

Abstract
In the present study, an effort was made to design prolonged release Eudragit nanoparticles of brimonidine tartrate by double emulsion–solvent evaporation technique for the treatment of open-angle glaucoma. The effect of various formulation variables like initial drug amount, lecithin proportion, phase volume and pH, secondary emulsifier and polymer proportion were studied. Various process variables like energy and duration of emulsification, lyophilization on the characteristics of nanoparticles and in vitro drug release profile were studied. The selected formulations were subjected to in vivo intraocular pressure-lowering efficacy studies by administering aqueous dispersion of nanoparticles into the lower cul de sac of glaucomatous rabbits. The prepared Eudragit-based nanoparticles were found to have narrow particle size range and improved drug loading. The investigated process and formulation variables found to have significant effect on the particle size, drug loading and entrapment efficiency, and in vitro drug release profile of nanoparticles. The selected formulations upon in vivo ocular irritability and tolerability tests were found to be well tolerated with no signs of irritation. In vivo pharmacodynamic efficacy studies revealed that the selected nanoparticle formulations significantly improved the therapy as area under the ∆IOP vs. time curve [AUC(∆IOP vs.t)] showed several fold increase in intensity and duration of intraocular pressure (IOP) decrease. All the selected nanoparticle formulations were found to prolong the drug release in vitro and prolong IOP reduction efficacy in vivo, thus rendering them as a potential carrier in developing improved drug delivery systems for the treatment of glaucoma.
KEY WORDS: brimonidine tartrate, Eudragit, glaucoma, intraocular pressure, nanoparticles
INTRODUCTION
The treatment of glaucoma requires the therapeutic agent to be present at the site of action at an optimal concentration for a prolonged period of time in order to control the increasing intraocular pressure to minimise the frequency of administration and to improve patient compliance. The topical administration of ophthalmic drugs in aqueous solution form, which are currently most accessible ocular formulations, results in extensive precorneal loss facilitated by the rapid tear fluid turnover (1). Typically less than 5% of topically applied drug penetrates the cornea and reaches the intraocular tissues (2). Many approaches have been investigated for the improvement of topical drug delivery for ocular therapeutics.
Nanoparticles, colloidal drug carriers, whose size range from 10 to 1,000 nm could be an attractive approach, which can deliver drug at the right dosage to the right target organ. The nanoparticles can prevent or minimise the entrapped drug from undergoing degradation, metabolism and cellular efflux in the course of drug delivery (3,4). Also, since they are present in the sub-micronic size range, they may be applied in liquid form just as eyedrops, avoiding the discomfort associated with the other dosage forms. Many polymers ranging from natural, semi synthetic, synthetic and biodegradable have been investigated in the preparation of ocular nanoparticles. Polymers investigated are bovine serum albumin (5), poly(lactide-co-glycolide) (6–8), chitosan (9–12), polymethacrylates (13), poly epsilon-caprolactone (14,15) and poly carboxylic acid (16,17).
Brimonidine tartrate (BRT) [5-bromo-6-(2-imidazolidinylideneamino) quinoxaline l-tartrate] is a selective alpha-2 adrenergic agonist, used as ocular hypotensive agent. Its ocular hypotensive effect is due its ability to decrease aqueous humour production and increase uveoscleral outflow (18,19). Its selectivity towards alpha-2 adrenergic receptors (19,20) and its neuroprotective activity on retinal ganglionic cells (20) makes it as an important therapeutic agent for the treatment of open-angle glaucoma (21). In few clinical trials, it has been shown to have comparable ocular hypotensive activity to that of timolol (22,23). In another clinical trial, it was found to have intraocular pressure (IOP)-lowering efficacy greater than that of betaxolol (24). It is also shown to have neuroprotective actions on retinal ganglionic cells of the retina in glaucoma (25–29) thus making it as an important addition to the class of antiglaucoma agents that is not contraindicated in patients with cardiopulmonary disease (21,22). The loading of the drug in the nanoparticles could result in slower release over several hours, thus prolonging the IOP reduction and improved neuroprotective actions of BRT on RGC cells.
Eudragit RL 100 (ERL 100) and Eudragit RS 100 (ERS 100) are inert polymeric resin copolymers of poly(ethacrylate, methylmetacrylate and chlorotrimethyl ammonioethyl methacrylayte) containing an amount of quaternary ammonium groups, ranging between 4.5–6.8% and 8.8–12.0% for RS and RL, respectively. Several reports are cited in the literature on the application of ERS 100 and ERL 100 as polymeric carriers for the delivery of drugs (30–33). Most of them are for the entrapment of water-insoluble/lipophilic drugs. No reports are available in the literature on the entrapment of water-soluble drug using ERL 100 and/or ERS 100 with better loading and entrapment efficiency. However in the present investigation, the authors have made no attempt to compare the relative efficacy of Eudragit to entrap water-soluble or insoluble molecules.
In the present study, BRT nanoparticles were prepared using ERL 100 and ERS 100 in combination by double emulsion–solvent evaporation method. The resultant nanoparticles were evaluated for particle size, drug loading, and entrapment efficiency and in vitro drug release profile. Selected formulations were evaluated in vivo for their IOP-lowering efficacy on alpha-chymotrypsin-induced glaucomatous rabbits in comparison to marketed preparation of BRT.
MATERIALS AND METHODS
Materials and Equipments
Brimonidine tartrate was obtained as a gift sample from FDC Ltd, Mumbai, India. Eudragit RL 100 and Eudragit RS 100 were obtained from Evonik Degussa, Mumbai, India. Soya lecithin, Pluronic F-68 (PF-68), dialysis membrane (10,000 Da) and alpha-chymotrypsin (type II, lyophilized powder, ≥40 units/mg protein) were purchased from Sigma Aldrich, Bangalore, India. Poly(vinyl) alcohol was purchased from SD Fine chemicals, Mumbai, India. Brimonidine tartrate eyedrops (Iobrim® E/D, FDC, Mumbai, India) was purchased randomly from Indian market. Each millilitre of Iobrim® E/D contains 2 mg of brimonidine tartrate equivalent to 1.3 mg of brimonidine base in aqueous-buffered vehicle. All other chemicals and reagents used were of analytical grade.
A five-digit analytical balance Mettler Toledo (AG135, Mettler, GMBH, Greifensee, Switzerland) was used for all weighing purposes. For dispersing polymer in suitable solvents, magnetic stirrer (Remi, Mumbai, India) was used. Probe sonicator (Microsons probe sonicator, Micronix Inc., NY, USA) was used in preparing the nanoparticles. Solvent removal was carried out using Rotavapor (Buchi® Rotavapor R-210, Switzerland). Centrifugation was carried out using centrifuge (Remi Compufuge CPR 42, Mumbai, India). Obtained nanoparticles were lyophilized using freeze dryer and vacuum centrifuge (Maxi Dry Lyo, Heto, Germany). For the characterisation of nanoparticles, Zetasizer (3000HS-Zetasizer, Malvern Instruments Inc., Malvern, UK), scanning electron microscopy (SEM; JSM 840A, Jeol, Japan) and transmission electron microscopy (200CX, Jeol, Japan) were used. In vitro release studies were carried out using the United States Pharmacopeia (USP) type I dissolution apparatus (basket type, Electrolab TDT-08 L, Mumbai, India). The IOP was measured using calibrated Schiotz tonometer (Scope medical, Mohali, India) provided with standard weights.
Preparation of Nanoparticles
Nanoparticles were prepared by double emulsion–solvent evaporation technique. The drug dissolved in aqueous phase was dispersed in dichloro methane (DCM) containing ERL 100 and ERS 100 and soya lecithin, under ultrasonication using a ultrasonicator (Microsons probe sonicator) at 10 kW energy for 2 min in pulsed mode (30 sec per cycle) under controlled temperature (4°C) for the formation of w/o emulsion. The primary emulsion was added rapidly into an aqueous phase: phosphate buffer (pH 7.4, 100 mM) containing poly(vinyl) alcohol (PVA) or PF-68 under vigorous stirring and ultrasonication (Microsons probe sonicator) at 10 kW energy for 2 min in pulsed mode (30 s/cycle) under controlled temperature (4°C) to form w/o/w emulsion. The double emulsion was stirred for 2 h for the removal of organic phase. The complete solvent removal was attained by using Rotavapor (Buchi® Rotavapor) at 25°C over a period of 30 min. The obtained nanoparticle dispersion was centrifuged (Remi Compufuge CPR 42) at 14,000 rpm at 4°C for 15 min, washed with distilled water and finally lyophilized. The samples for lyophilisation were pre-freezed at −20°C for 12 h. Lyophilisation was carried out after adding suitable cryoprotectants (1% w/v mannitol) in glass ampoules for 24 h at 1 mbar pressure and −110°C temperature (using Maxi Dry, Heto, Germany) to obtain free-flowing powder. The prepared nanoparticles were stored in tightly sealed containers under refrigeration.
The effect of initial drug amount (10, 20, 30, 40 and 60 mg), emulsifier type and proportion (lecithin: 0.05, 0.1, 0.15 and 0.2% w/v; PVA and PF-68 at 0.5%, 1.0%, 1.5% and 2.0% w/v, respectively), phase volume ratio [internal to external ratio (ml) of 1:30, 2:30, 3:30 and 4:30; 3:20, 3:30, 3:40 and 3:60, respectively], phase pH (internal: 2.0, 4.0, 6.0, 7.4 and 8.5; external: 6.0 7.4, 8.0 and 9.0) and polymer proportion on the characteristics of nanoparticles were extensively investigated. The composition of nanoparticle formulations prepared is shown in Table I.
Table I.
Formulation Composition for Eudragit-Based Brimonidine Tartrate Nanoparticle Formulations
| Formulation code | BRT (mg)a | Org. phase | ERS:ERL (mg) | LCT (% w/v) | PVA (% w/v) | PF-68 (% w/v) | Internal phase (ml) | External Phase (ml) | pH (internal phase/external phase) |
|---|---|---|---|---|---|---|---|---|---|
| Effect of initial drug amount | |||||||||
| BENP-D10 | 10 | EtOAc | 100:100 | 0.1 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-D20 | 20 | EtOAc | 100:100 | 0.1 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-D30 | 30 | EtOAc | 100:100 | 0.1 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-D40 | 40 | EtOAc | 100:100 | 0.1 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-D60 | 60 | EtOAc | 100:100 | 0.1 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| Effect of lecithin proportion | |||||||||
| BENP-L0.05 | 30 | EtOAc | 100:100 | 0.05 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-L0.10 | 30 | EtOAc | 100:100 | 0.15 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-L0.15 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-L0.20 | 30 | EtOAc | 100:100 | 0.20 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| Effect of internal phase volume | |||||||||
| BENP-IP1 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 1 | 30 | 7.4/7.4 |
| BENP-IP2 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 2 | 30 | 7.4/7.4 |
| BENP-IP3 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-IP4 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 4 | 30 | 7.4/7.4 |
| Effect of external phase volume | |||||||||
| BENP-EP20 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 20 | 7.4/7.4 |
| BENP-EP30 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-EP40 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 40 | 7.4/7.4 |
| BENP-EP60 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 60 | 7.4/7.4 |
| Effect of secondary emulsifier (PVA) | |||||||||
| BENP-PVA0.5 | 30 | EtOAc | 100:100 | 0.10 | 0.5 | – | 3 | 30 | 7.4/7.4 |
| BENP-PVA1.0 | 30 | EtOAc | 100:100 | 0.10 | 1.0 | – | 3 | 30 | 7.4/7.4 |
| BENP-PVA1.5 | 30 | EtOAc | 100:100 | 0.10 | 1.5 | – | 3 | 30 | 7.4/7.4 |
| BENP-PVA2.0 | 30 | EtOAc | 100:100 | 0.10 | 2.0 | – | 3 | 30 | 7.4/7.4 |
| Effect of secondary emulsifier (PF-68) | |||||||||
| BENP-PF0.5 | 30 | EtOAc | 100:100 | 0.10 | – | 0.5 | 3 | 30 | 7.4/7.4 |
| BENP-PF1.0 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-PF1.5 | 30 | EtOAc | 100:100 | 0.10 | – | 1.5 | 3 | 30 | 7.4/7.4 |
| BENP-PF2.0 | 30 | EtOAc | 100:100 | 0.10 | – | 2.0 | 3 | 30 | 7.4/7.4 |
| Effect of internal phase pH | |||||||||
| BENP-IpH2 | 30 | EtOAc | 100:100 | 0.10 | – | 0.5 | 3 | 30 | 2.0/7.4 |
| BENP-IpH4 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 4.0/7.4 |
| BENP-IpH6 | 30 | EtOAc | 100:100 | 0.10 | – | 1.5 | 3 | 30 | 6.0/7.4 |
| BENP-IpH7.4 | 30 | EtOAc | 100:100 | 0.10 | – | 1.5 | 3 | 30 | 7.4/7.4 |
| BENP-IpH8.5 | 30 | EtOAc | 100:100 | 0.10 | – | 2.0 | 3 | 30 | 8.5/7.4 |
| Effect of external phase pH | |||||||||
| BENP-EpH6.0 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/6.0 |
| BENP-EpH7.4 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-EpH6.0 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/8.0 |
| BENP-EpH8.5 | 30 | EtOAc | 100:100 | 0.10 | – | 1.0 | 3 | 30 | 7.4/9.0 |
| Effect of polymer proportion | |||||||||
| BENP-P50 | 30 | EtOAc | 1:1 (50) | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-P100 | 30 | EtOAc | 1:1 (100) | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-P510 | 30 | EtOAc | 1:1 (150) | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-P200 | 30 | EtOAc | 1:1 (200) | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-P100 | 30 | EtOAc | 1:2 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
| BENP-P100 | 30 | EtOAc | 2:1 | 0.10 | – | 1.0 | 3 | 30 | 7.4/7.4 |
BRT brimonidine tartrate, EtOAc ethyl acetate, ERS Eudragit RS 100, ERL Eudragit RL 100, LCT lecithin, PVA (poly)vinyl alcohol, PF-68 Poloxamer Pluronic 68
aAmount per 200 mg of polymer
Characterization of Nanoparticles
Drug Content Estimation
The drug entrapped in the nanoparticles was estimated by dispersing the weighed amount of particles in DCM (2 ml) by ultrasonication and followed by extraction of free drug into phosphate buffer pH 7.4 (2 ml). The extract was analysed for brimonidine spectrophotometrically at 248 nm after suitable dilution (34). The drug loading efficiency (LE) and drug entrapment efficiency (EE) was calculated using Eqs. 1 and 2, respectively.
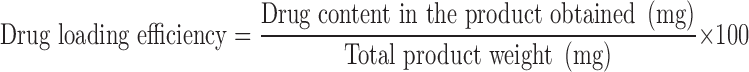 |
1 |
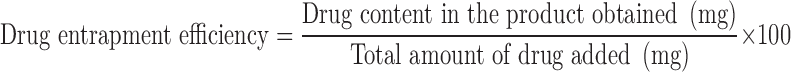 |
2 |
Particle Size
The particle size analysis of prepared nanoparticle formulations were performed by using photon correlation spectroscopy in Malvern Zeta Sizer equipped with He–Ne laser beam at a wavelength of 633 nm and 90° scattering angle. For measurement, about 5 mg of formulation was dispersed in 10 mL of Milli-Q® water. Obtained homogenous dispersion was immediately used for the determination of mean particle diameter. The data acquisition and processing was performed using PCS software (Malvern Instruments Inc.)
In Vitro Drug Release Studies
The in vitro release of BRT from the Eudragit nanoparticles was carried out at 37 ± 0.5°C using 25 ml of freshly prepared simulated tear fluid (STF; pH 7.4) using dialysis membrane pouches (dialysis membrane of 10,000 Da). Accurately weighed lyophilised nanoparticle formulations were dispersed in 1 ml of STF (pH 7.4) introduced into the pouches, which were maintained in USP dissolution apparatus (USP type I, basket type) agitated at 50 rpm. Samples were withdrawn at predetermined time intervals, diluted suitably and brimonidine was analysed spectrophotometrically at 248 nm (34). The percentage of drug released at each time interval was calculated as cumulative percent drug release. Percent drug release was determined based on the initial amount of nanoparticles taken for the studies and were plotted as a function of time (h). The drug release (60% w/w drug release) was then fitted into Korsmeyer–Peppas (KP) model (Eq. 3) to ascertain the release kinetics and mechanism of drug release. The KP model is given by,
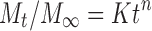 |
3 |
where, ‘K’ is kinetic constant incorporating structural and geometric characteristics of the matrix, Mt is the amount of drug released at time t, M∞ is the amount of drug released at infinite time, and n is the release exponent indicative of release mechanism. If n = 0.45, it indicates a Fickian diffusion-controlled release process. If n = 0.45–0.89, it indicates non-Fickian anomalous considered as combination of drug diffusion in the hydrated matrix and polymer relaxation and erosion. If n = 1.0, it indicates zero-order release and if n is more than 1.0, it indicates super-Case II release. The values of ‘n’, ‘K’ and ‘R2’ were used to determine the release rate mechanism and a best-fit model. Based on the regression analysis of log% drug released vs. log time, data using Eq. 3, the value of n, K and R2 were determined. Using n and K values, t10%, t50% and t90% (time for 10% w/w, 50% w/w and 90% w/w drug release, respectively) were calculated.
Stability Studies
The stability studies of selected nanoparticle formulations in dispersed and in freeze-dried state were carried out as per International Conference on Harmonisation guidelines after storage of the formulations for 24 months. The storage conditions employed were ambient (25°C ± 2°C/60 ± 5% RH), refrigeration (5 ± 3°C) and freeze (−20 ± 5°C). The required volume of nanoparticle dispersion was stored in closed glass bottles and sealed tightly. At predetermined time intervals, samples were withdrawn and studied for the characteristics such as loading and entrapment efficiency, particle size and in vitro drug release profile.
In Vivo Studies
Animals
New Zealand white rabbits weighing 2.5–3.5 kg were provided by the Central Animal House Facility of Birla Institute of Technology and Science (BITS), Pilani and were housed under controlled and standardised conditions. They were fed a normal pellet diet and water was given ad libitum. The animals were acclimatised to light and dark cycles for 12 h. All the animals met the following criteria: (1) both the eyes were completely healthy with no injury or history of injury, (2) the basal IOP was in the range of 22 ± 3 mm Hg, and (3) the IOP difference between contralateral eyes were not exceeding 2 mm Hg. Animal handling and studies were conducted in accordance with the Principles of Laboratory Animal Care (National Institutes of Health publication no 92–93, revised in 1985) and in conformation with Association for Research in Vision and Ophthalmology and was approved by the Institutional Animal Ethics Committee of BITS, Pilani (protocol no: IAEC/RES/12-04).
Ocular Irritation Studies
Ocular irritation studies were performed on selected formulations showing promising in vitro results, according to Draize technique (35) on healthy New Zealand white rabbits each weighing 2.5–3.5 kg, divided into following three groups. The solutions saline, marketed eyedrop (2–3 drops; Iobrim® E/D (containing 2 mg/ml drug), FDC Ltd, Mumbai, India) and selected nanoparticle formulations were administered as aqueous dispersion in phosphate-buffered saline (pH 7.4) once a day for a period of 7 days. At the time of formulation instillation, the animals were maintained in restrainer boxes but allowed to move their heads freely. The evaluation was performed according to the Draize technique (35) by periodically observing for ocular redness, swelling and watering conjunctival chemosis, discharge and corneal lesions. The standard scoring system was followed to ascertain the outcome of the experiment.
In Vivo Pharmcodynamic Studies
Induction of Glaucoma
Rabbits were anaesthetized by intramuscular injection of 4 mg/kg of Xylazine and 35 mg/kg of Ketamine. Chronic ocular glaucoma was induced by a single posterior injection of alpha-chymotrypsin (10 mg/ml, 0.1 ml) into posterior segment of eye in rabbits (36). Care was taken to avoid the contact of alpha-chymotrypsin with the surface of the eye. A daily ocular examination was followed for few days. After 2–3 days of injection, one drop of ciprofloxacin eyedrop (Ciplox® Cipla, India), dexamethasone eyedrop (Dexacip®, Cipla, India) and a drop of diclofenac sodium eyedrop (Voltaren®, Novartis, India) were instilled to prevent topical inflammation. Animals that showed cases of severe inflammation and erratic or inconsistent IOP increase were excluded from the study. When the IOP was stabilised to 39 ± 3 mm Hg, for three successive days, the pharmacodynamic response studies were initiated.
For IOP-lowering studies, the selected nanoparticles were administered as aqueous dispersions (2 mg/ml) prepared in phosphate-bufferred saline (pH 7.4). The corresponding reference treatment was conventional ophthalmic drops (Iobrim® E/D (containing 2 mg/ml drug), FDC Ltd, Mumbai, India). Both the test and reference products were instilled (two drops in each case) carefully into the lower cul de sac of the left eye of the rabbits (n = 3) while to the right eye, two to three drops of normal saline was administered. The saline-treated eye acted as control in the experiments. Immediately after instillation, eye lid was closed for 10 s in order to avoid spillage or movement of the preparation. IOP was measured by using calibrated Schiotz tonometer (Scope medical, Mohali, India) at different time intervals. The change in IOP (∆IOP) at each time point from the stabilised IOP (zero time) was determined by
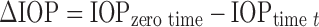 |
∆IOP is reported as mean (±SEM) of three animals (n = 3) for each treatment at each time point. The ∆IOP vs. time curve was plotted to compare the efficacy of prepared formulations with the conventional ophthalmic drops and the comparison was done in terms of (1) Imax, peak decrease in IOP; (2) tmax, time to reach peak IOP decrease; (3) area under the curve (AUC)(∆IOP vs.t), area under the ∆IOP vs. time curve; (4) duration of IOP decrease and (5) slope of terminal linear portion of the decrease in IOP vs. time curve (37). The AUC(∆IOP vs.t) of ∆IOP vs. time curve was calculated using trapezoid rule (also calculated using Graph Pad Prism 4 software). The AUCRel was calculated using the following equation
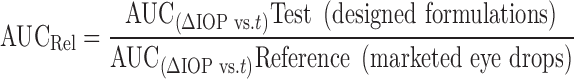 |
Data Analysis
The difference in the in vivo performance of different treatments formulations was compared using paired t test for means using Microsoft Office 2007 Excel package.
RESULTS AND DISCUSSION
The double emulsion–solvent evaporation technique was found to be suitable for the preparation of BRT-loaded ERS–ERL nanoparticles. The particles obtained with ERS–ERL were of low mean size, well suited for ocular application. The low drug incorporation may be attributed to the high water solubility of BRT. A rapid diffusion of the drug into the aqueous phase during secondary emulsion formation resulted in a low drug incorporation. The effect of various formulation variables like initial drug amount, lecithin proportion, phase volume and pH, secondary emulsifier and polymer proportion were studied. Various process variables like energy, duration of emulsification and lyophilization on the characteristics of nanoparticles and in vitro drug release profile has been elaborately discussed in the following sections.
Effect of Initial Amount of Drug
When the initial drug loading was increased from 10 to 30 mg the particle size (PS) and EE increased significantly (p < 0.01) but no statistically significant difference (p < 0.05) was observed in case of LE (Fig. 1). Further increase in initial drug loading from 30 to 60 mg significantly decreased the PS (p < 0.001), increased the LE (p < 0.001) and decreased the EE (p < 0.01).
Fig. 1.

Effect of initial drug amount on the characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from 10 mg initial drug amount (IDA; p < 0.01). **Statistically significant difference from 10 mg IDA (p < 0.05). #Statistically significant difference from 30 mg IDA (p < 0.001). ##Statistically significant difference from 30 mg IDA (p < 0.01)
Effect of Lecithin Proportion
The results of effect of varying lecithin proportion on the characteristics of nanoparticles are shown in Fig. 2. Lecithin as emulsifier for the preparation of nanoparticles at a concentration of 0.1% w/v of primary emulsion produced nanoparticles with an average particle size of 299 nm and LE and EE of 5.2% w/w and 34.6% w/w, respectively. Increase in lecithin proportion from 0.1% w/v to 0.2% w/v significantly (p < 0.001) decreased average particle size which could be due the stabilisation of primary emulsion droplets during emulsification process and also increase in viscosity of external phase. However with increase in lecithin concentration, the LE and EE decreased significantly (p < 0.001). With 0.15% w/v, the average particle size, LE and EE were found to be 254 nm, 4.8% and 20.6% w/w, respectively. Further increase in lecithin concentration (2% w/v) resulted in further decrease in particle size to 237 nm with LE of 2.5% w/w and EE of 18.2% w/w, respectively. The decrease in entrapment and loading efficiency upon increase in lecithin concentration can be attributed to slowing of the rate of formation of the nanoparticles because of decreased rate of evaporation of the organic layer due to increased viscosity of the organic layer in the presence of higher proportion of lecithin. Decrease in lecithin proportion to 0.05% w/v resulted in statistically significant increase in average particle size, LE and EE to 312 nm (p < 0.05), 7.6% w/w (p < 0.01) and 39.9% w/w (p < 0.01), respectively.
Fig. 2.

Effect of proportion of lecithin on the characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from lecithin conc. of 0.1% w/v (p < 0.001). #Statistically significant difference from lecithin conc. of 0.1% w/v (p < 0.01). ##Statistically significant difference from lecithin conc. of 0.1% w/v (p < 0.05)
Effect of Phase Volume
The internal and the external phase volume ratio employed at pH 7.4 for both phases during the emulsion preparation process greatly influenced the formation of emulsion and hence the characteristics of the nanoparticles obtained (Figs. 3 and 4).
Fig. 3.

Effect of varying internal phase volume on characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from internal phase volume of 4 ml (p < 0.001)
Fig. 4.

Effect of varying external phase volume on the characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from external phase volume of 20 ml (p < 0.001). #Statistically significant difference from external phase volume of 30 ml (p < 0.001). ##Statistically significant difference from external phase volume of 30 ml (p < 0.01). ###Statistically significant difference from external phase volume of 30 ml (p < 0.05)
Decrease in the internal aqueous phase volume of primary emulsion from 4 ml to 1 ml resulted in statistically significant decrease in average particle size from 311 nm to 220 nm (p < 0.001). A corresponding decrease in LE from 5.9% w/w to 2.0% w/w (p < 0.001) and EE from 40.1% w/w to 6.8% w/w (p < 0.001) was also observed. Higher internal phase volume results in lower drug concentration in the internal aqueous phase. This decrease in drug concentration decreases the loss of drug from the internal phase through drug diffusion process and thereby contributing to increase in LE and EE. Also during emulsification process, with constant energy and duration, decrease in internal phase volume resulted in decrease in droplet size of primary emulsion resulting in decreased average particle size. At lower internal phase volume, the increase in number of droplets with smaller size may also contribute to the increased loss of drug by diffusion resulting in lower LE and EE.
In the case of designed Eudragit-based nanoparticles of BRT, increasing the volume of the external phase decreased particle size, entrapment and loading efficiency. The formulation with 20 ml of external phase showed an average particle size of 358 nm, LE of 7.6% w/w and EE of 44.3% w/w. Upon increasing the external phase volume to 30 ml, there was a significant decrease (p < 0.001) in average particle size to 299 nm, LE to 5.2% w/w and EE to 34.6% w/w. Further increase in external phase volume to 60 ml decreased the average particle size to 234 nm (p < 0.001), LE to 4.1% w/w (p < 0.01) and EE to 29.9% w/w (p < 0.05). The decrease in average particle size could be due to the increase in external phase volume which could be due to non-agglomeration of formed nanoparticles in dispersed phase and better emulsification during secondary emulsion formation. The decrease in LE and EE upon increase in external phase volume is probably due to higher diffusion of drug during double emulsification process due to more favourable concentration gradient from internal phase to the external aqueous phase.
Effect of Aqueous Phase pH
The aqueous phase pH was found have a significant influence on nanoparticle characteristics. When the pH of the internal phase was increased from 2.0 to 7.4 (Fig. 5a) while keeping external phase pH at 7.4, the particle size, LE and EE significantly decreased (p < 0.0001). On the other hand, increase in the external phase pH from 6.0 to 9.0 significantly increased the particle size, LE and EE (p < 0.0001; Fig. 5b).
Fig. 5.

Effect of varying internal phase pH (a) and external phase pH (b) on characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from internal phase pH of 2.0 (p < 0.0001). #Statistically significant difference from external phase pH of 6.0 (p < 0.0001)
The pH of the solution governs the ionisation of the drug and hence its solubility. BRT, a weak basic drug with a pKa of 7.22 (34), exhibits predominately ionised form at acidic pH ranges where its solubility is higher and hence the drug loss due to rapid diffusion into external phase (pH 7.4) is less resulting in increased mechanical entrapment of drug into the nanoparticles. Increase in internal phase pH from 2.0 to 7.4 pH results in decrease in extent of ionisation and therefore resulted in a decrease in average particle size while drug loading and entrapment efficiency also decreased. The decrease in LE and EE would be due to the fact that increase in pH decreases the solubility of drug in the internal phase, hence higher trend for diffusion of drug into the external phase.
At higher pH values of external phase, an increase in average particle size, increase in drug loading and entrapment efficiency was observed due to the decreased solubility of the drug in external phase maintained at higher pH. The decrease in solubility in higher pH external phase presents with an unfavourable diffusion path from internal to external phase consequently LE and EE increased.
Effect of Secondary Emulsifier (Type and Proportion)
Upon increasing the proportion of PVA (as secondary emulsifier from 0.5% w/v to 2.0% w/v) particles size decreased significantly (p < 0.0001) whereas both LE and EE increased significantly (P < 0.0001 and p < 0.01, respectively, in case of LE and EE) as shown in Fig. 6a. Similar trend was observed when PF-68 was used as secondary emulsifier in the double emulsion (Fig. 6b). In the case of PF-68, as the secondary emulsifier proportion was increased from 0.5% w/v to 2.0% w/v, the particles size of the nanoparticles decreased from 368 to 221 nm (p < 0.001) whereas LE increased from 4.9% w/w to 11.0% w/w (p < 0.0001) and EE increased from 31.2% w/w to 49.6% w/w (p < 0.0001).
Fig. 6.

Effect of varying PVA proportion (a) and PF-68 proportion (b) on the characteristics of Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from PVA conc. of 0.5% w/v (p < 0.0001). **Statistically significant difference from PVA conc. of 0.5% w/v (p < 0.01). #Statistically significant difference from PF conc. of 0.5% w/v (p < 0.001). ##Statistically significant difference from PF conc. of 0.5% w/v (p < 0.0001)
The increased LE and EE in both cases can be attributed to the formation of stable secondary emulsion and formation of uniform dispersion which increased drug encapsulation efficiency and prevented drug loss. The average particle size decreased with the increased proportion of secondary emulsion, which could be due the stabilisation of primary emulsion droplets during emulsification process and also increase in viscosity of external phase. However, the effect was more pronounced in the case of PF-68 when compared to PVA because PF-68 when used as secondary emulsifier does not incorporate viscosity to the solution, hence net shear during emulsification would be higher than that of PVA formulations, which ultimately contributed to smaller as well as narrower average particle size when compared to PVA-based formulations.
Effect of Polymer Proportion
The average particle size increased with increasing total ERS100/ERL100 proportion (100–400 mg of 1:1 ERL 100 to ERS 100 ratio) in the formulation (Fig. 7). The particle size increased from 245 nm [BENP-1:1(50)] to 410 nm [BENP-1:1(200)] (p < 0.0001). This increase is attributed to the fact that increased polymer percentage resulted in increased extent of agglomeration of particles under constant net shear of emulsification. Net shear of emulsification is the total energy (in kilowatt) applied during ultrasonication and stirring process to the emulsion system in order to fabricate a stable emulsion. This energy varies with duration of emulsification/stirring. Formation of high viscous polymer solution and consequent increase in droplet size of emulsion caused an increase in particle size. The LE was decreased from 5.6% w/w [BENP-1:1(50)] to 3.4% w/w in case of BENP-1:1(200) with p < 0.001. The EE was increased significantly (p < 0.0001) from 30.1% w/w [BENP-1:1(50)] to 59.1% w/w [BENP-1:1(200)]. This could be due to increased polymer proportion and the extent of encapsulation of the drug while LE did not increase.
Fig. 7.

Effect of polymer proportion on characteristics Eudragit-based brimonidine tartrate nanoparticle formulations. Each data represents the average of two batches in triplicate with standard deviation. *Statistically significant difference from total polymer content of 100 mg (p < 0.0001). **Statistically significant difference from total polymer content of 100 mg (p < 0. 001). #Statistically significant difference from BENP-1:2 (p < 0.001). ##Statistically significant difference from BENP-1:2 (p < 0.1)
Statistically significant increase (p < 0.001) was observed in the average particle size when the relative proportion of ERL 100 to ERS 100 was reversed from 1:2 to 2:1 in the formulation with total polymer proportion kept constant at 300 mg. The corresponding EE values decreased from 51.2% w/w to 47.6% w/w for the two formulations (p < 0.1). However, statistically insignificant difference was observed in case of LE for the relative ERL100 to ERS 100 ratio of 1:2 and 2:1.
In vitro Drug Release Studies
The BRT release from the prepared nanoparticles showed varying profiles over a period of time depending on the composition of formulations, environment of preparation or process parameters.
The in vitro release profiles of varying proportions of PVA were shown in Fig. 8a. It is evident that the formulations with higher proportion of PVA resulted in higher initial burst release, and drug release was faster than the formulations with lower proportion of PVA. The higher burst release with increased PVA proportion could be due to the fact that increased PVA resulted in decreased average particle size, which increased the effective surface area exposed to the drug release media, subsequently resulting in increased initial release. The dissolution and release of surface-bound PVA molecules caused the initial high release. Also, increased PVA proportion increased LE in the nanoparticle formulation, which further contributed to increased burst release. The formulations with lower proportion of PVA showed higher average particle size, lower LE, hence decreased the burst release and a prolonged release of drug was observed.
Fig. 8.

In vitro release profiles of Eudragit-based brimonidine tartrate nanoparticle formulations prepared with different proportions of a PVA and b PF-68. Each datapoint represents the average of two batches in triplicate with standard deviation
With PF-68 as secondary emulsifier, the trend in drug release was found to be similar to that of PVA as emulsifier (Fig. 8b). The increased PF-68 proportion decreased average particle size and therefore, a higher burst release and a faster rate of drug release was observed.
The drug release from nanoparticle formulations with varying PVA and PF-68 proportion is shown in Table II. In case of formulations with varying PVA proportions, the release exponent ‘n’ varied from 0.60 to 0.67, suggesting a non-Fickian anamolous drug transport mechanism in the release of drug. Multiple mechanisms such as swelling, erosion, polymer relaxation, etc. might play a role in drug release. As the proportion of PVA was increased, a gradual decrease in release exponent values was observed. This could be because rapid dissolution of PVA from the surface of the nanoparticles created pores or channels and further drug release might have been through these pores or channels rather than by erosion.
Table II.
Results of Drug Release Kinetics Studies for the Eudragit-Based Brimonidine Tartrate Nanoparticle Formulations Fitted into Korsmeyer–Peppas Kinetics Model
| Batch code | K–P model | |||
|---|---|---|---|---|
| n | t 20% (h) | t 50% (h) | t 90% (h) | |
| Effect of varying proportions of PVA | ||||
| BENP-PVA0.5 | 0.66 | 3.2 | 12.1 | 42.2 |
| BENP-PVA1.0 | 0.59 | 2.8 | 9.2 | 34.4 |
| BENP-PVA1.5 | 0.59 | 2.2 | 7.9 | 30.2 |
| BENP-PVA2.0 | 0.57 | 1.9 | 6.6 | 27.3 |
| Effect of varying proportions of PF-68 | ||||
| BENP-PF0.5 | 0.63 | 2.9 | 13.0 | 41.0 |
| BENP-PF1.0 | 0.49 | 2.5 | 10.2 | 33.9 |
| BENP-PF1.5 | 0.68 | 2.1 | 8.3 | 31.1 |
| BENPPF2.0 | 0.58 | 1.2 | 6.8 | 25.8 |
| Effect of varying amount and proportions of polymer | ||||
| BENP-1:1(50) | 0.64 | 1.4 | 7.3 | 25.4 |
| BENP-1:1(100) | 0.49 | 2.5 | 10.2 | 33.9 |
| BENP-1:1(150) | 0.57 | 3.0 | 12.8 | 45.2 |
| BENP-1:1(200) | 0.58 | 3.7 | 17.3 | 59.4 |
| BENP-1:2 | 0.59 | 2.9 | 15.7 | 42.4 |
| BENP-2:1 | 0.55 | 2.7 | 14.1 | 41.3 |
n Release exponent indicator of drug release mechanism; t 20%, t 50%, and t 90% time taken (in h) for 20%, 50% and 90% drug release, respectively
Similar results were also observed in the case of formulations with varying PF-68 as secondary emulsifier. The drug release was by non-Fickian anamolous mechanism.
The in vitro drug release profile of formulations with varying polymer proportions showed a decreased rate of drug release with increase in total polymer proportion (Fig. 9). The polymer proportion played major role in determining the burst release, duration of release along with having impact on physicochemical characteristics of nanoparticles. Increase in the total proportion of ERL 100 and ERS 100 from 100 to 400 mg resulted in significant decrease in burst effect from 28.4% [BENP-1:1(50)] to about 15.0% in the case of formulation BENP-1:1(200). The t20% for BENP-1:1(50) was found to be 1.4 h while for the formulation BENP-1:1(200), it was found to be 3.7 h (Table II). At higher proportions of polymer, the formation of compact polymer matrix and higher degree of encapsulation of drug into the matrix would have resulted in the decreased burst release (Fig. 9).
Fig. 9.

In vitro release profiles of Eudragit-based brimonidine tartrate nanoparticle formulations prepared with varying proportions of polymer. Each datapoint represents the average of two batches in triplicate with standard deviation
The duration of drug release was also greatly affected by the proportion of polymers in the formulations. As the polymer quantity was increased, the drug release was found to be more sustained for a longer period of time. In the case of formulation BENP-1:1(50), the t90% value was found to be 25.4 h, while increasing the total polymer amount to 200 mg in BENP-1:1(100) resulted in increase of t90% to 33.9 h. Further increase in total polymer amount to 300 mg in BENP-1:1 (150) and to 400 mg in BENP-1:1 (200) resulted in further controlled release of the entrapped drug with t90% values of 45.2 and 59.4 h, respectively, with complete release by 72 h (Table II). Variations in the relative proportions of ERL 100 and ERS 100 (BENP-1:2 and BENP-2:1) did not alter the release profile significantly.
Formulations with varying amount and proportions of polymer showed drug release mechanism by non-Fickian anamolous transport. Increase in the polymer proportion did not alter the drug release mechanism.
Stability Studies
The stability studies performed at various storage conditions ambient (25°C ± 2°C/60 ± 5% RH), refrigeration (5 ± 3°C) storage in freezer (−20 ± 5°C) to determine the effect of these conditions on selected nanoparticles in terms of degree of aggregation, drug content, particle size and ease of redipersibility. Nanoparticle batches showed detectable aggregation at ambient conditions, while at refrigerated (5 ± 3°C) storage in freezer (−20 ± 5°C) conditions negligible aggregation was observed. The physicochemical characteristics were found to be unaltered upon storage under refrigerated and freeze-dried conditions, while at ambient condition, detectable changes in the physicochemical characteristics are observed after 6 months of storage.
In Vivo Studies
Ocular Irritation and Tolerability Studies
The results of ocular irritability and tolerability studies of selected nanoparticle formulations suggested that all the formulations investigated were well tolerated with no signs of any irritation or toxicity. The scores were found to be the same as that of marketed preparation, which shows the potential of the developed formulation as ocular delivery systems.
In vivo Pharmacodynamic Studies
The glaucoma induction by alpha-chymotrypsin is primarily because of lysis of zonular material and trabecular meshwork which serves to drain the aqueous humour in and out of the eye, lysis of which results in its accumulation and subsequent increase in IOP. This model has been found suitable for the studies involving comparison of drug effects on IOP reduction and can thus be extrapolated into human glaucoma (38,39). Both the test product (designed nanoparticle dispersions) and reference conventional eyedrop solution was administered only once to obtain relative comparison between single-dose administrations. In both the case, approximately 222 μg BRT was administered as single dose. The control saline treatment (right eye) IOP measurements were found to be statistically not different from baseline IOP values (p < 0.001).
The Eudragit RS100- and Eudragit RL100-based nanoparticle formulations with improved in vitro characteristics (BENP-D30, BENP-IP4, BENP-PF20 and BENP-1:1(150)) were selected for in vivo studies. The selected nanoparticle formulations showed narrow particle size, high drug loading, and efficiency and prolonged release of drug and were stable.
The nanoparticle formulations were found to decrease the elevated IOP in rabbits in glaucoma for a longer period of time when compared to conventional eyedrop formulation [p < 0.0001 for AUC(∆IOP vs. t)]. As shown in Fig. 10 and in Table III, the Imax for all the selected nanoparticles was lesser in comparison to eyedrop preparation. Imax was 7.77 mm Hg for BENP-D30, 7.97 mm Hg for BENP-IP4, 7.71 mm Hg for BENP-PF20 and 7.6 mm Hg for BENP-1:1(150) in comparison to 8.55 mm Hg for eyedrop preparation. The tmax was about 2–3 h for nanoparticles, while it was 1 h for marketed eyedrop preparation. This suggested that the drug has to get dissoluted and then absorbed into the eye while eyedrop preparation had drug in dissolved form, hence a rapid absorption occurred. The formulation BENP-D30 showed an AUC(∆IOP vs. t) of 204.93 h mm Hg. When the internal phase in the BENP-D30 formulation was increased to 4 ml (BENP-IP4), the AUC(∆IOP vs. t) was decreased to 151.73.
Fig. 10.

Comparative IOP reduction profile for the selected Eudragit (ERL 100 and ERS 100)-based brimonidine tartrate nanoparticle formulations in comparison with commercial eyedrops (Iobrim® E/D) in glaucomatous rabbits. Each ∆IOP data point represents the mean (± SEM) of three animals (n = 3). BRT Brimonidine tartrate (*amount per 200 mg of polymer), EtOAc ethyl acetate, ERS Eudragit RS 100, ERL Eudragit RL 100, LCT lecithin, PVA (poly)vinyl alcohol, PF-68 Ploxamer pluronic 68
Table III.
Results of In Vivo Pharmacodynamic Efficacy Studies of Selected Eudragit (ERL 100 and ERS 100)-Based Brimonidine Tartrate Nanoparticle Formulations in Comparison with Commercial BRT Eyedrops (Iobrim® E/D) in Glaucomatous Rabbits
| Formulation | I max (mm Hg) | t max (h) | AUC(∆IOP vs. t) (h mm Hg) | Slope | Duration (h) | AUCRel |
|---|---|---|---|---|---|---|
| Eyedrops (Iobrim® E/D) | 8.55 ± 0.21 | 1 | 38.40 ± 4.21 | 0.4763 | 6 | – |
| BENP-D30 | 7.77 ± 0.32 | 3 | 204.93 ± 5.33* | 0.1022 | 48 | 5.3 |
| BENP-IP4 | 7.97 ± 0.21 | 2–3 | 151.73 ± 4.98* | 0.1055 | 48 | 4.0 |
| BENP-PF2.0 | 7.71 ± 0.19 | 3 | 136.33 ± 5.12* | 0.1244 | 36 | 3.6 |
| BENP-1:1(150) | 7.69 ± 0.24 | 3 | 268.09 ± 4.89* | 0.0855 | 72 | 7.0 |
I max Maximum reduction in IOP (mm Hg), t max time taken for maximum reduction in IOP (h), AUC (∆IOP vs. t) area under the ∆IOP vs. time curve, slope slope of terminal linear portion of ∆IOP vs. time curve, AUC Rel ratio of AUC(∆IOP vs. t) test (designed formulations) to AUC(∆IOP vs. t) reference (marketed eyedrops). Each ∆IOP data point represents the mean (± SEM) of three animals (n = 3)
aStatistically significant difference from conventional eyedrop (p < 0.0001)
In case of formulation BENP-PF20, where the PF-68 proportion was 2.0% w/w compared to 1.0% w/w in case of BENP-D30, the particle size was found to decrease and with corresponding increase in drug loading, loading efficiency and in vitro drug release rate. In vitro observations correlated with in vivo effect as seen by reduced duration of IOP reduction effect to 36 h compared to 48 h in case of BNP-D30. The AUC(∆IOP vs. t) was found to be slightly decreased to 136.33 h mm Hg compared to 204.93 h mm Hg for BENP-D30.
When the polymer proportion was increased from 100: 100 (ERS: ERL; BENP-D30) to 150:150 (ERS: ERL; BENP-1:1(150)), the particle size was slightly increased. The in vitro drug release was more prolonged and the corresponding in vivo effect lasted for 72 h. The AUC was increased to 268.09. The AUCRel in comparison to eyedrop formulations was found to be 5.34 for BENP-D30, 3.95 for BENP-IP4, 3.55 for BENP-PF20 and significantly improved to 6.98 for the formulation BENP-1:1(150). The lower value of slope for the terminal portion of the ∆IOP vs. time curve for all the selected Eudragit nanoparticles compared to eyedrop preparations suggested that nanoparticle formulations were slowly eliminated compared to eyedrops.
CONCLUSIONS
The BRT nanoparticles were prepared using ERS 100 and ERL 100 polymers by double emulsion–solvent evaporation method. The double emulsion–solvent evaporation method was found to be suitable for the preparation of BRT nanoparticles of low average particle size, improved drug loading and entrapment efficiency, and prolonged and controlled release of BRT over a longer period of time. Various formulation and process variables were investigated to study the effect of these variables on the characteristics of nanoparticles. The effect of initial drug loading, phase volume ratio, surfactant concentration, aqueous phase pH, secondary surfactants and polymer proportion showed varying effects of particle size, drug loading and entrapment efficiency and in vitro drug release. The optimised formulations showed improved drug loading and entrapment efficiency and extended release of drug over 48–72 h which could be a promising tool for designing long-acting BRT ophthalmic formulations. The stability studies showed that the selected formulations were found to be more stable at freezer and refrigerator temperature conditions than at room temperature.
The nanoparticle formulations were found to decrease the elevated IOP in rabbits in glaucoma for a longer period of time. The AUC(∆IOP vs. t) for the selected formulations were about seven times higher than that of eyedrop preparations. The slow elimination of administered particles from the eye and controlled and prolonged release of encapsulated drug makes them a potential carrier for the delivery of drug in the treatment of glaucoma. Eudragit-based nanoparticles controlled the release of drug (in vitro) and reduced IOP for up to 72 h [BENP-1:1(150)] showing a significant increase in both extent and duration of IOP reduction. As the study was carried out on rabbits after glaucoma induction, the results can be extrapolated to humans as this model is shown to be identical to human glaucoma (38,39). The obtained results shows that the developed nanoparticle formulations have potential in improving BRT delivery to the eye in treating glaucoma and can potentially increase patient compliance by reducing the frequency of administration.
ACKNOWLEDGEMENTS
This work is a part of a grant support (no: SR/FTP/L-42/2005) provided to Dr. Sajeev Chandran by Department of Science and Technology, Ministry of Science and Technology, Govt. of India, New Delhi, India. Authors are highly grateful to Dr. Sushil Yadav, veterinarian, BITS Pilani and Mr. Dilip Pandey, BITS Pilani, for their valuable help in conducting the animal studies. Authors are also thankful to FDC Ltd. Mumbai, India for the generous gift sample of brimonidine tartrate.
REFERENCES
- 1.Lee VHL, Robinson JR. Mechanistic and quantitative evaluation of precorneal pilocarpine disposition in albino rabbits. J Pharm Sci. 1979;68:673–684. doi: 10.1002/jps.2600680606. [DOI] [PubMed] [Google Scholar]
- 2.Patton TF, Robinson JR. Quantitative precorneal disposition of topical applied pilocarpine nitrate in rabbit eyes. J Pharm Sci. 1976;65:1295–1301. doi: 10.1002/jps.2600650909. [DOI] [PubMed] [Google Scholar]
- 3.De S, Robinson D. Polymer relationships during preparation of chitosan–alginate and poly-l-lysine–alginate nanospheres. J Control Rel. 2003;89:101–112. doi: 10.1016/S0168-3659(03)00098-1. [DOI] [PubMed] [Google Scholar]
- 4.Gref R, Minamitake Y, Perracchia MT, Trubeskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 5.Merodio M, Espuelas MS, Mirshahi M, Arnedo A, Irache JM. Efficacy of Ganciclovir-loaded nanoparticles in human cytomegalovirus (HCMV)-infected cells. J Drug Target. 2002;10:231–238. doi: 10.1080/10611860290022660. [DOI] [PubMed] [Google Scholar]
- 6.Bourges JL, Gautier SE, Delie F, Bejjani RA, Jeanny JC, Gurny R, et al. Ocular drug delivery targeting the retina and retinal pigment epithelium using poly lactide nanoparticles. Invest Ophthalmol Vis Sci. 2003;44:3562–3569. doi: 10.1167/iovs.02-1068. [DOI] [PubMed] [Google Scholar]
- 7.Kompella UB, Bandi N, Ayalasomayajula SP. Subconjunctival nano- and microparticles sustain retinal delivery of budesonide, a corticosteroid capable of inhibiting VEGF expression. Invest Ophtholmol Vis Sci. 2003;44:1192–1200. doi: 10.1167/iovs.02-0791. [DOI] [PubMed] [Google Scholar]
- 8.Santos ALG, Bochot A, Doyle A, Tsapis N, Siepmann J, Siepmann F, et al. Sustained release of nanosized complexes of poly ethylenimine and anti-TGF-β2 oligonucleotide improves the outcome of glaucoma surgery. J Control Release. 2006;112:369–381. doi: 10.1016/j.jconrel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Calvo P, Vila-Jato JL, Alonso MJ. Evaluation of cationic polymer-coated nanocapsules as ocular drug carriers. Int J Pharm. 1997;153:41–50. doi: 10.1016/S0378-5173(97)00083-5. [DOI] [Google Scholar]
- 10.De Campos AM, Sanchez A, Alonso MJ. Chitosan nanoparticles: a new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to cyclosporine A. Int J Pharm. 2001;224:159–168. doi: 10.1016/S0378-5173(01)00760-8. [DOI] [PubMed] [Google Scholar]
- 11.De Campos AM, Diebold Y, Carvalho ELS, Sanchez A, Alonso MJ. Chitosan nano particles as new ocular drug delivery systems: in vitro stability, in vivo fate, and cellular toxicity. Pharm Res. 2004;21:803–810. doi: 10.1023/B:PHAM.0000026432.75781.cb. [DOI] [PubMed] [Google Scholar]
- 12.Motwani SK, Chopra S, Talegaonkar S, Kohli K, Ahmad FJ, Khar RK. Chitosan-sodium alginate nanoparticles as submicroscopic reservoirs for ocular delivery: formulation, optimisation and in vitro characterisation. Eur J Pharm Biopharm. 2008;68:513–525. doi: 10.1016/j.ejpb.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Langer K, Mutschler E, Lambrecht G, Mayer D, Troschau G, Stieneker F, et al. Methylmethacrylate sulfopropylmethacrylate copolymer nanoparticles for drug delivery. Part III: evaluation as drug delivery system for ophthalmic applications. Int J Pharm. 1997;158:219–231. doi: 10.1016/S0378-5173(97)00255-X. [DOI] [Google Scholar]
- 14.Masson V, Billardon P, Fessi H, Devissagaut JP, Puisieux F. Tolerance studies and pharrnacokinetic evaluation of indomethacin-loaded nanocapsules in rabbit eyes. Proc Int Symp Control Rel Bioact Mater. 1992;19:423. [Google Scholar]
- 15.Heussler ML, Fessi H, Devissaguet JP, Hoffman M, Maincent P. Colloidal drug delivery systems for the eye: a comparison of the efficacy of three different polymers: polyisobutyl cyanoacrylate, polylactic-co-glycolic acid, polyepsilon-caprolacton. STP Pharma Sci. 1992;2:98–104. [Google Scholar]
- 16.De TK, Rodman DJ, Holm BA, Prasad PN, Bergey EJ. Brimonidine formulation in polyacrylic acid nanoparticles for ophthalmic delivery. J Microencapsulation. 2003;20:361–374. doi: 10.1080/0265204031000093591. [DOI] [PubMed] [Google Scholar]
- 17.De TK, Bergey EJ, Chung SJ, Rodman DJ, Bharali DJ, Prasad PN. Polycarboxylic acid nanoparticles for ophthalmic drug delivery: an ex vivo evaluation with human cornea. J Microencapsulation. 2004;21:841–855. doi: 10.1080/02652040400008515. [DOI] [PubMed] [Google Scholar]
- 18.Munk SA, Harcourt D, Arasasingham P. Analogs of UK 14,304: structural features responsible for 2-adrenoceptor activity. Bioorg Med Chem. 1995;5:1745–1750. doi: 10.1016/0960-894X(95)00295-5. [DOI] [Google Scholar]
- 19.Torris CB, Camras CB, Yablonski ME. Acute versus chronic effects of brimonidine on aqueous humor dynamics in ocular hypertensive patients. Am J Ophthalmol. 1999;128:8–14. doi: 10.1016/S0002-9394(99)00076-8. [DOI] [PubMed] [Google Scholar]
- 20.Burke J, Manlapaz C, Kharlamb A, Runde E, Padillo E, Spada C. Therapeutic use of alpha-2-adrenoceptor agonists in glaucoma. In: Alpha-2-adrenergic receptors: structure, function and therapeutic implications. In: S. Lanier and Limbird (eds). Harwood Academic: Reading, UK. 1996. pp 179–187.
- 21.Burke J, Schwartz M. Preclinical evaluation of brimonidine. Surv Ophthalmol. 1996;41(suppl 1):S9–S18. doi: 10.1016/S0039-6257(96)82027-3. [DOI] [PubMed] [Google Scholar]
- 22.Cantor BL, Burke J. Drug evaluation: brimonidine. Expert Opin Investig Drugs. 1997;6:1063–1083. doi: 10.1517/13543784.6.8.1063. [DOI] [PubMed] [Google Scholar]
- 23.Konstas AGP, Stewart WC, Topouzis F, Tersis I, Holmes KT, Stangos NT. Brimonidine 0.2% given two or three times daily versus timolol maleate 0.5% in primary angle glaucoma. Am J Ophthalmol. 2001;131:729–733. doi: 10.1016/S0002-9394(01)00834-0. [DOI] [PubMed] [Google Scholar]
- 24.Serle JB. A comparison of the safety and efficacy of twice daily brimonidine 0.2% versus betaxolol 0.25% in subjects with elevated intraocular pressure. Surv Ophthalmol. 1996;41(Suppl 1):S39–S46. doi: 10.1016/S0039-6257(96)82030-3. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed FA, Hegazy K, Chaudhary P, Sharma SC. Neuroprotective effect of alpha-2 agonist (brimonidine) on adult rat retinal ganglion cells after increased intraocular pressure. Brain Res. 2001;913:133–139. doi: 10.1016/S0006-8993(01)02759-7. [DOI] [PubMed] [Google Scholar]
- 26.Wheeler LA, Tatton M, Elstner M, Chalmers-Redman RME, Tatton WG. Alpha-2 adrenergic receptor activation by brimonidine reduces neuronal apoptosis through Akt (protein kinase B) dependent new synthesis of bcl-2. Invest Ophthalmol Vis Sci. 2001;42:S441. [Google Scholar]
- 27.Wheeler L, WoldeMussie E, Lai R. Role of alpha-2 agonists in neuroprotection. Surv Ophthalmol. 2003;48(Suppl 1):S47–S51. doi: 10.1016/S0039-6257(03)00004-3. [DOI] [PubMed] [Google Scholar]
- 28.Lai R, Hassan D, Chun T. Neuroprotective effect of ocular hypotensive agent brimonidine, Xith Congress of the European Society of Ophthalmology. Budapest, 1997: 439–444.
- 29.Lafuente MP, Villegas-Perez MP, Mayor S, Aguilera ME, Miralles IJ, Vidal-Sanz M. Neuroprotective effects of brimonidine against ischemia induced retinal ganglion. Cell death: a dose response in vivo study. Exp Eye Res. 2002;74:181–189. doi: 10.1006/exer.2001.1122. [DOI] [PubMed] [Google Scholar]
- 30.Pignatello R, Bucolo C, Puglisi G. Ocular tolerability of Eudragit RS100 and RL100 nanosuspensions as carriers for ophthalmic controlled drug delivery. J Pharm Sci. 2002;91:2536–2641. doi: 10.1002/jps.10227. [DOI] [PubMed] [Google Scholar]
- 31.Pignatello R, Bucolo C, Spedalieri G, Maltese A, Puglisi G. Flurbiprofen-loaded acrylate polymer nanosuspensions for ophthalmic application. Biomaterials. 2002;23:3247–3255. doi: 10.1016/S0142-9612(02)00080-7. [DOI] [PubMed] [Google Scholar]
- 32.Al-Kassas R. Design and in vitro evaluation of gentamicin–Eudragit microspheres intended for intra-ocular administration. J Microencapsulation. 2004;21:71–81. doi: 10.1080/02652040310001619992. [DOI] [PubMed] [Google Scholar]
- 33.Krinzar B, Mateovic T, Bogataj M, Mrhar A. The influence of chitosan on in vitro properties of eudragit RS microspheres. Chem Pharm Bull. 2003;51:359–364. doi: 10.1248/cpb.51.359. [DOI] [PubMed] [Google Scholar]
- 34.Bhagav P, Deshpande P, Pandey S, Chandran S. Development and validation of stability indicating UV spectrophotometric method for the estimation of brimonidine tartrate in pure form, formulations and preformulation studies. Der Pharmacia Lettre. 2010;2(3):106–122. [Google Scholar]
- 35.Draize JH, Woodard G, Calvery HO. Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J Pharmacol Exp Ther. 1944;82:377–390. [Google Scholar]
- 36.Percicot CL, Schnell CR, Debon C, Hariton C. Continuous intraocular pressure measurement by telemetry in alpha-chymotrypsin-induced glaucoma model in the rabbit: effects of Timolol, Dorzolamide, and Epinephrine. J Pharmacol Toxicol Methods. 1996;36:223–228. doi: 10.1016/S1056-8719(96)00130-X. [DOI] [PubMed] [Google Scholar]
- 37.Anumolu SS, Singh Y, Gao D, Stein S, Sinko JP. Design and evaluation of novel fast forming pilocarpine-loaded ocular hydrogels for sustained pharmacological response. J Control Release. 2009;137:152–159. doi: 10.1016/j.jconrel.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez DR, Ramirez JM, Trivino A, Sanchez D, Paraiso P, Garcia DLM, et al. Experimental glaucoma significantly decreases atrial natriuretic factor (ANF) receptors in the ciliary processes of the rabbit eye. Exp Eye Res. 1991;53:591–596. doi: 10.1016/0014-4835(91)90217-3. [DOI] [PubMed] [Google Scholar]
- 39.Himber J, De Burlet G, Andermann G. Effects of adrenergic agents on alpha-chymotrypsin induced ocular hypertension in albino and pigmented rabbits: a comparative study. J Ocul Phurmacol. 1989;5:93–98. doi: 10.1089/jop.1989.5.93. [DOI] [PubMed] [Google Scholar]