

Abstract
p16, a nuclear protein encoded by the p16INK4a gene, is a regulator of cell cycle regulation. Previous studies have shown that expression of p16 in tissue biopsies of patients with ductal carcinoma in situ (DCIS) is associated with increased risk of breast cancer, particularly when considered in combination with other markers such as Ki67 and COX2. Here we evaluated how expression of p16 in breast tissue biopsies of women with atypical hyperplasia (AH), a putative precursor lesion to DCIS, is associated with subsequent development of cancer. p16 expression was assessed by immunohistochemistry in archival sections from 233 women with AH diagnosed at the Mayo Clinic. p16 expression in the atypical lesions was scored by percent of cells positive and intensity of staining. We also studied coexpression of p16, with Ki67 and COX2, biomarkers of progression in AH. Risk factor and follow-up data were obtained via study questionnaire and medical records. Forty-seven patients (20%) developed breast cancer with a median follow-up of 14.5 years. Staining of p16 was increased in older patients relative to younger patients (p=0.0025). While risk of developing breast cancer was not associated with increased p16 expression, joint overexpression of Ki67 and COX2 was found to convey stronger risk of BC in the first 10 years after diagnosis as compared to one negative marker (p<0.01). However, the addition of p16 levels did not strengthen this association. p16 overexpression, either alone or in combination with COX2 and Ki67, does not significantly stratify breast cancer risk in women with AH.
Keywords: atypical hyperplasia, breast cancer risk, p16, immunohistochemistry
INTRODUCTION
Atypical hyperplasia (AH) of the breast is diagnosed in approximately 40,000–50,000 US women/year, as it is found in about 4–5% of the 1,000,000 benign breast biopsies performed annually (1, 2). Women diagnosed with AH have a substantial risk of subsequent development of breast cancer, with a cumulative incidence of 30% at 25 years (3). AH is thought to represent a precursor stage to in situ and invasive carcinoma: biopsies of breast tissue removed prophylactically from BRCA1/2 carriers contain various proliferative benign lesions including AH in over 50% of cases (4, 5), and AH is frequently found in random periareolar fine needle aspirations from high risk women compared to normal risk women (6). AH is closely related to low-grade in situ carcinoma (7–9), with the distinction based mainly upon the extent of the lesion. We have previously investigated processes associated with breast cancer progression as biomarkers of breast cancer risk for AH patients, finding that increased expression of the proliferation marker Ki67 (10) or the inflammatory/invasive marker cyclooxygenase-2 (COX2) (11) are each associated with a higher risk of developing cancer, and that expression of estrogen receptor (ER) is not associated with subsequent cancer development (12). Previous studies investigating ductal carcinoma in situ (DCIS) have found that expression of Ki67 and/or COX2 in combination with expression of p16INK4a (p16) are associated with subsequent cancer incidence (13, 14); here we assessed whether these markers had prognostic significance for patients with AH.
The p16 protein acts as a negative regulator of cell proliferation by inhibiting the phosphorylation of retinoblastoma (Rb) family members by cyclin-dependent kinases CDK4/6 (reviewed in (15)). Cells which have been stimulated to proliferate rapidly by oncogenic stimuli can activate an antiproliferative stress-associated senescence program, leading to increased expression of p16 and consequent maintenance of Rb in a hypophosphorylated state, inducing G1 cell cycle arrest (15). p16 expression is also increased in an age-associated fashion (i.e. replicative senescence) in tissues which have undergone repeated proliferative cycles over extended periods of time (16).
Conversely in the setting of deregulation of Rb, p16 expression can be elevated in proliferating cells as the growth-suppressive effects of p16 are abrogated downstream of Rb. p16 overexpression is therefore conditional on the functional state of Rb. p16 overexpression in the setting of Rb deregulation is indicative of cancer progression and, in mammary carcinoma, is a hallmark of the basal-like molecular subtype (13, 17). When assayed in pre-malignant lesions of the breast (i.e. DCIS), increased expression of p16 has been found to be associated with increased risk of subsequent development of cancer when expressed in combination with Ki67 and/or COX-2 (13, 14).
As AH is thought to represent a precursor to DCIS (7, 18, 19), we hypothesized that combinatorial analysis of p16, Ki67, and COX-2 expression in AH might stratify risk for subsequent development of DCIS or invasive BC. To test this hypothesis, we first measured the expression of p16 in 233 women with AH and determined association with patient age, type of AH, and risk of subsequent development of cancer. We then studied the risk of subsequent tumor development with concurrent expression of p16, Ki67 and COX-2 in AH.
METHODS
Study population
Entry criteria for this study have been described previously (2, 3). Briefly, the samples used for this study were derived from a Mayo Benign Breast Disease (BBD) cohort of 9376 women ages 18–85 who had excisional breast biopsies with benign findings at the Mayo Clinic in Rochester, MN between January 1, 1967 and December 31, 1991 (2). Women were excluded from the study if there was cancer found in any of the specimen. Of the 331 women within this cohort with atypical hyperplasia, paraffin-embedded, formalin-fixed tissue for p16 staining was available for 233 of them. All protocol procedures and patient contact materials were reviewed and approved by the Institutional Review Board of the Mayo Clinic.
Follow-up and risk factor data
Follow-up for breast cancer events and risk factor information was obtained through Mayo medical records and a study questionnaire (2, 3). Family history was classified as negative, weak, or strong. Criteria for a strong family history were: at least one first-degree relative with breast cancer before the age of 50 years or two or more relatives with breast cancer, with at least one being a first degree relative. Any lesser degree of family history was considered to be weak. Eligible cancer events include women with either DCIS or invasive cancer diagnoses.
Immunostaining
Immunostaining of COX-2 and Ki67 was described previously (10, 11). p16 (16P07 – Neo Markers, MS1064-P) was assessed by immunohistochemistry using 5 µm formalin-fixed, paraffin-embedded tissue sections. Samples were deparaffinized with three changes of xylene and rehydrated in a series of alcohols (100, 95, then 70% EtOH) and rinsed well in running distilled water. Slides were then placed in a preheated 0.1 mM EDTA, pH 8.0 retrieval buffer for 30 minutes and then cooled in the buffer for 5 minutes followed by a 5 minute rinse in running distilled water. After the HIER (heat-inactivated epitope retrieval) step, slides were placed on the DAKO Autostainer for the following procedure (at room temperature). Sections were incubated with 3% H2O2 in ethanol for 5 minutes to inactive the endogeneous peroxides. Sections were incubated in p16 antibody at 1/80 dilution for 30 minutes at room temperature. Sections were rinsed with Tris-buffered saline/Triton-X100 (TBST) wash buffer. Secondary incubation was with DAKO DUAL+, horseradish peroxidase (HRP) for 15 minutes. The slides were rinsed with TBST wash buffer. Sections were then incubated in 3,3’-diaminobenzidine (DAB+) (K3467, DAKO) for 5 minutes, counterstained with modified Schmidt’s hematoxylin for 5 minutes, followed by a 3 minute tap water rinse to blue sections, dehydrated through graded alcohols and cleared in three changes of xylene and mounted with permanent mounting media. Using light microscopy, stained tissue sections were inspected by the study pathologist (HB). Cytoplasmatic immunopositivity with occasional nuclear staining was identified by the brown chromogen diaminobenzidine. Colon cancer tissue was used as positive control, and no primary antibody was used as negative control. Representative p16 staining is illustrated in Figure 1.
Figure 1.

Staining levels of p16 in AH. Samples of atypical ductal hyperplasia (ADH, left column) and atypical lobular hyperplasia (ALH, right column), showing fractional staining levels of <1% cells staining positively for p16 (top row), 1–10% p16+ cells (second row), 11–50% p16+ cells (third row), 51–90% p16+ cells (fourth row), and >90% p16+ cells (bottom row).
Statistical analysis
Data were summarized descriptively using frequencies and percentages for categorical variables and medians and interquartile ranges (IQRs) for continuous variables. Among the 331 women with atypical hyperplasia, we compared distributions of demographic and clinical variables by tissue availability for p16 staining using chi-square tests of significance for categorical variables and Wilcoxon rank sum tests for continuous variables.
The length of follow-up for each woman in the study was defined as the number of days from her benign biopsy to the date of her breast cancer diagnosis, death or last contact. In addition, women with prophylactic mastectomy, or with a diagnosis of LCIS or DCIS, were censored at the corresponding date of occurrence. We estimated relative risks, overall and by strata of p16, Ki67, and COX-2 staining levels, with standardized incidence ratios (SIRs) by dividing the observed numbers of incident breast cancer by population-based expected values. The approach allowed us to compare rates of breast cancer in our cohort with that of the general population rather than an internal referent group, recognizing that all women in our cohort were at some increased risk of breast cancer from their diagnosis of atypical hyperplasia. Expected values were calculated by apportioning each woman’s person-years of follow-up into 5-year age and calendar-period categories, thereby accounting for any age or cohort period effects on the risk of cancer, and multiplying these by the corresponding breast cancer incidence rates from the Iowa Surveillance, Epidemiology, and End Results (SEER) registry. This reference population was chosen because of its demographic similarities to the Mayo Clinic population (80% of cohort members reside in the upper Midwest). Separate analyses were carried out for percent staining and intensity of staining. Potential heterogeneity in standardized incidence ratios across p16, Ki-67 and COX2 staining levels, as well as combinations of the two or three variables, was assessed by use of Poisson regression analysis, with the log-transformed expected event rate for each individual modeled as the offset term. We visually displayed observed and expected event rates using cumulative incidence curves, while accounting for the effects of death as a competing risk (20). Expected events were calculated for each one-year follow-up interval in a manner similar to that used for determining SIRs. A modified Kaplan-Meier approach was used to accumulate expected incidence over these intervals. The expected curve was then smoothed using linear interpolation. All statistical tests were two-sided, and all analyses were conducted using the SAS (SAS Institute, Inc., Cary, NC) software system.
RESULTS
Patient characteristics
Within our AH cohort of 331 women, samples from 233 patients were available to be stained for p16. We compared these 233 subjects to the remaining 98 and saw no significant differences in terms of distributions of case status, age at AH, family history of breast cancer, and for breast cancer patients, time to diagnosis (p>0.05 for each attribute). The characteristics of the subjects included in this study are presented in Table 1. Median post-biopsy follow-up was 14.5 years for the 233 women, 47 of whom (20.2%) have developed breast cancer. Of the 47 women in this study who developed breast cancer, 34 had documented invasive cancer, 10 had DCIS, and 3 had confirmed breast cancer but with unknown tumor stage.
Table 1.
Association of p16 expression (% cells positive) with demographic and clinical variables among women with atypia
| p16 percent cells/positive | |||
|---|---|---|---|
| 0–10% (N=158) | 11+% (N=75) | p value1 | |
| Age at BBD diagnosis | 0.0025 | ||
| <30 | 3 (100%) | 0 (%) | |
| 30 – 39 | 8 (88.9%) | 1 (11.1%) | |
| 40 – 49 | 47 (82.5%) | 10 (17.5%) | |
| 50 – 59 | 40 (58%) | 29 (42%) | |
| 60 – 69 | 40 (72.7%) | 15 (27.3%) | |
| 70+ | 20 (50%) | 20 (50%) | |
| Year of Benign Biopsy | 0.3588 | ||
| 1967–1971 | 8 (88.9%) | 1 (11.1%) | |
| 1972–1976 | 18 (78.3%) | 5 (21.7%) | |
| 1977–1981 | 18 (62.1%) | 11 (37.9%) | |
| 1982–1986 | 45 (70.3%) | 19 (29.7%) | |
| 1987–1991 | 69 (63.9%) | 39 (36.1%) | |
| Indication for Biopsy2 | 0.1738 | ||
| Lump | 70 (72.9%) | 26 (27.1%) | |
| Mammogram | 87 (64.4%) | 48 (35.6%) | |
| Number of Atypical Foci | 0.6472 | ||
| 1 Focus | 90 (70.3%) | 38 (29.7%) | |
| 2 Foci | 42 (65.6%) | 22 (34.4%) | |
| 3+ Foci | 26 (63.4%) | 15 (36.6%) | |
| Involution3 | 0.2132 | ||
| None | 12 (85.7%) | 2 (14.3%) | |
| Partial | 124 (67.8%) | 59 (32.2%) | |
| Complete | 19 (59.4%) | 13 (40.6%) | |
| Family History4 | 0.5063 | ||
| No Family Hx | 91 (68.4%) | 42 (31.6%) | |
| Weak Family Hx | 22 (59.5%) | 15 (40.5%) | |
| Strong Family Hx | 36 (70.6%) | 15 (29.4%) | |
| Type of AH | 0.4596 | ||
| ADH | 63 (65.6%) | 33 (34.4%) | |
| ALH | 71 (67%) | 35 (33%) | |
| Both ADH and ALH | 24 (77.4%) | 7 (22.6%) | |
| Follow-Up in Years | 0.5163 | ||
| Median (IQR) | 14.5 (9.0 – 18.1) | 14.7 (9.7 – 18.6) |
Wilcoxon rank sum test for follow-up in years, chi-square tests of statistical significance for all other variables.
Indication for Biopsy was available for 231 of the 233 women.
Involution status was available for 229 of the 233 women.
Family history of breast cancer was available for 221 of the 233 women.
p16 immunostaining of AH samples
In the 233 samples, we analyzed both percent cells positive for p16 and intensity of staining. For extent of p16 positivity, we used the following categories: 0, 1–10%, 11–50%, 51–90%, and > 90% atypical cells positive (Figure 1). Intensity was categorized as 0 (negative), 1+ (weak), 2+ (moderate), and 3+ (strong) staining. The correlation between these two measures was 0.76 (p-value 0.001). Because of this strong correlation, and because results using intensity of staining were similar to those using percents, all subsequent results focus on percent cells positive. For comparisons with clinicopathological features, we combined the p16-expression categories into two groups: p16 low-expressing (0–10% cell positive) and p16 high-expressing (11+% cells positive), as using an 11%+ threshold separates individuals into groups of approximately two-thirds with less than 11% positive cells and one-third with 11% or more. The positive effects of this categorization are two-fold. First, sub-stratifying women who already have a relatively uncommon condition in the population (women with atypical hyperplasia compose only 3–4% of the total number of women with clinically-confirmed BBD) into categories smaller than one-third of all AH results in subgroups representing less than 1% of all women with BBD, which limits our ability to assess public health significance. Second, due to sample size restrictions, sub-stratifying P16 into smaller categories prohibited the examination of combinations of P16 with Ki67 and COX-2 shown in Table 3. Using these measures, we found a significant (p=0.0025) association of greater p16 expression with increasing age at BBD diagnosis (Table 1). We also examined the data categorizing women into 0% cells positive and 1%+, which also separated women into groups of approximately two-thirds and one-third. This threshold yielded similar risk estimate results as with the 11% threshold, but attenuated the association with age at BBD, which was no longer statistically significant. We saw no association between level of p16 expression and year of benign biopsy, indication for biopsy, number of atypical foci, lobular involution status, family history, type of AH, and time of followup (Table 1).
Table 3.
Risk of breast cancer by p16 expression with Ki67 and COX-2
| Staining category | N | Events | Expected Events |
SIR (95% CI)5 | P- value6 |
|---|---|---|---|---|---|
| p162 | 0.99 | ||||
| 0–10% | 158 | 30 | 7.7 | 3.89 (2.72 – 5.56) | |
| 11+% | 75 | 17 | 4.4 | 3.91 (2.43 – 6.28) | |
| p16/Ki673 | 0.92 | ||||
| 0–10%/2+ | 32 | 3 | 1.6 | 1.86 (0.60 – 5.78) | |
| 0–10%/<2 | 84 | 14 | 4.4 | 3.20 (1.90 – 5.41) | |
| 11+%/2+ | 19 | 5 | 1.1 | 4.47 (1.86 – 10.7) | |
| 11+%/<2 | 37 | 6 | 2.3 | 2.66 (1.20 – 5.93) | |
| p16/COX-24 | 0.03 | ||||
| 0–10%/0 or 1+ | 73 | 8 | 3.9 | 2.08 (1.04 – 4.15) | |
| 0–10%/2+ or 3+ | 60 | 12 | 3 | 4.01 (2.28 – 7.07) | |
| 11+%/0 or 1+ | 37 | 4 | 2.2 | 1.78 (0.67 – 4.74) | |
| 11+%/2+ or 3+ | 32 | 9 | 1.8 | 5.03 (2.62 – 9.66) |
p16 results based on 233 subjects with p16 results; p16/Ki67 combinations based 172 subjects with both p16 and Ki67 results; P16/COX-2 combinations based on 202 subjects with both p16 and COX-2 results.
p16 staining is dichotomized as percent cells stained, 0–10% vs 11+%.
Ki67 staining is dichotomized as percent cells stained, 0 or 1 vs 2 or higher (10).
COX-2 staining is dichotomized by general staining intensity, 0 or 1+ vs 2+ or 3+ (11).
Standardized incidence ratios (SIR) and corresponding 95% confidence intervals, comparing the observed number of breast cancer events to those expected based on incidence rates from Iowa SEER data. Analyses account for the effects of age and calendar period.
Test for heterogeneity of the standardized incidence ratio across staining levels.
Association of p16 expression with breast cancer risk
We found no association between extent of p16 expression and risk of subsequent breast cancer (Table 2, Figure 2). The overall risk of cancer in the p16 low-expressing group was 3.89 (95% CI=2.72 to 5.56) and RR=3.91 (95% CI=2.43 to 6.28) in the high-expressing group. Similar results were found for cells with low intensity staining (0–1+) vs. 2–3+ staining (data not shown). We also examined time to breast cancer from AH biopsy and found no association with p16 expression (Figure 2). The median time to breast cancer in the low p16 group was 12.9 years, vs. 13.5 years in the high p16 group.
Table 2.
Risk of breast cancer by p16 expression in AH
| Percent Cells Positive for p16 |
N | Events | Expected Events |
SIR (95% CI)1 | P-value2 |
|---|---|---|---|---|---|
| 0.88 | |||||
| 0 | 80 | 18 | 4.2 | 4.24 (2.67 – 6.73) | |
| 1–10 | 78 | 12 | 3.5 | 3.45 (1.96 – 6.08) | |
| 11–50 | 45 | 11 | 2.6 | 4.15 (2.30 – 7.50) | |
| 51+ | 30 | 6 | 1.7 | 3.52 (1.58 – 7.84) |
Standardized incidence ratios and corresponding 95% confidence intervals, comparing the observed number of breast cancer events to those expected based on incidence rates from Iowa SEER data. Analyses account for the effects of age and calendar period.
Test for heterogeneity of the standardized incidence ratio across staining levels.
Figure 2.
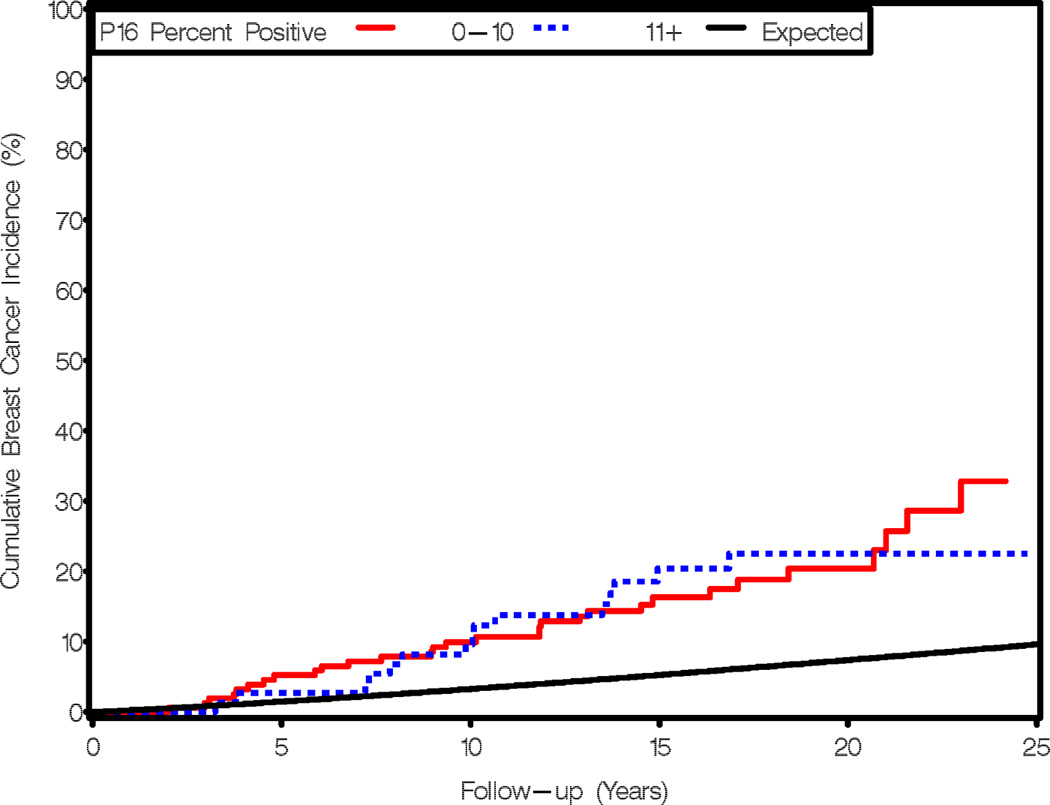
Expression of p16 is not related to cumulative breast cancer incidence or time to breast cancer for women with AH. Observed and expected events are cumulated after accounting for death as a competing risk and are plotted as a function of follow-up interval and stratified by p16 expression levels. Red line, 0–10% cells staining for p16; blue line, 11+% cells staining for p16; black line, expected breast cancer incidence according to Iowa Surveillance, Epidemiology, and End Results survey.
Co-expression of p16 with Ki67 and COX-2
We evaluated the effect of coexpression of p16 with proliferative (Ki67) and protumorigenic (COX2) markers. We have previously evaluated Ki67 staining for 172 (10) and COX-2 for 158 (11) of these 233 women. Staining of p16, Ki67, and COX-2 on sequential slides from a single block has been performed in 158 women. We did not find that addition of Ki67 or COX-2 expression levels modified the association of p16 with cancer risk (Table 3). We previously showed that higher Ki67 levels in AH are associated with breast cancer risk (10). Adding p16 to this analysis did not strengthen the association (not shown). Overexpression of both Ki67 plus COX2 showed an increased breast cancer risk among patients in the first 10 years (SIR 6.70, 95% CI 3.19–14.1) as compared to one negative marker. Further sub-stratification into women with high expression levels of all three biomarkers (N=13) resulted in an SIR of 9.38 (95% CI 3.52–25.0). Although seemingly higher, the addition of P16 did not result in a statistically significant increase in risk over and above the contribution of just Ki67 and COX2, perhaps in part due to the limited sample size in this group of women. Thus, results for this triple high expression group should be interpreted with caution.
DISCUSSION
We quantified p16 expression in a well-characterized cohort of 233 women with AH in which 47 patients had developed breast cancer with a median follow-up of 14.5 years. We found that 32% of the biopsies showed more than 10% atypical cells staining positive for p16. We found that p16 expression levels correlated with age at biopsy (p=0.0025), but not with other demographic and clinical features. We found that patients showing increased expression of p16 in AH had no significant risk of subsequent development of breast cancer, even when considered in combination with expression of Ki-67 and COX2.
p16 is a biomarker of two opposing phenotypes – overexpression of p16 can indicate either activation of a senescence program or loss of growth control through deregulation of Rb signaling. Studies in cultured primary human breast epithelial cells suggest that these phenotypes can be distinguished through combined measurement of p16 and proliferation rate (21). In invasive breast cancer, combined overexpression of p16 and increased proliferation rate is unique to a subset of tumors of the basal-like molecular subtype (13). It has also been observed that a subset of DCIS lesions show combined increase in p16 and proliferation (assessed by Ki67). This appears biologically significant as DCIS lesions with increased p16/Ki67 show increased risk for subsequent tumor development (13, 14). In these DCIS studies, the threshold of Ki67 positivity was set at 10%. The median Ki67 level in the series of DCIS is higher than in the series of AH; in the AH cohort, the median Ki67 is 1% and the a cutoff of 2% cells positive for Ki67 was used to dichotomize groups of individuals with higher or lower degrees of staining (10). The relative lower proliferation rate in AH may suggest that the high risk p16 phenotype is uncommon in AH. Alternatively, deregulation of Rb in a subset of AH may not correspond to high proliferation and expression of other biomarkers (i.e. E2F targets) may be more informative when combined with p16. Finally, to the extent that p16 expression in AH reflects activation of cellular senescence, prevalence of p16 in AH suggests a substantial percentage of these lesions maintain intact growth regulation.
Women diagnosed with AH are at significantly elevated risk for subsequent development of cancer, but methods for estimating risk for individual women with AH are lacking. We have found that risk models commonly used for women with atypia, which rely primarily on clinical and epidemiologic features, perform no better than chance alone for individual women with atypia (22, 23). Instead, we have found that direct examination of pathological characteristics of the breast tissue biopsy can be used to better stratify cancer risk for women with AH. For example, we have shown that delayed or reduced lobular involution (24), the presence of multiple atypical foci (3), and elevated expression of COX-2 (11) and Ki67 (10) are all associated with increased risk for subsequent development of cancer. We predict that a risk model that integrates morphologic and molecular features with clinical and epidemiologic information will improve breast cancer risk assessment.
We found a significant association between p16 expression in the atypical lesions and increasing age at biopsy (p=0.0025). p16 levels have been shown to rise with age in many different tissue types (16, 25–27), although association of p16 with age in breast tissue has not been described previously to our knowledge. The consequences of p16 expression in aging tissue is not known. Some evidence suggests that increased expression of p16 directly leads to tissue degeneration associated with aging (28): deletion of p16 in a genetic model of premature aging results in attenuated phenotype and extended lifespan (29), and caloric restriction that retards aging phenotypes in experimental models also attenuates the accumulation of p16-positive cells (16). Activation of p16 in aging tissues has been proposed to be a response to extended replication and potentially to serve a tumor suppressive function; however, cells in which p16 expression is maintained for extended periods of time are known to secrete proteins that alter tissue structure and function and promote tumor development (30, 31). In this cohort of women with atypia, we saw no evidence of reduced or increased risk of breast cancer with higher p16 expression. Investigations with larger cohorts and further analysis of senescence-associated biomarkers may help to determine whether increased p16 expression in older women reflects activation of a tumor-suppressive senescence program or a tumor-potentiating phenotype.
Strengths of our study include central pathologic review of atypia samples and long term post-biopsy follow-up. Our access to extensive medical record information permits assessment of a number of potential covariates; in combination with our previous characterization of this cohort we could examine associations within subsets of concomitant pathologic features. Limitations of the study include the semiquantitative nature of tissue-based biomarker studies, which is further complicated by the generally weak character of p16 immunoreactivity in AH. Furthermore, while to our knowledge this is the largest examination of biomarker information in women with AH to date, we had relatively low statistical power to detect heterogeneity in standard incidence ratios, particularly when including Ki-67 and COX-2 as biomarkers. Investigation of larger or additional cohorts may be required to determine whether p16 expression levels can provide additional discriminatory power when combined with other biomarkers.
Conclusions
p16 has emerged as a marker and mediator of cellular senescence, a key step in the process of preventing tumor progression. Previous studies have found that patients with DCIS with increased p16 expression, in combination with Ki67 (indicating active proliferation) or with COX2 (indicating activation of inflammatory and invasive phenotypes), show significantly higher likelihood of progression to breast cancer, and that the sustained activation of p16 in these cases could be due to bypass of normal senescence responses. Here, we find that expression of p16 in patients with AH is not associated with future cancer incidence, even when combined with expression of Ki67 and/or COX2, which may indicate that the p16-associated senescence pathway remains intact in AH. Comparison of p16 expression with other patient characteristics revealed a significant correlation only for the increased proportion of p16-expressing lesions in older patients (P=0.0025), consistent with previous studies showing age-dependent increases in p16 expression in other tissues. Determining whether the increased abundance of p16-positive cells in AH of older women is associated with tumor protective or promoting effects of senescence activation will require further investigation.
Acknowledgements
This work is supported by NCI grants CA122086, 132789, and by the Mayo Clinic Breast SPORE CA116201. We thank Linda Murphy for immunostaining, and Patricia Haugen for her perspective as a patient advocate.
REFERENCES
- 1.Ghosh K, Melton LJ, 3rd, Suman VJ, Grant CS, Sterioff S, Brandt KR, et al. Breast biopsy utilization: a population-based study. Arch Intern Med. 2005;165:1593–1598. doi: 10.1001/archinte.165.14.1593. [DOI] [PubMed] [Google Scholar]
- 2.Hartmann LC, Sellers TA, Frost MH, Lingle WL, Degnim AC, Ghosh K, et al. Benign breast disease and the risk of breast cancer. N Engl J Med. 2005;353:229–237. doi: 10.1056/NEJMoa044383. [DOI] [PubMed] [Google Scholar]
- 3.Degnim AC, Visscher DW, Berman HK, Frost MH, Sellers TA, Vierkant RA, et al. Stratification of breast cancer risk in women with atypia: a Mayo cohort study. J Clin Oncol. 2007;25:2671–2677. doi: 10.1200/JCO.2006.09.0217. [DOI] [PubMed] [Google Scholar]
- 4.Hoogerbrugge N, Bult P, de Widt-Levert LM, Beex LV, Kiemeney LA, Ligtenberg MJL, et al. High prevalence of premalignant lesions in prophylactically removed breasts from women at hereditary risk for breast cancer. Journal of Clinical Oncology. 2003;21:41–45. doi: 10.1200/JCO.2003.02.137. [DOI] [PubMed] [Google Scholar]
- 5.Kauff ND, Brogi E, Scheuer L, Pathak DR, Borgen PI, Hudis CA, et al. Epithelial lesions in prophylactic mastectomy specimens from women with BRCA mutations. Cancer. 2003;97:1601–1608. doi: 10.1002/cncr.11225. [DOI] [PubMed] [Google Scholar]
- 6.Fabian CJ, Kamel S, Zalles C, Kimler BF. Identification of a chemoprevention cohort from a population of women at high risk for breast cancer. J Cell Biochem Suppl. 1996;25:112–122. [PubMed] [Google Scholar]
- 7.Allred DC, Mohsin SK, Fuqua SA. Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001;8:47–61. doi: 10.1677/erc.0.0080047. [DOI] [PubMed] [Google Scholar]
- 8.Arpino G, Laucirica R, Elledge RM. Premalignant and in situ breast disease: biology and clinical implications. Ann Intern Med. 2005;143:446–457. doi: 10.7326/0003-4819-143-6-200509200-00009. [DOI] [PubMed] [Google Scholar]
- 9.Guray M, Sahin AA. Benign breast diseases: classification, diagnosis, and management. Oncologist. 2006;11:435–449. doi: 10.1634/theoncologist.11-5-435. [DOI] [PubMed] [Google Scholar]
- 10.Santisteban M, Reynolds C, Barr Fritcher EG, Frost MH, Vierkant RA, Anderson SS, et al. Ki67: a time-varying biomarker of risk of breast cancer in atypical hyperplasia. Breast Cancer Res Treat. 2010;121:431–437. doi: 10.1007/s10549-009-0534-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Visscher DW, Pankratz VS, Santisteban M, Reynolds C, Ristimaki A, Vierkant RA, et al. Association between cyclooxygenase-2 expression in atypical hyperplasia and risk of breast cancer. J Natl Cancer Inst. 2008;100:421–427. doi: 10.1093/jnci/djn036. [DOI] [PubMed] [Google Scholar]
- 12.Barr Fritcher EG, Degnim AC, Hartmann LC, Radisky DC, Boughey J, Anderson S, et al. Estrogen receptor expression in atypical hyperplasia: lack of association with breast cancer. Cancer Prev Res (Phila) 2011 doi: 10.1158/1940-6207.CAPR-10-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gauthier ML, Berman HK, Miller C, Kozakeiwicz K, Chew K, Moore D, et al. Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell. 2007;12:479–491. doi: 10.1016/j.ccr.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, et al. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst. 2010;102:627–637. doi: 10.1093/jnci/djq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim WY, Sharpless NE. VHL inactivation: a new road to senescence. Cancer Cell. 2008;13:295–297. doi: 10.1016/j.ccr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 16.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10:R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Garcia MA, Geyer FC, Lacroix-Triki M, Marchio C, Reis-Filho JS. Breast cancer precursors revisited: molecular features and progression pathways. Histopathology. 2010;57:171–192. doi: 10.1111/j.1365-2559.2010.03568.x. [DOI] [PubMed] [Google Scholar]
- 19.Santen RJ, Mansel R. Benign breast disorders. N Engl J Med. 2005;353:275–285. doi: 10.1056/NEJMra035692. [DOI] [PubMed] [Google Scholar]
- 20.Gooley TA, Leisenring W, Crowley J, Storer BE. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18:695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 21.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Boughey JC, Hartmann LC, Anderson SS, Degnim AC, Vierkant RA, Reynolds CA, et al. Evaluation of the Tyrer-Cuzick (International Breast Cancer Intervention Study) model for breast cancer risk prediction in women with atypical hyperplasia. J Clin Oncol. 2010;28:3591–3596. doi: 10.1200/JCO.2010.28.0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pankratz VS, Hartmann LC, Degnim AC, Vierkant RA, Ghosh K, Vachon CM, et al. Assessment of the accuracy of the Gail model in women with atypical hyperplasia. J Clin Oncol. 2008;26:5374–5379. doi: 10.1200/JCO.2007.14.8833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milanese TR, Hartmann LC, Sellers TA, Frost MH, Vierkant RA, Maloney SD, et al. Age-related lobular involution and risk of breast cancer. J Natl Cancer Inst. 2006;98:1600–1607. doi: 10.1093/jnci/djj439. [DOI] [PubMed] [Google Scholar]
- 25.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 27.Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, et al. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- 28.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederlander NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]