preformed surface AChRs. These experiments demonstrate an essential role for Tid1S in the induction and maintenance of high-density AChR clusters.
Both the amount and localization of Tid1 are under neural regulation. Tid1 increases in amount after innervation and during muscle development. Most strikingly, chronic denervation results in increased expression of Tid1 and a marked dispersal of Tid1 from the endplate membrane, an event parallel to, or slightly preceding, the disassembly of AChR clusters. Tid1 also appears to influence presynaptic morphology. When Tid1 is reduced in adult muscle fibers, endplates disperse into small aggregates of AChRs, with a corresponding rearrangement of synaptic terminals. The resulting reorganization of endplate structure is similar to that seen after αBTX blockade (Balice-Gordon and Lichtman, 1994).
The localization of Tid1S to the motor endplate is apparently mediated by MuSK, a membrane protein. Tid1S has no transmembrane sequence, but is found in both the membrane and cytosol of myotubes (Fig. 2). In other cells, Tid1 has been localized to the cytosol and mitochondria (Lu et al., 2006). In wildtype myotubes, 30% of total Tid1S is bound to the membrane, but in MuSK−/−myotubes, only 8% is bound. Clustered Tid1 is also not observed on the surface of MuSK−/− myotubes, either in the presence or absence of agrin (Fig. 2). The binding of Tid1S to MuSK appears to be independent of MuSK activation, as agrin does not change the amount of Tid1S in membrane fractions of wildtype myotubes (Fig. 2) and agrin does not increase the amount of Tid1S that is co-immunoprecipitated with MuSK (Fig. 8C). The observed binding of Tid1S to the juxtamembrane region of MuSK in the bacterial two-hybrid system is also consistent with the idea that MuSK phosphorylation and activation is not required for Tid1S binding.
Most likely, the C-terminal domain of Tid1S, which differs from that of Tid1L (Fig. 1A, Supplementary Fig. 1C), contains a region responsible for MuSK binding. All of the clones isolated from the two-hybrid screens contain the C-terminal sequence, as well as the cysteine-rich domain, but none of them contain the N-terminal third of the protein (i.e. aa 1–146), which includes the DnaJ domain (Fig. 1A). The idea that the C-terminal domain mediates binding is consistent with studies in other cells showing that this region mediates the binding of Tid1 to other RTKs, including ErbB2 and the Trk family (Kim et al., 2004; Liu et al., 2005), whose activities Tid1 regulates.
The functional activity of Tid1S, however, appears to reside in its N-terminal domain. The N-terminal half of Tid1, Tid1(1–222), when introduced into either wildtype or MuSK−/− myotubes, led to robust clustering of AChRs, whereas other fragments of Tid1S did not (Fig. 4). Moreover, silently-mutated human Tid1(1–222) rescued both spontaneous (data not shown) and agrin-induced AChR clustering after shRNA-mediated knockdown of endogenous Tid1 in myotubes (Supplementary Fig. 6D). Tid1(1–222) induced AChR clustering without activating MuSK. Interestingly, full-length Tid1S was unable to induce AChR clustering, suggesting that the N-terminal domain may be in an inactive conformation in the native molecule. Activation of MuSK may change the conformation of bound Tid1S to a form in which the N-terminal domain is exposed and thus activated. Activation-dependent conformational changes are characteristic of hsp-like proteins, including Tid1’s co-chaperone, hsp70 (Liberek et al., 1991). The ability of full-length Tid1S to rescue spontaneous and agrin-induced AChR clustering after shRNA inhibition and its inability to induce clustering when overexpressed could be explained if it is assumed that exogenously-added Tid1S becomes associated with MuSK in experiments in which endogenous levels are low, but that when overexpressed, exogenously-added Tid1S does not become associated with MuSK, because MuSK is fully occupied with endogenous Tid1S.
The DnaJ domain of Tid1, contained in the N-terminal half, binds to and activates hsp70 in other cells (Liberek et al., 1991). An H121Q mutation in the DnaJ domain prevents activation, but not binding, of Tid1 to hsp70 (Syken et al., 1999) and, in breast cancer cells, interferes with the ability of Tid1 to down-regulate the ErbB2 receptor (Kim et al., 2004). In myotubes, the H121Q mutation abolishes the ability of both human Tid1S and the Tid1 N-terminus to rescue clustering from Tid1 shRNA inhibition. The DnaJ domain may thus regulate AChR clustering by mediating the activation of hsp70 by Tid1. Hsp90, known to bind to hsp70 and to mediate signal transduction (Pratt and Toft, 2003), has also been isolated from the Torpedo electric organ, which is rich in cholinergic synapses (Balasubramanian et al., 1998). How Tid1S interacts with these proteins to orchestrate clustering remains an important question for the future.
In fibroblasts, Tid1 binds to a RasGAP protein, which also interacts with the adaptor molecule Dok (Trentin et al., 2001). The major form of Dok in muscle is Dok-7, a cytoplasmic protein essential for mediating MuSK-dependent AChR clustering (Okada et., 2006). Dok-7 binds to MuSK through a phosphotyrosine binding (PTB) domain and is required for MuSK activation by agrin. Our experiments indicate an intricate interplay between MuSK, Tid1S and Dok-7 in myotubes. We find that agrin-induced Dok-7 binding to MuSK is diminished when Tid1 is reduced by knockdown; Tid1S binding to MuSK is, however, unaffected by reduction of Dok-7 (Fig. 8). These results suggest that Tid1S binds to MuSK in the absence Dok-7 and that during activation of MuSK, Dok-7 binds to the MuSK/Tid1S complex. Knockdown and rescue experiments showed that full induction of clustering by Dok-7 requires Tid1S (Fig. 7A). Knockdown of Dok-7, however, had no effect on the ability of Tid1(1–222) to induce AChR phosphorylation and clustering. Thus, Tid1S appears to act downstream of Dok-7 in the signaling pathway responsible for AChR clustering. Taken together, our results suggest a model in which the role of Dok-7 is to mediate between the activation of MuSK and the activation of Tid1S. We propose that during activation of MuSK, the MuSK/Tid1S complex binds Dok-7, which initiates a conformational change in Tid1S that activates the N-terminal domain (Fig. 8D). The requirement for activation is obviated when only the N-terminal domain of Tid1S is introduced directly into muscle cells.
The model explains why Dok-7 is required for agrin-induced clustering but not for that induced by Tid(1–222). It also explains the inability of full-length Tid1S to initiate clustering when overexpressed, as Tid1S activation depends on binding to MuSK and Dok-7. The model does not explain, however, why Tid1S is required for spontaneous AChR clustering in the absence of agrin. Such clusters could occur through a low level of spontaneous activation of MuSK in the absence of agrin (Burden, 2002) or a low level of spontaneous activation of Tid1S in the absence of MuSK phosphorylation and Dok-7 binding.
How Tid1S acts downstream of MuSK and Dok-7 to implement AChR clustering is unknown. Cytoskeletal rearrangements are thought to be crucial for AChR clustering (Luo et al., 2003). We have shown that Tid1S is required for agrin-induced AChR phosphorylation, important for linking AChRs to the cytoskeleton (Friese et al., 2007). We previously reported that Adenomatous Polyposis Coli (APC), an important regulator of the cytoskeleton, binds to the β subunit of the AChR and may play a role in AChR cluster formation (Wang et al., 2003). As Tid1 and APC may interact (Polakis et al. 1997), Tid1 could link AChRs to the MuSK scaffold through APC. We have shown that Tid1 regulates the activity of RhoA and Rac-1 (Fig. 6) and, through the H121Q mutation studies, that Tid1 likely activates heat shock proteins, each of which can influence cytoskeletal dynamics (Weston et al., 2000, 2003; Liang and MacRae, 1997). Tid1 is known to bind to heat shock cognate protein 70 (hsc70) (Lu et al., 2006), which negatively regulates the activation of Rac1, Cdc42, and RhoA (Kauppinen et al., 2005). Activation of Tid1 at developing endplates could release hsc70, disinhibiting the downstream GTPases. The interaction between Tid1/hsp70 and RasGAP may also promote the formation of a signaling complex which influences the cytoskeleton (Trentin et al., 2001). Indeed, RasGAP binds p190 (Settleman et al., 1992), a Rho/Rac GTPase activating protein. Interaction between Tid1 and RasGAP and subsequent activation of p190 could thus provide an alternate, or parallel, mechanism for small GTPase activation downstream of MuSK (Supplementary Fig. 9).
Tid1 may have additional roles at the NMJ other than those mentioned above. For instance, Tid1 has been shown to regulate the cellular signaling of ErbB2 and the Trk family of RTKs (Kim et al., 2004; Liu et al., 2005). TrkB appears to play a role in the maintenance of AChR localization at the NMJ (Gonzalez et al., 1999) and Erb may regulate the transcription of subsynaptic NMJ proteins (Rimer, 2007). Through a complex interplay with MuSK, TrkB, and ErbB, Tid1 may unite these multiple RTK signaling programs to coordinate the synaptic localization and maintenance of AChRs. How and when these different pathways converge during neuromuscular synapse development will be an important question for the future. Our identification of Tid1S as a critical component of the postsynaptic apparatus emphasizes the important role of heat shock proteins in synaptogenesis at the NMJ.
Experimental Procedures
Materials
The BacterioMatch II Two-Hybrid System and BacterioMatch II Rat Skeletal Muscle cDNA Library were from Stratagene (La Jolla, CA). cDNAs encoding human Dok-7 and mouse MuSK were amplified from cDNA libraries of human and mouse skeletal muscles (Stratagene), respectively, and confirmed by DNA sequencing. The pDsRed Express and RNAi-Ready pSIREN-DNR-DsRed-Express cloning vectors were from Clontech (Mountain View, CA). The Effectene transfection reagent was from Qiagen (Valencia, CA). Neural and muscle agrin were produced as described by Ferns et al. (1992).
A rabbit polyclonal antibody against [pan-]Tid1 (anti-Tid1) and its associated pre-immune serum were generous gifts from J.-D. Lee (Scripps Research Institute, La Jolla, CA). The Tid1 short-specific polyclonal antibody (anti-Tid1S) was generated by immunizing a rabbit with the last 20 residues in the C-terminus of mouse Tid1S (VEGTVNGVTHTSTGKRSTGN) (Genemed Synthesis, San Francisco, CA). The rabbit antibody against MuSK was a gift from Zach Hall (Fuhrer et al., 1997; Sugiyama et al., 1997). Rabbit anti-Dok-7 (H-77 and H-284) were from Santa Cruz Biotech (Santa Cruz, CA) and anti-Rac and anti-Rho monoclonal antibodies were from Pierce (Rockford, IL). MuSK-knockout and wildtype control myoblasts were gifts from Dr. Yancopoulos (Regeneron Pharmaceuticals, Tarrytown, NY). See supplementary materials for additional details.
Bacterial Two-Hybrid Screens
Bacterial two-hybrid screens were performed according the manufacturer’s protocols. The cytoplasmic (aa 515–869) and juxtamembrane (aa 515–692) domains of MuSK were subcloned into the bait vector, pBT, and the commercial (E19) rat skeletal muscle cDNA library was subcloned into the target vector, pTRG.
cDNA Constructs
Full-length Tid1S (residues 1–453), Tid1S lacking the DnaJ domain (residues 151–453), Tid1S lacking the DnaJ and cysteine-rich domains (residues 304–453), the C-terminus of Tid1S (residues 428–453), and the N-terminal half of Tid1 (residues 1–222) were amplified from the rat muscle cDNA library by PCR and subcloned into the StuI site in the mammalian expression vector pCS2+MT (from D.L. Turner, University of Michigan, MI). Constructs were confirmed by DNA sequencing.
Transfection and Cell Lysis
C2C12 myoblasts were grown, fused into myotubes, and extracted as detailed by Jacobson, et al. (1998). Fusion media was changed daily. Myoblasts were transfected at ~60% confluence with Effectene according to the manufacturer. Extraction buffer consisted of 1% NP-40, 0.5% deoxycholic acid, 50 mM Tris-HCl pH 8.0, 50 mM NaCl, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO4 with a protease inhibitor cocktail (1%; Sigma catalog number P3840) and PMSF (4 mM).
Denervation and Muscle Extraction
The Institute Animal Care and Use Committee (IACUC) of University of Southern California approved all animal experiments. ~5 mm of the sciatic nerve was removed from the right hindlimb of adult C57/BL6 mice under anesthesia with sodium pentobarbital. For immunostaining, hindlimb muscles were quickly dissected from euthanized animals, embedded in OCT medium, flash-frozen in dry ice-cooled isopentane, and sectioned with a cryostat (Wu et al., 1985). For biochemical assays, fresh muscles were minced with scissors and homogenized in a glass douncer with extraction buffer (1.25% Triton X-100, 50 mM Tris-HCl pH 7.4, 50 mM NaCl, 1 mM EDTA, 1 mM EGTA) supplemented with protease inhibitors. Protein concentrations of the cleared homogenates were determined by bicinchoninic acid (BCA) assays (Pierce Biotechnology, Rockford, IL).
Immunoprecipitation and Immunoblotting
Detergent extracts of myotubes or skeletal muscles were incubated overnight at 4°C with antibodies followed by Protein-A Sepharose beads for 1 h. Beads were spun down and washed with extraction buffer; bound proteins were eluted by heating the beads at 95°C for 5 min in NuPAGE (Invitrogen) LDS Sample Buffer with reducing agents. They were then separated on 4–12% NuPAGE Novex Bis-Tris gels, and electroblotted onto nitrocellulose membranes. Membranes were blocked for 2 h at room temperature in 5% milk in Tris-buffered saline, incubated with primary antibodies diluted in blocking buffer plus 0.1% Tween-20 overnight at 4°C, followed by Alexa Fluor dye-conjugated secondary antibodies for 1 h at room temperature. The membranes were scanned on an Odyssey Infrared Imaging System (LI-COR, Lincoln, Nebraska). Some blots were stripped (buffer: 0.2 M glycine pH 2.5, 0.05% Tween-20) for 30 min at room temperature, washed, blocked, and reprobed. Band intensity was semi-quantified by using the ImageJ software package (National Institutes of Health, Bethesda, MD).
Immunofluorescent Staining
C2C12 myotubes grown in 35 mm culture dishes or cryosections of skeletal muscles were fixed in 4% paraformaldehyde for 5 min, washed with PBS, and permeabilized with 0.3% Triton X-100 in PBS. The samples were blocked with 1% bovine serum albumin (BSA) for 30 min and incubated with primary antibodies overnight at 4°C, followed by Alexa Fluor-conjugated secondary antibodies for 1 h at room temperature. Purified rabbit IgG (2 μg/100 μl), rat serum, or pre-immune sera were used as negative controls. Sections were mounted in a glycerol mixture (4% n-propyl gallate, 86% glycerol, and 10% PBS), viewed under an epifluorescence microscope, and photographed with a cooled CCD camera.
Cell fractionation
Myotubes were fractionated as detailed by Kadoya et al. (2005).
Quantification of AChR Clusters
AChR clusters in C2C12 myotubes were viewed with a Nikon E600 epifluorescent microscope and were quantified as described either by Jacobson et al. (1998) or Okada et al. (2006).
Radioligand Binding Assay
Surface expression of AChRs was determined by incubating C2C12 myotubes for 90 min with 5 nM 125I-αBTX in growth medium (Wang et al., 2003). Nonspecific binding was measured by pre-incubation with an excess amount of unlabeled αBTX (500 nM). The cells were washed with PBS 3 times, solubilized in 0.2M NaOH, and bound 125I-αBTX was measured in a gamma counter.
RNA Interference
Tid1-specific shRNA (GATCCAGATGATTATTACCAGATCTTCAAGAGAG ATCTGGTAATAATCATCTTTA (sense) and AGCTTAAAGATGATTATTACCAGA TCTCTCTTGAAGATCTGGTAATAATCATCTG (antisense)), targeting base pairs 347–361 of mouse Tid1 were subcloned into the RNAi vector, pSIREN-DNR-DsRed-Express (Clontech), as detailed in the supplementary materials. The control was a pSIREN vector containing a scrambled shRNA sequence. The shRNA constructs were transfected into C2C12 myoblasts with Effectene. ~24 h later, the cells were switched to fusion medium for ~72 h. Myotubes were fixed in 2% paraformaldehyde and then immunofluorescently stained.
For the rescue experiments, 2 nucleotides in the shRNA-targeted region of human Tid1 cDNA were changed by silent mutation. The final sequence of this region was AAGAAGACTATTATCAGATA (silent mutations in bold). 3 other nucleotides (underlined) in this region of human Tid1 differ from those in mouse Tid1 cDNA, preventing targeting by the mouse-specific shRNA. Site-directed mutagenesis added a SmaI site immediately before the DsRed sequence in the Tid1-targeted shRNA vector described above. cDNAs encoding the silently-mutated human Tid1S, Tid1 long (Tid1L), or the N-terminal half of human Tid1 were subcloned into the SmaI site, thus expressing Tid1 and DsRed as a fusion protein.
SiRNA to mouse Dok-7 (5′-CACCACTATGACACACCTCGA-3′) and the control siRNA (5′-AATTCTCCGAACGTGTCACGT-3′) were obtained from Qiagen and used as according to Okada et al. (2006).
In vivo RNA Interference using the Tid1-targeted and scrambled shRNA (5 μg DNA in 30 μl 0.9% NaCl, injected into TA muscles of adult C57/BL6 mice) was performed as described by Kong et al. (2004) and Sadasivam et al. (2005). 5.5 weeks after electroporation, the animals were anesthetized, perfused with 2% paraformaldehyde, and electroporated muscles were dissected and immunostained. Fluorescent images were taken using an Olympus FluoView-1000 confocal microscope with three non-overlapping laser lines.
Electrophysiology
5 weeks post-electroporation, electrophysiological recordings were made from positively-transfected myofibers superfused with Kreb’s solution (95% O2 and 5% CO2), as detailed by Wang et al. (2005). 30s of equilibration was allowed; up to 30 mEPPs followed by up to 30 EPPs (stimulated at 1 Hz) were recorded (at a 10 kHz sampling frequency) per endplate for offline analysis.
Rac/Rho GTPase Activation Assays
The EZ-Detect Rac or Rho activation kits were used according to the manufacturer (Pierce Chemical, Rockford, IL). Total Rac and Rho were immunoprecipitated with anti-Rac and anti-Rho antibodies, also used for immunoblotting. Relative band intensity normalized to time 0 was quantified (ImageJ software). Control and experimental conditions were compared at each time point.
Statistical Analysis
All quantitative data were analyzed using Origin 8.0. Statistically-significant differences between two groups were determined by two-way Student’s T-tests for independent populations. Differences between multiple groups were analyzed by one-way ANOVA.
Supplementary Material
{kind=link}
Acknowledgments
We thank the Zilkha Neurogenetic Institute of USC and the MSTP of the University of Pittsburgh for their support during the final preparation of the manuscript for publication. We thank Dr. Zach Hall for his help in the final revision of the manuscript, and Dr. Michael Ferns for his suggestions. We thank Dr. Daniel Altschuler for advice and reagents for the shRNA experiments, and Drs. Sidney Pestka and Karl Münger for providing human Tid1 plasmid cDNAs. We are grateful to Drs. Willi Halfter, Donald DeFranco, Elias Aizenman, and Guillermo Romero for helpful discussions. This work was supported by research grants from the National Science Foundation (NSF, IOS-0641660), National Institutes of Health (NIH, RO1-NS038301), and Muscular Dystrophy Association (MDA) to Z.Z.W, as well as predoctoral training grants from NIGMS (T32 GM08424) and NINDS (F30 NS053013) to J. L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson MJ, Cohen MW, Zorychta E. Effects of innervation on the distribution of acetylcholine receptors on cultured muscle cells. J Physiol. 1977;268:731–756. doi: 10.1113/jphysiol.1977.sp011879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balasubramanian S, Fung ET, Huganir RL. Characterization of the tyrosine phosphorylation and distribution of dystrobrevin isoforms. FEBS Lett. 1998;432:133–140. doi: 10.1016/s0014-5793(98)00804-7. [DOI] [PubMed] [Google Scholar]
- 3.Balice-Gordon RJ, Lichtman JW. Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature. 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]
- 4.Bowen DC, Park JS, Bodine S, Stark JL, Valenzuela DM, Stitt TN, Yancopoulos GD, Lindsay RM, Glass DJ, DiStefano PS. Localization and regulation of MuSK at the neuromuscular junction. Dev Biol. 1998;199:309–319. doi: 10.1006/dbio.1998.8936. [DOI] [PubMed] [Google Scholar]
- 5.Burden S. Building the vertebrate neuromuscular synapse. J Neurobiol. 2002;53:501–511. doi: 10.1002/neu.10137. [DOI] [PubMed] [Google Scholar]
- 6.Christian CN, Daniels MP, Sugiyama H, Vogel Z, Jacques L, Nelson PG. A factor from neurons increases the number of acetylcholine receptor aggregates on cultured muscle cells. PNAS. 1978;75:4011–4015. doi: 10.1073/pnas.75.8.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferns M, Hoch W, Campanelli JT, Rupp F, Hall ZW, Scheller RH. RNA splicing regulates agrin-mediated acetylcholine receptor clustering activity on cultured myotubes. Neuron. 1992;8:1079–1086. doi: 10.1016/0896-6273(92)90129-2. [DOI] [PubMed] [Google Scholar]
- 8.Finn AJ, Feng G, Pendergast AM. Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat Neurosci. 2003;6:717–723. doi: 10.1038/nn1071. [DOI] [PubMed] [Google Scholar]
- 9.Frank E, Gautvik K, Sommerschild H. Persistence of junctional acetylcholine receptors following denervation. Cold Spring Harb Symp Quant Biol. 1976;40:275–281. doi: 10.1101/sqb.1976.040.01.028. [DOI] [PubMed] [Google Scholar]
- 10.Friese MB, Blagden CS, Burden SJ. Synaptic differentiation is defective in mice lacking acetylcholine receptor beta-subunit tyrosine phosphorylation. Development. 2007;134:4167–4176. doi: 10.1242/dev.010702. [DOI] [PubMed] [Google Scholar]
- 11.Fuhrer C, Sugiyama JE, Taylor RG, Hall ZW. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 1997;16:4951–4960. doi: 10.1093/emboj/16.16.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaestel M. Molecular chaperones in signal transduction. Handb Exp Pharmacol. 2006;172:93–109. doi: 10.1007/3-540-29717-0_4. [DOI] [PubMed] [Google Scholar]
- 13.Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD. Agrin acts via a MuSK receptor complex. Cell. 1996;85:513–523. doi: 10.1016/s0092-8674(00)81252-0. [DOI] [PubMed] [Google Scholar]
- 14.Godfrey EW, Nitkin RM, Wallace BG, Rubin LL, McMahan UJ. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J Cell Biol. 1984;99:615–627. doi: 10.1083/jcb.99.2.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of TrkB-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- 16.Hall ZW, Sanes JR. Synaptic structure and development: the neuromuscular junction. Cell. 1993;72(Suppl):99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 17.Herbst R, Burden S. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. EMBO J. 2000;19:67–77. doi: 10.1093/emboj/19.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson C, Montanaro F, Lindenbaum M, Carbonetto S, Ferns M. Alpha-dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling. J Neurosci. 1998;18:6340–6348. doi: 10.1523/JNEUROSCI.18-16-06340.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joung J, Ramm E, Pabo C. A bacterial two-hybrid system for studying protein-DNA and protein-protein interactions. PNAS. 2000;97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kauppinen K, Duan F, Wels J, Manor D. Regulation of the Dbl proto-oncogene by heat shock cognate protein 70 (Hsc70) J Biol Chem. 2005;280:21638–21644. doi: 10.1074/jbc.M413984200. [DOI] [PubMed] [Google Scholar]
- 21.Kim SW, Chao TH, Xiang R, Lo JF, Campbell MJ, Fearns C, Lee JD. Tid1, the human homologue of a Drosophila tumor suppressor, reduces the malignant activity of ErbB-2 in carcinoma cells. Cancer Research. 2004;64:7732–7739. doi: 10.1158/0008-5472.CAN-04-1323. [DOI] [PubMed] [Google Scholar]
- 22.Kadoya T, Khurana A, Tcherpakov M, Bromberg KD, Didier C, Broday L, Asahara T, Bhoumik A, Ronai Z. JAMP, a Jun N-terminal kinase 1 (JNK1)-associated membrane protein, regulates duration of JNK activity. Mol Cell Biol. 2005;25:8619–8630. doi: 10.1128/MCB.25.19.8619-8630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong XC, Barzaghi P, Ruegg M. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Reports. 2004;5:183–188. doi: 10.1038/sj.embor.7400065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang P, MacRae TH. Molecular chaperones and the cytoskeleton. J Cell Science. 1997;110:1431–1440. doi: 10.1242/jcs.110.13.1431. [DOI] [PubMed] [Google Scholar]
- 25.Liberek K, Skowyra D, Zylicz M, Johnson C, Georgopoulos C. The Escherichia coli DnaK chaperone, the 70-kDa heat shock protein eukaryotic equivalent, changes conformation upon ATP hydrolysis, thus triggering its dissociation from a bound target protein. J Biol Chem. 1991;266:14491–14496. [PubMed] [Google Scholar]
- 26.Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 2001;410:1057–1064. doi: 10.1038/35074025. [DOI] [PubMed] [Google Scholar]
- 27.Liu HY, MacDonald JIS, Hryciw T, Li C, Meakin SO. Human tumorous imaginal disc 1 (TID1) associates with Trk receptor tyrosine kinases and regulates neurite outgrowth in nnr5-TrkA cells. J Biol Chem. 2005;280:19461–19471. doi: 10.1074/jbc.M500313200. [DOI] [PubMed] [Google Scholar]
- 28.Lo JF, Hayashi M, Woo-Kim S, Tian B, Huang JF, Fearns C, Takayama S, Zapata JM, Yang Y, Lee JD. Tid1, a cochaperone of the heat shock 70 protein and the mammalian counterpart of the Drosophila tumor suppressor l(2)tid, is critical for early embryonic development and cell survival. Mol Cell Biol. 2004;24:2226–2236. doi: 10.1128/MCB.24.6.2226-2236.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu B, Garrido N, Spelbrink JN, Suzuki CK. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J Biol Chem. 2006;281:13150–13158. doi: 10.1074/jbc.M509179200. [DOI] [PubMed] [Google Scholar]
- 30.Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- 31.Luo ZG, Wang Q, Dobbins GC, Levy S, Xiong WC, Mei L. Signaling complexes for postsynaptic differentiation. J Neurocyt. 2003;32:697–708. doi: 10.1023/B:NEUR.0000020618.65271.63. [DOI] [PubMed] [Google Scholar]
- 32.Merlie JP, Isenberg KE, Russell SD, Sanes JR. Denervation supersensitivity in skeletal muscle: analysis with a cloned cDNA probe. J Cell Biol. 1984;99:332–335. doi: 10.1083/jcb.99.1.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mittaud P, Marangi PA, Erb-Vogtli S, Fuhrer C. Agrin-induced activation of acetylcholine receptor-bound Src family kinases requires rapsyn and correlates with acetylcholine receptor clustering. J Biol Chem. 2001;276:14505–14513. doi: 10.1074/jbc.M007024200. [DOI] [PubMed] [Google Scholar]
- 34.Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- 35.Polakis P. The adenomatous polyposis coli (APC) tumor suppressor. Biochemica et Biophysica Acta. 1997;1332:F127–F147. doi: 10.1016/s0304-419x(97)00008-5. [DOI] [PubMed] [Google Scholar]
- 36.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 37.Rimer M. Neuregulins at the neuromuscular synapse: Past, present, and future. J Neurosci Res. 2007;85:1827–1833. doi: 10.1002/jnr.21237. [DOI] [PubMed] [Google Scholar]
- 38.Sadasivam G, Willmann R, Lin S, Erb-Vogtil S, Kong XC, Ruegg MA, Fuhrer C. Src-family kinases stabilize the neuromuscular synapse in vivo via protein interactions, phosphorylation, and cytoskeletal linkage of acetylcholine receptors. J Neurosci. 2005;25:10479–10493. doi: 10.1523/JNEUROSCI.2103-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanes JR, Lichtman RW. Induction, assembly, maturation, and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 40.Sarkar S, Pollack BP, Lin KT, Kotenko SV, Cook JR, Lewis A, Pestka S. hTid-1, a human DnaJ protein, modulates the interferon signaling pathway. J Biol Chem. 2001;276:49034–49042. doi: 10.1074/jbc.M103683200. [DOI] [PubMed] [Google Scholar]
- 41.Settleman J, Narasimhan V, Foster LC, Weinberg RA. Molecular cloning of cDNAs encoding the GAP-associated protein p190: implications for a signaling pathway from ras to the nucleus. Cell. 1992;69:539–549. doi: 10.1016/0092-8674(92)90454-k. [DOI] [PubMed] [Google Scholar]
- 42.Sugiyama JE, Glass DJ, Yancopoulos GD, Hall ZW. Laminin-induced acetylcholine receptor clustering: an alternative pathway. J Cell Biol. 1997;139:181–191. doi: 10.1083/jcb.139.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syken J, De-Medina T, Münger K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. PNAS. 1999;96:8499–8504. doi: 10.1073/pnas.96.15.8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trentin GA, Yin X, Tahir S, Lhotak S, Farhang-Fallah J, Li Y, Rozakis-Adcock M. A mouse homologue of the Drosophila tumor suppressor l(2)tid gene defines a novel Ras GTPase-activating protein (RasGAP)-binding protein. J Biol Chem. 2001;276:13087–13095. doi: 10.1074/jbc.M009267200. [DOI] [PubMed] [Google Scholar]
- 45.Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nunez L, Park JS, Stark JL, Gies DR, Thomas S, Le Beau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 46.Wang J, Jing Z, Zhang L, Zhou G, Braun J, Yao Y, Wang ZZ. Regulation of acetylcholine receptor clustering by the tumor suppressor APC. Nat Neurosci. 2003;6:1017–1018. doi: 10.1038/nn1128. [DOI] [PubMed] [Google Scholar]
- 47.Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, Zheng H. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watty A, Neubauer G, Dreger M, Zimmer M, Wilm M, Burden SJ. The in vitro and in vivo phosphotyrosine map of activated MuSK. PNAS. 2000;97:4585–4590. doi: 10.1073/pnas.080061997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weston C, Yee B, Hod E, Prives J. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases Rac and Cdc42. J Cell Biol. 2000;150:205–212. doi: 10.1083/jcb.150.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weston C, Gordon C, Teressa G, Hod E, Ren XD, Prives J. Cooperative regulation by Rac and Rho of agrin-induced acetylcholine receptor clustering in muscle cells. J Biol Chem. 2003;278:6450–6455. doi: 10.1074/jbc.M210249200. [DOI] [PubMed] [Google Scholar]
- 51.Wu JS, Hogan GR, Morris JD. Modified methods for preparation of cryostat sections of skeletal muscle. Muscle Nerve. 1985;8:664–666. doi: 10.1002/mus.880080807. [DOI] [PubMed] [Google Scholar]
- 52.Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001;30:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.