

Abstract
It has long been known that tumour necrosis factor (TNF)/TNFRSF1A signalling is involved in the pathophysiology of multiple sclerosis (MS). Different genetic and clinical findings over the last few years have generated renewed interest in this relationship. This paper provides an update on these recent findings. Genome-wide association studies have identified the R92Q mutation in the TNFRSF1A gene as a genetic risk factor for MS (odds ratio 1·6). This allele, which is also common in the general population and in other inflammatory conditions, therefore only implies a modest risk for MS and provides yet another piece of the puzzle that defines the multiple genetic risk factors for this disease. TNFRSF1A mutations have been associated with an autoinflammatory disease known as TNF receptor-associated periodic syndrome (TRAPS). Clinical observations have identified a group of MS patients carrying the R92Q mutation who have additional TRAPS symptoms. Hypothetically, the co-existence of MS and TRAPS or a co-morbidity relationship between the two could be mediated by this mutation. The TNFRSF1A R92Q mutation behaves as a genetic risk factor for MS and other inflammatory diseases, including TRAPS. Nevertheless, this mutation does not appear to be a severity marker of the disease, neither modifying the clinical progression of MS nor its therapeutic response. An alteration in TNF/TNFRS1A signalling may increase proinflammatory signals; the final clinical phenotype may possibly be determined by other genetic or environmental modifying factors that have not yet been identified.
Keywords: genetic factors, multiple sclerosis, TNF, TNFR1, TNFRSF1A R92Q mutation
A brief introduction to the genetics of MS
Multiple sclerosis (MS) is a chronic recurrent inflammatory and neurodegenerative disorder. The genetic determinants of susceptibility to this disease are highly complex. For many years, the major histocompatibility complex (MHC) has been the only region of the genome found to be associated consistently with the disease. In particular, the DR15 haplotype (DRB1*1501-DQB1*0602) is a constant finding in northern European populations [1,2], with an odds ratio of 2·75 [3].
In recent years, genome-wide association (GWA) studies have identified other loci outside the MHC showing solid associations with MS, albeit with reduced genetic effects. These loci act as genetic markers, regardless of whether or not they have functional effects themselves [4] and include, among others, genes encoding cytokine receptors such as the interleukin (IL)-2 receptor (IL-2RA) and the IL-7 receptor (IL-7R) [5]. In a recent meta-analysis of three large MS cohorts, the combined analysis led to the identification of three new loci associated with MS genetic risk, TNFRSF1A (tumour necrosis factor receptor superfamily 1A), IRF8 (interferon regulatory factor 8) and CD6[3].
The association between MS and the TNFRSF1A gene (chromosome 12p13) encoding the type 1 tumour necrosis factor (TNF)-α receptor (TNFR1) is particularly intriguing, as the TNF-α signalling pathway has major implications in the pathophysiology of MS. It could be speculated that allelic variant-mediated alterations in TNFRSF1A function might have an impact on the TNF-α signalling pathways, and increase proinflammatory signals.
Two polymorphisms have been detected in this gene that are associated with MS: rs1899693, located in intron 6 of the gene, which is highly frequent but of a little effect, and rs4149584, positioned in exon 4 of the gene. The latter polymorphism is associated with an arginine-to-glutamine substitution at position 92 (R92Q), with a lower frequency than the first one, but with greater genetic effect [3]. Although more in-depth research is needed to characterize the effects of this polymorphism and others in the TNFRSF1A gene in MS, this meta-analysis provides the first definitive evidence that TNFRSF1A is an MS susceptibility locus [3].
Objective and method
The aim of this paper is to address the issue related to the potential involvement of TNF/TNFRSF1A signalling mechanisms in the pathophysiology of MS, its relationship to the TNF receptor-associated periodic syndrome (TRAPS) and the possibility of a genetic link between both disorders through the R92Q mutation.
To this purpose we will review the biology of this cytokine, the data that provide evidence of the implication of TNF–TNFR1 signalling in MS, and will make a brief description of the diseases associated with the R92Q mutation, mainly TRAPS. We will also comment on the scarce studies to date that address the clinical relationship between MS and TRAPS, and some questions that remain unsolved. All relevant original articles and reviews in English were taken into account to complete this paper.
Biology and functions of TNF-α
TNF-α is a proinflammatory cytokine produced by immune cells, mainly macrophages and lymphocytes, in response to a great variety of stimuli. It exerts pleiotropic effects on immunity, inflammation, cell proliferation, differentiation and apoptosis, and also acts as an endogenous pyrogen [6–9]. Two forms of TNF-α are biologically active: transmembrane TNF (tmTNF) and soluble TNF (sTNF) [9].
TNF interacts with two receptors (TNFR), TNFR1 and TNFR2, in a wide range of cell types. TNFRs are transmembrane glycoproteins characterized by four cysteine-rich extracellular domains (CRDs), which can be cleaved proteolytically to generate soluble receptor fragments that may function as natural TNF-α antagonists [10,11].
TNFR1 is expressed constitutively in all cell types except erythrocytes, while TNFR2 is inducible and expressed preferentially by endothelial and immune cells. sTNF binds preferentially to TNFR1 versus TNFR2 [12].
TNFRs lack their own enzyme activity and their signalling depends on the association of adaptor proteins that bind to the cytoplasmic domains of the receptors. The cytoplasmic tail of TNFR1 contains a death domain that couples this receptor to one of two distinct signalling pathways. One pathway leads to activation of nuclear factor kappa-B (NK-κB) and subsequently transcription of proinflammatory and anti-apoptotic genes, whereas a different pathway leads to apoptosis via activation of caspases 8 and 10. Therefore, TNFR1 will activate proinflammatory or apoptotic signals depending mainly on the cell type and the state of the cell [13] (Fig. 1).
Fig. 1.
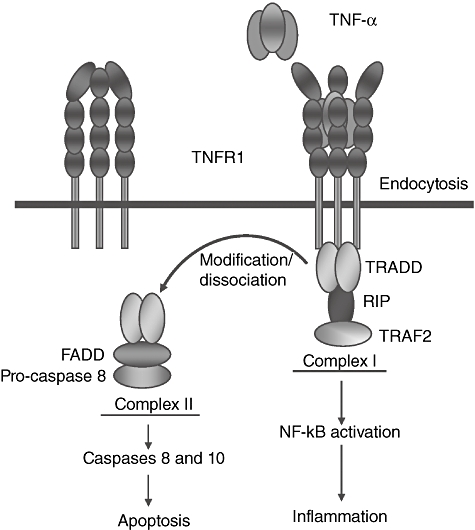
Tumour necrosis factor receptor 1 (TNFR1) and its signalling pathways. TNFR1 has four extracellular cysteine-rich domains (CRDs). Signalling via TNFR1 occurs by TNF-induced recruitment of monomeric membrane-bound TNFR1 molecules to form a homotrimer configuration which, when bound to ligand, causes a three-dimensional conformational change in the extracellular domain that is then transduced intracellularly to activate signalling pathways. The cytoplasmic tail of TNFR1 contains a TNF receptor type 1-associated death domain (TRADD) that couples this receptor to one of two distinct signalling pathways. The default pathway activates nuclear factor kappa-B (NF-κB) and other non-apoptotic pathways (not shown). This is mediated by the formation of complex I, comprising TNFR1 and NF-kB activating signalling components such as receptor interacting protein (RIP) and TNF receptor-associated factor 2 (TRAF2). Formation of complex I is transient and TNFR1 can be internalized after TNF binding leading to dissociation of the TRADD/TRAF2/RIP complex and association of FADD, recruitment of pro-caspase 8 and formation of complex II, which then recruits the apoptotic machinery. If NF-kB activation triggered by complex I is successful, cellular FLIPLlevels (see below) are sufficiently elevated to block apoptosis and cells survive. The ovals in the figure represent different activating signalling components. FADD: FAS-associated death domain. FLIPL: FLICE-like inhibitory protein long, the long form of caspase 8 inhibitor.
TNFR2-mediated signalling produces far fewer biological effects than TNFR1 signalling. Such signals lead to the activation of NF-kB, promoting tissue survival, although in some cases the receptor could also collaborate with its counterpart, contributing to cell death [14]. Although the activation of this receptor is also important in mediating TNF-α functions in different pathological conditions, such as MS, a description of TNFR2 signalling and its implications in MS falls outside the scope of the present review.
TNF-α and TNFRSF1A in MS
TNF-α signalling is implicated in the immunopathogenesis of MS [15]. Autopsy studies in MS patients have revealed high levels of TNF-α in active MS lesion sites [16]. The TNF-α levels in cerebrospinal fluid (CSF) are increased in MS patients versus controls, and these levels in turn are correlated with the severity of the lesions, the severity of the disease, the rate of neurological deterioration and a poorer prognosis after 24 months of follow-up [17]. In peripheral blood, the mononuclear cells of patients with MS secrete large amounts of TNF-α prior to the exacerbation of symptoms, compared with the cells of the same patients during periods of disease remission [18].
TNF-α involvement in the pathogenesis of MS has also been suggested in the experimental allergic encephalitis (EAE) animal model [19], as well as in the cuprizone model of toxic demyelination [20]. Most biological responses attributed to TNF-α are due to its action on TNFR1, because signalling through this receptor plays an important role in the onset of inflammation and the extension of demyelination in the acute phases of the disease [21]. TNF has also shown immunosuppressive properties during the later phases of the disease and, for instance, in the absence of TNF, autoreactive T cells fail to revert and expansion of activated T cells appears to be abnormally prolonged [22,23]. These immunosuppressive properties were shown to be independent of TNFR1 signalling and probably mediated through TNFR2 [22–24]. Therefore, it is believed that divergent roles exist between the two receptors in CNS autoimmunity, with TNFR1 playing roles in CNS inflammation and demyelination and TNFR2 apparently acting to limit pathology, possibly by elimination of autoreactive CD4+ T cells and macrophages and/or by playing roles in remyelination.
TRAPS: a genetically defined heterogeneous entity
TRAPS is an autosomal-dominant (AD) autoinflammatory disorder caused by heterozygous mutations in the TNFRSF1A gene [25,26]. It belongs to the group of ‘hereditary recurrent fever syndromes’[25]. It is characterized by recurrent, unprovoked, self-limiting attacks of fever, abdominal pain, myalgia, rash, arthralgia/arthritis, rhinopharingolaryngeal involvement and conjunctivitis/periorbital oedema. The severest complication of TRAPS is renal amyloidosis. TRAPS episodes can last from a few days to weeks (mean 21 days), and on average they recur every 5–6 weeks. Clinical symptoms can vary greatly from one individual to another and even from one episode to another. Mean age of onset is 10 years, but ranges from 22 weeks of life to age 53 years [25]. Table 1 shows the diagnostic indicators of TRAPS as proposed by Hull et al. [25]. Many other symptoms may be part of the clinical phenotype of TRAPS, although less often (vasculitis, sacroileitis, splenomegaly, neurological manifestations, etc.) [26–28].
Table 1.
Diagnostic indicators of traps (modified from Hull KM et al.) [25].
| 1. Recurrent episodes of inflammatory symptoms spanning a period longer than 6 months (several symptoms will generally occur simultaneously) |
| 1.1 Fever |
| 1.2 Gastrointestinal involvement (diarrhoea, abdominal pain, etc.) |
| 1.3 Myalgia (migratory) |
| 1.4 Rash (erythematous macular rash occurs with myalgia) |
| 1.5 Conjunctivitis/periorbital oedema |
| 1.6 Chest pain |
| 1.7 Arthralgia or monoarticular synovitis |
| 1.8 Lymphadenopathy |
| 2. Episodes last more than 5 days on average (variable) |
| 3. Responsive to glucocorticosteroids but not colchicine |
| 4. Affected family members (may not always be present) |
| 5. Any ethnicity may be affected |
During TRAPS exacerbations, acute-phase reactants are elevated [25,26]. Autoantibodies are either not detected or are found at very low levels [25,26].
Inflammation responds to glucocorticoids but not to colchicine. The response to anti-TNF-α drugs is variable, and there are reports of response to anakinra (a recombinant IL-1 receptor antagonist) [25,29].
Genetics
To date, more than 60 mutations have been reported in association with TRAPS in coding and non-coding regions of the TNFRSF1A gene (for details, refer to the INFEVERS database http://fmf.igh.cnrs.fr/ISSAID/infevers) [30]. The majority of these mutations affect the extracellular domain of the receptor (Fig. 2), leading to modifications in ligand binding (CRD2) or alterations in ligand-independent receptor assembly and activation (CRD1) [11,31–33].
Fig. 2.
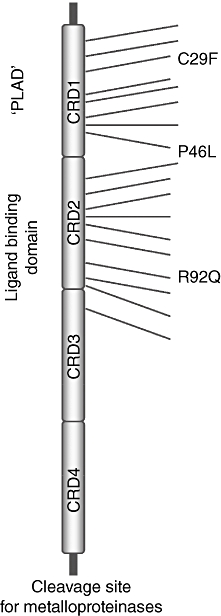
Tumour necrosis factor receptor 1 (TNFR1) mutations in TNF receptor-associated periodic syndrome (TRAPS) [32]. The figure illustrates the ectodomain of the TNFR1 with the location of the TRAPS mutations, clustered mainly in cysteine-rich domains (CRD1 and CRD2). There are more than 60 known mutations. Only a few are shown in this diagram. R92Q is located at CRD2. PLAD: preligand assembly domain.
Two other rare variants in the gene coding sequence have been described that alter single amino acids and do not affect cysteine residues, P46L and R92Q. These variants have also been identified in asymptomatic relatives of an index case, in patients without family history (sporadic forms), in other inflammatory conditions and also infrequently in healthy populations (1–5% in the general population; P46L substitution is found in up to 10% of the West African population) [34–36]. The increased frequencies of these two substitutions in patients with TRAPS, as well as functional studies of TNFRSF1A in vitro, suggest that these are low-penetrance mutations rather than benign polymorphisms [34]. It is therefore possible that they could synergize with other genetic-immune defects to cause a milder form of TRAPS with low penetrance [37].
Nevertheless, not all familial or sporadic cases of patients with clinical symptoms compatible with TRAPS have TNFRSF1A gene mutations, so there is still much to be learned about the genetics of these diseases.
There are no phenotype–genotype correlations [25], with two exceptions: (i) mutations in cysteine residues have a higher clinical penetrance than non-cysteine residues substitutions (93% versus 82%) and they increase the probability of developing amyloidosis (24% versus 2%) [25,34,38,39]; and (ii) patients with R92Q and P46L mutations have more heterogeneous and benign manifestations of the condition.
R92Q mutation and TRAPS
This substitution is located in the CRD2 (Fig. 2). It is considered to be a low-penetrance mutation [34] and the most frequent genetic defect observed in TRAPS patients. Onset occurs at a later age and presentation in adulthood is fairly common. Episodes are shorter-lasting, the rash is less typical, abdominal pain is less common, and in general these individuals require less steroid or immunosuppressive therapy. The majority of patients with this mutation are sporadic cases [34,35,40]. This mutation was observed in 3·3% of 274 independent chromosomes in individuals with clinical symptoms suggestive of TRAPS (1% in control chromosomes in an Irish and North American population of European ancestry) [34].
From genetics to pathogenesis
Altered receptor cleavage (‘shedding hypothesis’) was considered initially to be a key process in this disease [31], allowing repeated stimulation of membrane receptors and a diminished pool of potentially inhibitory soluble receptors. However, defective shedding has not been demonstrated in many TNFRSF1A mutations associated with TRAPS, including the R92Q mutation [34].
The mutations affecting cysteine residues (structural mutations) [41] cause changes in the three-dimensional structure of the TNFR1 ectodomain, leading to accumulation of the mutated receptor in the endoplasmic reticulum [42]. The mutant receptors self-interact and trigger an inflammatory response [38,41,43]. They are also unable to activate the TNF-α-induced NF-kB transcription pathway, which leads to a reduction in apoptosis [44] and prolonged survival of the activated inflammatory cells, contributing to the inflammatory phenotype of the disease [45,46].
Conversely, the so-called non-structural variants, with fairly conservative amino acid substitutions – including the R92Q mutation – have shown normal behaviour in terms of receptor trafficking and TNF-α binding ability on the cell surface [38,43]. It has been observed that neutrophils of patients carrying this mutation are resistant to TNF-α-induced apoptosis [44]; defective shedding is not found in all cases, and only subtle abnormalities have been described in surface receptor expression in the endothelial cell line [26,34]. Much work is under way to try to understand the pathophysiology of the disease in patients with this substitution. It has been suggested that the R92Q mutation does not cause the disease directly, but acts in synergy with other genetic or environmental factors, triggering the autoinflammatory phenotype [44].
R92Q mutation and diseases other than TRAPS
The R92Q mutation has also been associated with clinical phenotypes other than TRAPS, such as early arthritis [34], Behçet's disease with extracranial deep vein thrombosis [47], atherosclerosis [48] or recurrent pericarditis [49]. These findings support the known fact that different inflammatory diseases share genetic susceptibility loci.
Thus, unlike the causal effect of high-penetrance mutations that behave like specific monogenic disorders, there are other milder, lower-penetrance mutations, such as the R92Q mutation, which may have a broader influence on susceptibility to inflammation or may contribute to amplifying inflammatory responses in multi-factorial diseases [25]. Considering the hypothesis that complex genetic diseases arise from a combination of mutations or common low-penetrance polymorphisms [50], R92Q mutation carriers would be more likely to develop a wide range of inflammatory and autoimmune diseases, and the final phenotypic expression may depend possibly on other genetic or environmental modifying factors, as yet unidentified [51].
TRAPS and neurological disease
There are reports of patients with TRAPS who have CNS involvement (CNS–TRAPS) in the form of headache, meningitis, encephalitis, psychosis, memory impairment, behavioural changes and inflammatory symptoms (optic neuritis/papillitis, posterior fossa involvement, etc.) [31,40,52–57]. In three of these cases abnormalities were found in neuroimaging studies with brain magnetic resonance imaging (MRI) [54–57], and in two of them they were also found through brain biopsy [54,55,57]. A 38-year-old male with clinical symptoms of TRAPS and the TNFRSF1A C55R mutation presented with posterior fossa symptoms and had a single gadolinium-enhancing lesion in the cerebellum that contained inflammatory cells, but showed no demyelination on histopathological examination. Furthermore, oligoclonal bands were not found in CSF. The patient improved with an anti-TNF agent (etanercept) [54,55]. In two other patients, one with the T50K mutation [56] and the other with the R92Q substitution [57], disease progression, brain MRI and CSF findings were consistent with MS. A brain biopsy performed in one case showed both inflammation and demyelination [57].
It is not known how many TRAPS patients have symptoms suggestive of CNS inflammatory disease. There is some debate as to whether the CNS disease in these cases is, in fact, CNS–TRAPS (neurological inflammatory symptoms that form part of the clinical phenotype of TRAPS) or whether TRAPS and MS co-exist in some patients, either by coincidence or because there is some genetic or pathophysiological connection between both diseases [57–59]. From a pathogenic viewpoint, increased TNF signalling through the TNFR1 receptor – which is typical in TRAPS – may also be involved in the CNS inflammatory symptoms in these patients.
Inflammatory CNS disease has also been reported in other autoinflammatory syndromes, such as familial Mediterranean fever (FMF). In a recent study conducted in an Israeli population, the frequency of MS in FMF patients was reported to be two times higher than expected in the general population [60]. However, the opposite situation has also been reported and Akman-Demir et al., for instance, observed a fourfold higher prevalence of FMF among patients with definite MS compared with the general Turkish population [61]. Distinguishing between CNS–TRAPS and MS plus TRAPS is not only important from a nosological standpoint, but also because the therapeutic approach differs and is actually contradictory. The clinical presentation of MS and TRAPS may be similar, both disorders manifesting with variable and episodic symptoms. MS is usually defined according to the McDonald criteria (2010 revisions) [62], which require dissemination in time and space clinically and on MRI, and exclusion of other possible causes of CNS disease; typical clinical episodes involve the optic nerve, brainstem and/or spinal cord, and it is often difficult to make the diagnosis in the first event, despite the use of paraclinical tests. Regarding CNS–TRAPS, the diagnosis is not so well established due to the small number of patients published to date, their short-term follow-up and the inherent difficulty in obtaining brain biopsies/autopsies. Therefore, a careful investigation, including disease course, laboratory and CSF examinations, evoked potentials and cerebral and spinal MRI, are the main tools to help us distinguish between these two entities. In fact, many R92Q mutation carriers with different neuropsychiatric manifestations did not fulfil the diagnostic criteria for MS.
In the case where CNS involvement is part of TRAPS, anti-TNF agents may be a therapeutic option [54], but if an individual has TRAPS and MS, these drugs would be contraindicated for treating TRAPS symptoms because they can worsen MS clinical progression [63,64].
TRAPS, R92Q and MS
In two cohorts of unselected samples of patients with MS or clinically isolated syndromes, the R92Q mutation was observed in 4·66% [65] and 5·5% [66] of patients versus 2·95% and 1·5%, respectively, in Caucasian control populations. Differences were not statistically significant, although a trend towards association was observed in both studies [65,66].
The carrier frequency of the R92Q mutation in a selected group of MS patients with additional TRAPS-like symptoms was 13%, a frequency higher than those reported for other groups of patients [67] (the incidence of the R92Q mutation was found to be approximately 5% in patients with early arthritis [34] and also in patients with a clinically suspected inherited autoinflammatory syndrome [26]). This high frequency in a selected group of MS patients could be explained partially because the selection of patients for the genetic study was based on family history and on a clinical picture, which only required the presence of recurrent attacks with at least two autoinflammatory symptoms of those listed in Table 1 (sections 1·1–1·8), fever not being essential.
The majority of patients with MS, TRAPS and the R92Q mutation developed oligosymptomatic and late forms of TRAPS, with combinations of the following symptoms: arthralgia/arthritis, myalgias, urticarial rashes, pharyngitis and fatigue; however, fever and acute-phase reactants were not always present [66,67]. Therefore, if we use less rigid diagnostic criteria, it is possible that the prevalence of TRAPS is higher than considered hitherto. Nevertheless, and although the R92Q mutation is the most common mutation associated with TRAPS, other mutations in the TNFRSF1A gene or mutations in different genes may be responsible for TRAPS or TRAPS-like symptoms in any population group. In almost all these patients, the start of the systemic symptoms of inflammation preceded the symptoms of MS, thus contributing to the diagnostic difficulty of a first inflammatory process of the CNS in a patient with TRAPS (CNS–TRAPS versus TRAPS plus MS) and to the choice of a suitable treatment which does not cause adverse reactions.
In a German cohort [67], the patients with MS, clinical TRAPS and the R92Q mutation presented adverse reactions to the use of disease-modifying drugs (interferons, glatiramer acetate, natalizumab or mitoxantrone) with great frequency; in addition, the reactions were more severe than normal, in many cases forcing the withdrawal of treatment. In contrast, this was not seen in an Argentine cohort [66].
The clinical course of MS in these patients with TRAPS and the R92Q mutation was otherwise considered normal [66,67], except for the presence of intense fatigue [67] and for the greater frequency of clinical brainstem symptoms (frequently with facial palsy) as the starting point of the MS (15% compared to 5% in other small case series of MS patients [66,68]). These data, together with the results of a recent GWA study [69], have allowed us to reach the conclusion that the R92Q mutation does not have an influence on the general clinical characteristics of the disease [66], nor on its severity [measured by the Multiple Sclerosis Severity Scale (MSSS)][69] or on the therapeutic response [66].
Conversely, the penetrance of TRAPS in patients with MS and the R92Q mutation is similar to that found for patients without MS in the general population (approximately 82%) [33,35,36]. In the Argentine cohort, this penetrance was 80% [68]. In the German cohort it was somewhat higher, and 88% of family members of patients with MS and clinical TRAPS who were carriers of the mutation had clinical symptoms of TRAPS [67].
It is not known how the R92Q mutation can cause disease and how it can fit into the pathogenesis of MS. Further studies are required to define the functionally relevant variants in the identified region and to investigate their effects at molecular and cellular level.
Conclusions
MS has a strong genetic component. Most of its risk variants are located at or near genes with central immunological functions also associated with other autoimmune diseases [3]. Many of the immunological effects of TNF in MS are thought to be delivered through the TNFR1. Thus, there is a biological plausibility for this association.
Variants within previously confirmed MS susceptibility loci do not appear to influence severity as measured by the MSSS [69], and this is also the case for the R92Q mutation; nor have clinical studies made it possible to demonstrate that this mutation modifies the clinical course or therapeutic response in MS [67].
The R92Q substitution is the most frequent mutation identified in patients suffering from TRAPS. Some clinical observations have identified MS patients carrying the R92Q mutation who have additional late onset and milder forms of TRAPS [65–67]. Thus, this disorder should be considered in MS patients who report symptoms such as arthralgias/arthritis, myalgias, urticarial rash and severe fatigue [65]. Nevertheless, the clinical course and severity of the MS were no different from those of patients without clinical TRAPS.
The TRAPS phenotype is associated with multiple mutations in TNFRSF1A gene and different clinical phenotypes have been associated with the presence of the R92Q mutation. Given the rare nature, autosomal-dominant mode of inheritance and the varied clinical presentation of TRAPS, it is difficult to conclude that TRAPS itself is involved directly in the great majority of MS cases. The co-existence of TRAPS and MS is possible, but is likely to be a rare occurrence.
As things stand today, we do not know the clinical and/or therapeutic implications that the presence of this mutation may have on patients with MS. Further studies are warranted to clarify its importance and its usefulness as a marker of some aspect of this complex disease.
Disclosure
The authors have no disclosures in relation to the article.
References
- 1.Olerup O, Hillert J. HLA class II-associated genetic susceptibility in multiple sclerosis: a critical evaluation. Tissue Antigens. 1991;38:1–15. doi: 10.1111/j.1399-0039.1991.tb02029.x. [DOI] [PubMed] [Google Scholar]
- 2.Barcellos LF, Sawcer S, Ramsay PP, et al. Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet. 2006;15:2813–24. doi: 10.1093/hmg/ddl223. [DOI] [PubMed] [Google Scholar]
- 3.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–82. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins FS, Guyer MS, Charkravarti A. Variations on a theme: cataloging human DNA sequence variation. Science. 1997;278:1580–1. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- 5.Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet. 2008;9:516–26. doi: 10.1038/nrg2395. [DOI] [PubMed] [Google Scholar]
- 6.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu Rev Immunol. 1999;17:331–67. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 7.Kluger MJ, Kozak W, Leon LR, Soszynski C, Conn CA. Cytokines and fever. Neuroimmunomodulation. 1995;2:216–23. doi: 10.1159/000097199. [DOI] [PubMed] [Google Scholar]
- 8.McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:1–13. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beutler B, Cerami A. The biology of cachectin/TNF – a primary mediator of the host response. Annu Rev Immunol. 1989;7:625–55. doi: 10.1146/annurev.iy.07.040189.003205. [DOI] [PubMed] [Google Scholar]
- 10.Naismith JH, Devine TQ, Kohno T, Sprang SR. Structures of the extracellular domain of the type I tumor necrosis factor receptor. Structure. 1996;4:1251–62. doi: 10.1016/s0969-2126(96)00134-7. [DOI] [PubMed] [Google Scholar]
- 11.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Leonardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–4. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 12.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA. 1998;95:570–5. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tracey D, Klareskog L, Sasso EJ, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–79. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–32. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–71. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- 16.Hofman FM, Hinton DR, Johnson K, Merrill JE. Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med. 1989;170:607–12. doi: 10.1084/jem.170.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharief MK, Hentges R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med. 1991;325:467–72. doi: 10.1056/NEJM199108153250704. [DOI] [PubMed] [Google Scholar]
- 18.Rieckmann P, Albrecht M, Kitze B, et al. Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann Neurol. 1995;37:82–8. doi: 10.1002/ana.410370115. [DOI] [PubMed] [Google Scholar]
- 19.Kassiotis G, Pasparakis M, Kollias G, Probert L. TNF accelerates the onset but does not alter the incidence and severity of myelin basic protein-induced experimental autoimmune encephalomyelitis. Eur J Immunol. 1999;29:774–80. doi: 10.1002/(SICI)1521-4141(199903)29:03<774::AID-IMMU774>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–16. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Probert L, Eugster HP, Akassoglou K, et al. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain. 2000;123:2005–19. doi: 10.1093/brain/123.10.2005. [DOI] [PubMed] [Google Scholar]
- 22.Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. 2001;193:427–34. doi: 10.1084/jem.193.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barret TA, Miller SD. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG (35-55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000;205:24–33. doi: 10.1006/cimm.2000.1706. [DOI] [PubMed] [Google Scholar]
- 24.Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alfa promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. 2001;4:1116–22. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 25.Hull KM, Drewe E, Aksentijevic I, et al. The TNF receptor-associated periodic syndrome (TRAPS). Emerging concepts of an autoinflammatory disorder. Medicine. 2002;81:349–68. doi: 10.1097/00005792-200209000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 2003;48:2632–44. doi: 10.1002/art.11215. [DOI] [PubMed] [Google Scholar]
- 27.Gertz MA, Petitt RM, Perrault J, Kyle RA. Autosomal dominant familial Mediterranean fever-like syndrome with amyloidosis. Mayo Clin Proc. 1987;62:1095–100. doi: 10.1016/s0025-6196(12)62502-6. [DOI] [PubMed] [Google Scholar]
- 28.McDermott EM, Smillie DM, Powell RJ. Clinical spectrum of familial Hibernian fever: a 14-year follow-up study of the index case and extended family. Mayo Clin Proc. 1997;72:806–17. doi: 10.4065/72.9.806. [DOI] [PubMed] [Google Scholar]
- 29.Church LD, Churchman SM, Hawkins PN, McDermott MF. Hereditary auto-inflammatory disorders and biologics. Springer Semin Immunopathol. 2006;27:494–508. doi: 10.1007/s00281-006-0015-6. [DOI] [PubMed] [Google Scholar]
- 30.Touitou I, Lesage S, McDermott M, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24:194–8. doi: 10.1002/humu.20080. [DOI] [PubMed] [Google Scholar]
- 31.McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndrome. Cell. 1999;97:133–44. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 32.Kimberley FC, Lobito AA, Siegel RM, Screaton GR. Falling into TRAPS-receptor misfolding in the TNF receptor 1-associated periodic fever syndrome. Arthritis Res Ther. 2007;9:1–8. doi: 10.1186/ar2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastne DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol. 2000;12:479–86. doi: 10.1016/s0952-7915(00)00124-2. [DOI] [PubMed] [Google Scholar]
- 34.Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet. 2001;69:301–14. doi: 10.1086/321976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumor necrosis factor superfamily 1A gene. Ann Rheum Dis. 2006;65:1158–62. doi: 10.1136/ard.2005.048611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tchernitchko D, Chiminqgi M, Galactéros F, et al. Unexpected high frequency of P46L TNFRSF1A allele in sub-Saharan West African populations. Eur J Hum Genet. 2005;13:513–5. doi: 10.1038/sj.ejhg.5201344. [DOI] [PubMed] [Google Scholar]
- 37.Gattorno M, Sormani MP, D'Osualdo A, et al. A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 2008;58:1823–32. doi: 10.1002/art.23474. [DOI] [PubMed] [Google Scholar]
- 38.Lobito AA, Kimberley FC, Muppidi JR, et al. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in the TNFR1-associated periodic fever syndrome (TRAPS) Blood. 2006;108:1320–7. doi: 10.1182/blood-2005-11-006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–8. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dodé C, André M, Bienvenu T, et al. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2002;46:2181–8. doi: 10.1002/art.10429. [DOI] [PubMed] [Google Scholar]
- 41.Simon A, Park H, Maddipati R, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci USA. 2010;25:9801–6. doi: 10.1073/pnas.0914118107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rebelo SL, Bainbridge SE, Amel-Kashipaz MR, et al. Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 2006;54:2674–87. doi: 10.1002/art.21964. [DOI] [PubMed] [Google Scholar]
- 43.Todd I, Radford PM, Daffa N, Bainbridge SE, Powell RJ, Tighe PJ. Mutant tumor necrosis factor receptor associated with tumor necrosis factor receptor-associated periodic syndrome is altered antigenically and is retained within patients' leukocytes. Arthritis Rheum. 2007;56:2765–73. doi: 10.1002/art.22740. [DOI] [PubMed] [Google Scholar]
- 44.D'Osualdo A, Ferlito F, Prigione I, et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumour necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 2006;54:998–1008. doi: 10.1002/art.21657. [DOI] [PubMed] [Google Scholar]
- 45.Sieber F, Fielding CA, Williams BD, Brennan P. Mutation of the extracellular domain of tumour necrosis factor receptor 1 causes reduced NF-kappaB activation due to decreased surface expression. FEBS Lett. 2005;579:5193–8. doi: 10.1016/j.febslet.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 46.Siebert S, Amos N, Fielding CA, et al. Reduced tumor necrosis factor signalling in primary human fibroblasts containing a tumor necrosis factor receptor superfamily 1A mutant. Arthritis Rheum. 2005;52:1287–92. doi: 10.1002/art.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amoura A, Dodé C, Hue S, et al. Association of the R92Q TNFRSF1A mutation and extracranial deep vein thrombosis in patients with Behcet's disease. Arthritis Rheum. 2005;52:608–11. doi: 10.1002/art.20873. [DOI] [PubMed] [Google Scholar]
- 48.Poirier O, Nicaud V, Gariepy J, et al. Polymorphism R92Q of the tumor necrosis factor receptor 1 gene is associated with myocardial infartion and carotid intima-media thickness – the ECTIM, AXA, EVA and GENIC studies. Eur J Hum Genet. 2004;12:213–19. doi: 10.1038/sj.ejhg.5201143. [DOI] [PubMed] [Google Scholar]
- 49.Cantarini L, Lucherini OM, Cimaz R, et al. Idiopathic recurrent pericarditis refractory to colchicine treatment can reveal tumor necrosis factor receptor-associated periodic syndrome. Int J Immunopathol Pharmacol. 2009;22:1051–8. doi: 10.1177/039463200902200421. [DOI] [PubMed] [Google Scholar]
- 50.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–17. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 51.Dipple KM, McCabe ER. Phenotypes of patients with ‘simple’ Mendelian disorders are complex traits: thresholds, modifiers and systems dynamics. Am J Hum Genet. 2000;66:1729–35. doi: 10.1086/302938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosen-Wolff A, Kreth HW, Hofmann S, et al. Periodic fever (TRAPS) caused by mutations in the TNF-alfa receptor 1 (TNFRSF1A) gene of three German patients. Eur J Haematol. 2001;67:105–9. [PubMed] [Google Scholar]
- 53.Hurst M, Hull K, Nicholls D, Ameratunga R. Hereditary periodic fever syndrome sans fever or distinct periodicity presenting with psychosis. J Clin Rheumatol. 2005;11:329–30. doi: 10.1097/01.rhu.0000195106.34348.59. [DOI] [PubMed] [Google Scholar]
- 54.Rudofsky G, Hoffmann F, Muller K, et al. A nephrotic patient with tumor necrosis factor receptor-associated syndrome, IgA nephropathy and CNS involvement. Nephrol Dial Transplant. 2006;21:1109–12. doi: 10.1093/ndt/gfk098. [DOI] [PubMed] [Google Scholar]
- 55.Wildemann B, Rudofsky G, Jr, Kress B, Jarius S, Konig F, Schwenger V. The tumor necrosis factor-associated periodic syndrome, the brain, and tumor necrosis factor-alfa antagonists. Neurology. 2007;68:1742–4. doi: 10.1212/01.wnl.0000260226.21010.2b. [DOI] [PubMed] [Google Scholar]
- 56.Minden K, Aganna E, McDermott MF, Zing A. Tumor necrosis factor receptor associated periodic syndrome (TRAPS) with central nervous system involvement. Ann Rheum Dis. 2004;63:1356–7. doi: 10.1136/ard.2003.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmann LA, Lohse P, König FB, Feneberg W, Hohlfeld R, Kümpfel T. TNFRSF1A R92Q mutation in association with a multiple sclerosis-like demyelinating syndrome. Neurology. 2008;70:1155–6. doi: 10.1212/01.wnl.0000296279.98236.8a. [DOI] [PubMed] [Google Scholar]
- 58.Herndon RM. Multiple sclerosis mimics. Adv Neurol. 2006;98:161–6. [PubMed] [Google Scholar]
- 59.Sriram S. TRAPS and MS: two diseases of an MS mimic? Neurology. 2008;70:1077–8. doi: 10.1212/01.wnl.0000307670.62365.08. [DOI] [PubMed] [Google Scholar]
- 60.Yahalom G, Kivity S, Lidar M, et al. Familial Mediterranean fever (FMF) and multiple sclerosis: an association study in one of the world's largest FMF cohorts. Eur J Neurol. 2011;18:1146–50. doi: 10.1111/j.1468-1331.2011.03356.x. [DOI] [PubMed] [Google Scholar]
- 61.Akman-Demir G, Gul A, Gurol E, et al. Inflammatory/demyelinating central nervous system involvement in familial Mediterranean fever (FMF): coincidence or association. J Neurol. 2006;253:928–34. doi: 10.1007/s00415-006-0137-8. [DOI] [PubMed] [Google Scholar]
- 62.Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69:292–302. doi: 10.1002/ana.22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Oosten BW, Barkhof F, Truyen L, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47:1531–4. doi: 10.1212/wnl.47.6.1531. [DOI] [PubMed] [Google Scholar]
- 64.Lenercept Multiple Sclerosis Study Group; University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology. 1999;53:457–65. [PubMed] [Google Scholar]
- 65.Kümpfel T, Hoffmann LA, Rübsamen H, et al. Late-onset tumor necrosis factor receptor-associated periodic syndrome in multiple sclerosis patients carrying the TNFRSF1A R92Q mutation. Arthritis Rheum. 2007;56:2774–83. doi: 10.1002/art.22795. [DOI] [PubMed] [Google Scholar]
- 66.Kauffman MA, Gonzalez-Morón D, Garcea O, Villa AM. TNFSFR1A R92Q mutation, autoinflammatory symptoms and multiple sclerosis in a cohort from Argentina. Mol Biol Rep. 2011 doi: 10.1007/s11033-011-0716-3. doi: 10.1007/s11033-011-0716-3 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 67.Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation. Clinical characteristics of 21 cases. Neurology. 2008;71:1812–20. doi: 10.1212/01.wnl.0000335930.18776.47. [DOI] [PubMed] [Google Scholar]
- 68.Fukazawa T, Moriwaka F, Hamada K, Hamada T, Tashiro K. Facial palsy in multiple sclerosis. J Neurol. 1997;244:631–3. doi: 10.1007/s004150050158. [DOI] [PubMed] [Google Scholar]
- 69.International Multiple Sclerosis Genetics Consortium. Briggs FB, Shao X, Goldstein BA, Oksenberg JR, Barcillos LF, De Jager PL. Genome-wide association study of severity in multiple sclerosis. Genes Immun. 2011 doi: 10.1038/gene.2011.34. 9 June 2011. doi: 10.1038/gene.2011.34 (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]