
Table 1. Data statistics.
Values in parentheses are for the highest resolution shell.
| PDB entry | 3vi6 |
| Data collection | PF-AR NW12 |
| Wavelength (Å) | 1.000 |
| Resolution range (Å) | 50–1.59 (1.62–1.59) |
| Space group | H32 |
| Unit-cell parameters (Å) | a = b = 56.3, c = 203.6 |
| No. of observations | 181997 |
| No. of unique reflections | 16978 (814) |
| Completeness (%) | 99.6 (98.3) |
| Multiplicity | 10.7 (10.1) |
| Average I/σ(I) | 39.0 (8.2) |
| Rmerge† | 0.052 (0.310) |
| Refinement | |
| No. of reflections | 15242 |
| Protein atoms | 753 |
| Formate atoms | 3 |
| Water atoms | 83 |
| Resolution range (Å) | 50–1.59 (1.64–1.59) |
| Rwork‡ | 0.192 (0.217) |
| Rfree§ | 0.216 (0.257) |
| Mean B factor (Å2) | 19.0 |
| Overall anisotropic B factors (Å2) | |
| B11, B22, B33 | 0.79, 0.79, −1.18 |
| B12, B12, B23 | 0.39, 0.00, 0.00 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.157 |
†
R
merge = 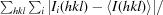
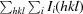 , where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
, where 〈I(hkl)〉 is the mean intensity of symmetry-equivalent reflections.
‡
R
work = 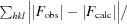
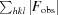 , where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
, where F
obs and F
calc are the observed and calculated structure-factor amplitudes, respectively.
§
The R free value was calculated as for the R factor but using only a test set of reflections (5% of the total) that were not used in the refinement.