

Abstract
Live attenuated oral enterotoxigenic Escherichia coli (ETEC) vaccines have been demonstrated to be safe and immunogenic in human volunteers and to provide a viable approach to provide protection against this important pathogen. This report describes the construction of new ETEC vaccine candidate strains from recent clinical isolates and their characterization. All known genes for ETEC toxins were removed, and attenuating deletion mutations were made in the aroC, ompC, and ompF chromosomal genes. An isolate expressing coli surface antigen 2 (CS2), CS3, heat-labile toxin (LT), heat-stable toxin (ST), and enteroaggregative Escherichia coli heat-stable toxin 1 (EAST1) was attenuated to generate ACAM2007. The subsequent insertion of the operon encoding CS1 created ACAM2017, and this was further modified by the addition of an expression cassette containing the eltB gene, encoding a pentamer of B subunits of LT (LTB), to generate ACAM2027. Another isolate expressing CS5, CS6, LT, ST, and EAST1 was attenuated to generate ACAM2006, from which a lysogenic prophage was deleted to create ACAM2012 and an LTB gene was introduced to form ACAM2022. Finally, a previously described vaccine strain, ACAM2010, had the eltB gene incorporated to generate ACAM2025. All recombinant genes were incorporated into the chromosomal sites of the attenuating mutations to ensure maximal genetic stability. The expression of the recombinant antigens and the changes in plasmids accompanying the deletion of toxin genes are described. Strains ACAM2025, ACAM2022, and ACAM2027 have been combined to create the ETEC vaccine formulation ACE527, which has recently successfully completed a randomized, double-blind, placebo-controlled phase I trial and is currently undergoing a randomized, double-blind placebo-controlled phase II challenge trial, both in healthy adult volunteers.
INTRODUCTION
Enterotoxigenic Escherichia coli (ETEC) is the second most frequent cause of severe dehydrating diarrheal disease after rotavirus in developing countries worldwide and is estimated to account for 280 million to 400 million diarrheal cases and the deaths of 300,000 to 500,000 children under the age of 5 years every year, with an additional 100 million and 400 million cases in the 5- to 14-year-old and 15-year-old-and-over age groups, respectively (2). ETEC is also a major cause of diarrhea in travelers to developing countries, including both civilian and military personnel, estimated to cause 10 million cases annually at present.
For general information about ETEC, the reader is referred to two excellent reviews (18, 23). ETEC expresses fimbrial colonization factor antigens (CFAs), which enable the organism to adhere to the small intestine mucosa, where it secretes toxins, causing the symptoms of diarrhea. The colonization factor antigens CFA/I, CFA/II, and CFA/IV are the most prevalent ones expressed by ETEC (35). CFA/II consists of three different fimbriae, called coli surface antigens (CSs), CS1, CS2, and CS3. Natural CFA/II ETEC strains express CS3 alone or with CS1 or CS2. Similarly, CFA/IV strains express CS4, CS5, and CS6, with CS6 being expressed alone or with either CS4 or CS5. ETEC expresses heat-stable toxin (ST) and/or heat-labile toxin (LT). In addition, many strains have been found to possess the gene coding for enteroaggregative Escherichia coli heat-stable toxin 1 (EAST1) (16, 26). ST and EAST1 are short polypeptides and are poorly immunogenic. LT is similar to cholera toxin (CT), consisting of a pentamer of B subunits (LTB), which acts as a carrier for a single, toxic subunit, subunit A (24). Like cholera toxin, LTB without the A subunit is nontoxic but is immunogenic and is therefore a good candidate for inclusion in a vaccine against ETEC (9).
A number of different approaches to develop an effective vaccine against ETEC targeting either or both of the two classes of virulence determinants (enterotoxins and colonization factors) have been explored, as reviewed recently (33). In order to provide maximum efficacy against the diverse range of ETEC strains globally, a vaccine should provide as broad a range of protective antigens as possible and be simple and cheap to produce and administer. To this end, an oral, whole-cell, live attenuated approach has been explored. ETEC is adapted to survive passage through the stomach acid barrier, and vaccination using live attenuated strains avoids the need for exogenous adjuvants. This has the dual advantages of presenting newly synthesized antigens in their native conformation to the immune system as the vaccine organisms colonize and undergo limited replication in the gastrointestinal tract as well as providing a large number of surface molecules, in addition to the known colonization factors which may additionally contribute to protection. It has previously been shown that live, toxin-negative ETEC derivatives that have had deletion mutations introduced into the aroC, ompC, and ompF genes, expressing CFA/I (5, 31) and CFA/II (5, 15, 32), are safe and immunogenic when administered orally to human volunteers. A vaccine containing strains expressing CFA/I, CFA/II, and CFA/IV plus LTB may provide protection against up to 90% of ETEC strains in most areas (23, 35).
This report describes in detail the construction and characterization of six ETEC vaccine candidate strains, two of which (ACAM2007 and ACAM2017) have previously been shown to be well tolerated and immunogenic in humans (5). Three new strains (ACAM2022, ACAM2025, and ACAM2027) have additionally been engineered to express LTB. These preferred three candidates together express CFA/I, CS1, CS2, CS3, CS5, CS6, and LTB and comprise the vaccine formulation ACE527, which has recently successfully completed a phase I clinical trial demonstrating them to be well tolerated and to induce in human subjects immune responses to the LTB and CFAs expressed by all three strains (12), ACE527 is currently undergoing phase II testing for efficacy in an experimental human challenge model.
MATERIALS AND METHODS
Strains.
The strains and plasmids used in this work are described in Table 1. ACAM2008 is a streptomycin-sensitive derivative of PTL003 (CS1 CS3 ΔaroC ΔompC ΔompF) (32). ETEC clinical isolates were kindly provided by the United States Naval Medical Research Unit 3 (NAMRU3), Cairo, Egypt, and were collected as part of a large epidemiological study of diarrhea in children residing in Abu Homos, Egypt (21).
Table 1.
Bacterial strains and plasmidsa
| Strain or plasmid | Relevant genotype, phenotype, or descriptionb | Source or referencec |
|---|---|---|
| Strains | ||
| DH5α | For general DNA manipulations and detection of bacteriophage | Stratagene |
| TG1 | For general DNA manipulations and detection of bacteriophage | Stratagene |
| SM10λpir | pir traRP4 | 33, IAH |
| WS-2773E | O141:H5 CS5 CS6 LT ST EAST1 (clinical isolate) | NMRC |
| ACAM2006 | O141:H5 CS5 CS6 ΔastA ΔeltAB ΔestA ΔompC ΔompF ΔaroC | This work |
| ACAM2012 | O141:H5 CS5 CS6 ΔastA ΔeltAB ΔestA ΔompC ΔompF ΔaroC Δphage | This work |
| ACAM2022 | O141:H5 CS5 CS6 ΔastA ΔeltAB ΔestA ΔompF::Ptac-LTB ΔaroC Δphage | This work |
| WS-3504D | O39:H12 CS2 CS3 LT ST EAST1 Apr (clinical isolate) | NMRC |
| ACAM2007 | O39:H12 CS2 CS3 ΔastAΔeltAB ΔestA ΔompC ΔompF ΔaroC | This work |
| ACAM2017 | O39:H12 CS2 CS3 ΔastA ΔeltAB ΔestA ΔompC::CS1 ΔompF ΔaroC | This work |
| ACAM2027 | O39:H12 CS2 CS3 ΔastA ΔeltAB ΔestA ΔompC::CS1 ΔompF::Ptac-LTB ΔaroC | This work |
| E1392/75 | O6:H16 CS1 CS3 ST LT Sur Smr | 5 |
| E1392/75-2A | O6:H16 CS1 CS3 Sur Smr | 5 |
| PTL003 | O6:H16 CS1 CS3 Sur Smr ΔaroC ΔompC ΔompF | 32 |
| ACAM2008 | O6:H16 CS1 CS3 ΔaroC ΔompC ΔompF | This work |
| WS-1858B | O71:H− CFA/I ST EAST1 Apr Tpr Sur (clinical isolate) | NMRC, 31 |
| ACAM2010 | O71:H− CFA/I ΔastA ΔestA ΔaroC ΔompC ΔompF | 31 |
| ACAM2025 | O71:H− CFA/I ΔastA ΔestA ΔaroC ΔompC ΔompF::pLLTB | This work |
| Plasmids | ||
| RP4 | 60-kb plasmid, covalently closed circular DNA size standard | 31 |
| pvirF98 | ∼90-kb plasmid from Salmonella enterica serovar Typhimurium F98, covalently closed circular DNA size standard | 31 |
| ColVIk94 | ∼127-kb plasmid, covalently closed circular DNA size standard | 31 |
| pKK223-3 | Expression vector, source of Ptac and rrnB transcription terminator | Amersham-Pharmacia |
| pPCR-Script Amp SK+ | Plasmid vector | Stratagene |
| pPCRProm | Native LT promoter in pPCR-Script | This work |
| pACYC184 | Plasmid vector | 3 |
| pACYC-Tc | ori15A Tcr | 31 |
| pACYC-CS1 | CS1 operon in pACYC184 | This work |
| pACYC-csaA | CS1 csaA gene in pACYC184 | This work |
| pACYC-PtacLTB | Ptac-LTB construct in pACYC184 | This work |
| pACYC-PtacLTBrrnB | Ptac-LTB-rrnB transcription terminator construct in pACYC184 | This work |
| pACYC-ΔompC | This work | |
| pACYC-ΔompC::cooBA | This work | |
| pJCB12 | oriR6K mobRP4 sacB cat | 31 |
| pJCB12-ΔeltA | 658-bp deletion construct of LTA gene in pJCB12 | This work |
| pJCB12- ΔestAE | ST gene deletion fragment in pJCB12 | This work |
| pJCB12-ΔastA | 286-bp fragment with 42-bp defined deletion in EAST1 gene in pJCB12 | 31 |
| pJCB12-ΔompC | 1,016-bp fragment with deletion of the whole ompC gene in pJCB12 | 31 |
| pJCB12-ΔompF | 943-bp fragment with deletion of the whole ompF gene in pJCB12 | 31 |
| pJCB12-ΔompF::LTB | This work | |
| pJCB12-ΔompF::PtacLTB | This work | |
| pJCB12-ΔompF::PtacLTBrrnB | LTB expressed from tac promoter, incorporating rrnB transcriptional terminator and flanked by ΔompF nucleotide sequences in pJCB12 | This work |
| pJCB12-ΔompF::LLTB | This work | |
| pJCB12-ΔompC::CS1 | CS1 operon flanked by ΔompC nucleotide sequences in pJCB12 | This work |
| pJCB12-ori15A | pACYC184 replication origin in pJCB12 | 31 |
| pTETnir15 | Inducible expression vector | 4 |
| pLLTB | pTETnir15 expressing LTB from its native promoter | This work |
For the sake of brevity, some strains are not listed.
Antibiotic resistance: Nalr, nalidixic acid; Apr, ampicillin; Tpr, trimethoprim; Smr, streptomycin; Sur, sulfamethoxazole; Tcr, tetracycline.
IAH, provided by P. Barrow, Institute for Animal Health, Compton, United Kingdom; NMRC, provided by S. Savarino, Naval Medical Research Center, Silver Spring, MD.
Growth media and conditions.
Bacteria were cultured in LB broth (Sigma) or in LB broth prepared from soy peptone A3 (10 g/liter; Organotechnie), yeast extract (5 g/liter; Sigma), and NaCl (10 g/liter; BDH). For solid media, broth media were supplemented with 15 g/liter Bacto agar (BD). Strains were routinely grown at 37°C. For identification of antibiotic-sensitive derivatives, colonies were replica plated from nonselective LB agar onto LB agar supplemented with appropriate antibiotics. When expression of CS was required, ETEC strains were grown on CFA agar (10 g/liter Casamino Acids [BD], 20 g/liter Noble agar [BD], 1.5 g/liter yeast extract [Sigma], 0.05 g/liter MgSO4 [Sigma], 0.005 g/liter MnCl2 [Sigma] [8]). When required, media were supplemented with 0.15% sodium desoxycholate (Sigma) and aromix (phenylalanine, 40 μg/ml; tryptophan, 40 μg/ml; p-aminobenzoic acid, 10 μg/ml; and 2,3-dihydroxybenzoic acid, 10 μg/ml [final concentrations]). Antibiotics were used at the following concentrations: tetracycline (Sigma), 15 μg/ml; streptomycin (Sigma), 20 μg/ml; chloramphenicol (Sigma), 15 μg/ml; or ampicillin (Sigma), 200 μg/ml. Conjugation for the transfer of suicide vector derivatives into ETEC strains was performed as described previously (32). When counterselecting against the sacB gene, NaCl was omitted from LB medium and 5% sucrose (Sigma) was added (13).
Analysis of expressed CS proteins by heat extraction and SDS-PAGE.
Bacterial strains were grown on CFA agar supplemented with aromix for aroC mutants, antibiotics, and/or sodium desoxycholate (Sigma) as required, until growth was confluent. Bacterial growth was suspended in phosphate-buffered saline (PBS), and 109 cells were harvested by centrifugation and then resuspended in 10 μl PBS. This sample was heated to 65°C for 10 min and centrifuged at 13,000 rpm for 5 min, and the supernatant was added to 10 μl 2× Novex Tris-Gly sample buffer (Invitrogen) containing 2 μl 1 M dithiothreitol. Samples were heated at 95°C for 5 min and then analyzed by SDS-PAGE on 14% Tris-Gly gels using an XCell II electrophoresis tank (Invitrogen). Gels were stained using Coomassie or SeeBlue stain (Invitrogen).
Detection of proteins by Western blotting.
Following electrophoresis, proteins were transferred to nitrocellulose membranes and visualized using a SuperSignal West Pico chemiluminescent substrate kit (Pierce). To obtain CS-specific antisera for use in Western blotting, CSs were prepared as described above, separated on 14-cm SDS-polyacrylamide gels, and stained with KCl, and a slice of the gel containing the CS was cut out. Gel slices were emulsified and used to immunize rabbits, from which sera were subsequently obtained (Abcam Ltd., Cambridge, United Kingdom). LTB was detected in bacterial culture supernatant or cell lysates using a rabbit polyclonal antibody specific for cholera toxin (C3062; Sigma) with an anti-rabbit horseradish peroxidase (HRP) conjugate (A4914; Sigma).
Antibiotic resistance.
Antibiotic resistance profiles for ampicillin, tetracycline, streptomycin, trimethoprim, and sulfamethoxazole were determined using antibiotic-impregnated paper discs (Oxoid).
Toxin assays and LTB quantitation.
The presence or absence of ST and LT in bacterial culture supernatants was confirmed using transepithelial electrical resistance (TEER) of CaCo-2 cells monitored using chopstick electrodes (World Precision Instruments, Stevenage, United Kingdom) which can distinguish between ST and/or LT (31). The absence of LT expression in toxin-negative strains was also confirmed using a Y1 adrenal cell toxicity assay (6). LTB expression was quantitated using a modified GM1-binding enzyme-linked immunosorbent assay (ELISA) (1).
DNA manipulations.
DNA manipulations were performed using standard procedures (25). Restriction enzymes were from New England BioLabs (Herts, United Kingdom). Plasmid DNA was prepared using plasmid purification kits from Qiagen (Crawley, United Kingdom), and DNA fragments were isolated from agarose gels using a QIAquick gel extraction kit from Qiagen. Genomic DNA was prepared using a Qiagen genomic buffer set and 100-gauge tips. PCR was performed using Taq DNA polymerase (Invitrogen), except when the products were for the construction of recombinant DNA molecules, when PfuTurbo DNA polymerase (Stratagene) was used. Electrotransformation of bacteria was performed as described previously (7) using a Bio-Rad Gene Pulser II apparatus connected to a Bio-Rad capacitance extender and pulse controller. Southern hybridizations were performed using an enhanced chemiluminescence (ECL) random prime labeling and detection system (version II), an ECL direct nucleic acid labeling and detection system, or an alkaline phosphatase direct labeling kit with chemiluminescent detection using the CDP-Star detection reagent (Amersham-Pharmacia). Southern hybridization probes were prepared by PCR using appropriate DNA templates and oligonucleotides (Table 2).
Table 2.
Oligonucleotides used
| Name | Nucleotide sequence (5′ → 3′) | Target locus and use |
|---|---|---|
| 4746 | CGGCATGCCGCAATTGAATTGGGGG | Ptac-LTB construction |
| 4746 | CGGCATGCCGCAATTGAATTGGGGG | eltB; construction of ΔLTA |
| 4749 | GGCGTCGACGAAAATGAAGGGGCGAAGTTC | astA; construction of EAST1 deletion, amplification of probe for Southern blotting |
| 4750 | ATGACACGAATGTTGATGGCATCCGGGAAGC | Construction of EAST1 deletion |
| 4751 | GCCATCAACATTCGTGTCATGGAAGGACTAC | Construction of EAST1 deletion |
| 4752 | GGCGCATGCAAGATTCGGCCAGTTAGCC | Construction of EAST1 deletion, amplification of probe for Southern blotting |
| 4764 | AATATTACTATGCTCTTCGTAGCGG | Construction of ST deletion mutation, amplification of probe for Southern blotting |
| 4772 | CCGTCGACTAAAAATCACCACCACTTC | Construction of ΔLTA |
| 4773 | ATTCATCCTCCTTATATATCATACAAGAAGACAATCC | Construction of ΔLTA |
| 4774 | GATATATAAGGAGGATGAATTATGAATAAAGTAAAATTT | Construction of ΔLTA |
| 4935 | TTGTGCAGGTGGATCCGGAGCTTATCGACTGC | Ptac-LTB promoter construction |
| 47112 | GGTCAGCCGGAATACGCGTT | Construction of pJCB12-ΔSTIE |
| 47120 | CATCAGAATCACTATTCATGCTTTCAGGACCAC | Construction of pJCB12-ΔSTIE |
| 47121 | CATGAATAGTGATTCTGATGATGTCTGTAACG | Construction of pJCB12-ΔSTIE |
| 47183 | CGTCGCATGCGGTGTCTTTATGTGTCTGC | Construction of ompF-LTB, includes SphI site |
| 47184 | GGCGTCGACGCTATGGACGTTCTGGCTCC | Construction of ompF-LTB, includes SalI site |
| 47192 | GGCGTCGACCCCATCACCGTCGATGA | Prophage deletion |
| 47193 | GGTATCCGTTGCTGTTTCATATCAAAACTCCC | Prophage deletion |
| 47194 | ATATGAAACAGCAACGGATACCACCGACGCAG | Prophage deletion |
| 47195 | GCCGCATGCATTCAGCCGCCATGTCAC | Prophage deletion |
| 47229 | GCGGATCCGAGCTCGACATTCAGAAATGAATGACGG | Construction of pJCB-ΔompC::CS1 |
| 47230 | GCTTAGCGCTGCAGGTTATTAACCCTCTGTTATATGCC | Construction of pJCB-ΔompC::CS1 |
| 47231 | AATAACCTGCAGCGCTAAGCCTCGAGTAATC | Construction of pJCB-ΔompC::CS1 |
| 47232 | CGGTCGACGTTAAAGCGCATCAGCGCGG | Construction of pJCB-ΔompC::CS1 |
| 47235 | GATTCCTGTGTGAAATTGTTATCCGATCATAATTCCACACATTATACGAGCC | Ptac-LTB construction |
| 47236 | GGATAACAATTTCACACAGGAATCTAGAATGAATAAAGTAAAATTTTATGTTTT | Ptac-LTB construction |
| 47227 | GTTACGGAATTGTTGACAATTAATCATCGGC | Ptac-LTB construction |
| 47228 | ATTGTCAACAATTCCGTAACAGGATGATCG | ompF-Ptac-LTB construction |
| 47255 | GCCACTAGTAATTTCCCCATGCGAGAGTAGG | rrnB terminators, includes SpeI site |
| 47256 | GCGACTAGTGGGATGGCTTGTAGATATGACG | rrnB terminators, includes SpeI1 site |
| 47257 | GGGCAAGCTTATTGCGGATTAGTTTTTTCTTAGC | ompF-Ptac-LTB construction |
| 47258 | CTAATCCGCAATAAGCTTGCCCCCCCAGCCTAG | ompF-Ptac-LTB construction |
| pFOR | CCGGTACCATGATTCAATGTACACC | Amplification of native LT promoter |
| pREV | ACGTAGATCTACTTATATATCATACAAG | Amplification of native LT promoter |
| Bfor | ACGTAGATCTTTATGAATAAAGTAAAATTTTATG | Cloning eltB, includes BglII site |
| Brev | GTACGCTAGCCATGTATCTCATTAGCTG | Cloning eltB, includes NheI site |
Isolation of CS1 operon.
The nucleotide sequence of the CS1 operon (10, 20, 27) indicates that it is within an EcoRV and BglII restriction fragment. DNA fragments generated by digestion of plasmid DNA from ETEC strain E1392/75 with EcoRV and BglII were ligated to pACYC184 digested with EcoRV and BamHI and then transformed into strain DH5α. A colony harboring a recombinant plasmid incorporating the CS1 operon was identified, and the recombinant plasmid was called pACYC-CS1.
Characterization and removal of bacteriophage from WS-2773E.
Culture supernatants from deletion-derivative strains were screened for plaque generation with E. coli strains DH5α and TG1. Bacteriophage was harvested from culture supernatants of WS-2773E, and DNA was isolated using the methods described for M13 phage (25). Digestion of the isolated double-stranded DNA with SphI generated 6 fragments of approximately 15, 6, 5, 3, 2, and 1.8 kb. The 15-kb phage-derived fragment was ligated into plasmid pACYC184, and the nucleotide sequences of each end were determined (data not shown). To generate a deletion construct, DNA fragments of approximately 350 bp from the sequenced ends of the 15-kb phage-derived fragment were amplified using primers 47192 with 47193 and 47194 with 47195, and the two resulting fragments were fused by overlap extension PCR. This construct was ligated into suicide vector pJCB12. This construct is predicted to remove 15 kb of the prophage genome.
Generation of a Ptac promoter/LTB gene construct.
The Ptac promoter was amplified as a 268-bp fragment from plasmid pKK223-3 (Amersham-Pharmacia) in such a way as to introduce point mutations into the lacO operator sequence (from AATTGTGAGGGGATAA to AATTATGATCGGATAA) with oligonucleotides 4935 and 47235. The eltB coding region was amplified from WS-2773E plasmid DNA using oligonucleotides 47236 and 4746 and fused to the Ptac fragment by overlap extension PCR using oligonucleotides 4935 and 4746. The resulting product was cloned into pACYC184 using BamHI and SphI sites incorporated into primers 4935 and 4746, respectively, giving pACYC-PtacLTB.
Generation of Plt promoter/LTB gene construct transcribed from its native promoter.
The native LT promoter was amplified from the wild-type ETEC strain WS-2773E using primers pFOR and pREV. The forward primer annealed ∼200 bp upstream of the LTA gene. A KpnI restriction site was included in the primer to facilitate cloning into expression vectors. The reverse primer annealed just upstream of the start codon of the LTA gene and was designed to introduce a BglII restriction site to allow correct positioning of the promoter fragment with respect to the LTB gene. The gel-purified promoter fragment was ligated into pPCR-Script Amp SK+ (Stratagene), resulting in plasmid pPCRProm. The eltB gene was cloned by PCR from the wild-type ETEC strain WS-2773E using primers Bfor, which incorporates a BglII site 8 bases 5′ prime of the ATG start codon, and Brev, which was designed to amplify from 200 bases downstream of the stop codon, such that any transcription terminators would be included in the PCR product. Gel-purified LTB fragment was ligated into pPCR-Script Amp SK+ (Stratagene), resulting in plasmid pPCRLTB. The LTB gene was transferred into a modified expression vector derived from pTETnir15 (3) containing the inducible nirB promoter. The nirB promoter was excised from the pTETnir15 derivative by digestion with KpnI and BglII, and the remaining vector fragment was isolated and purified. Plasmid pPCRProm was digested with KpnI and BglII, and the LT promoter fragment was isolated and purified. The vector and promoter fragments were ligated to yield the construct pLLTB.
Construction of suicide vectors for deletion of astA, eltAB, estA, aroC, ompC, and ompF.
The plasmid constructs used are listed in Table 1, and many have been described previously (31, 32). Plasmid pJCB12-ΔestAE incorporates a defined deletion mutation of the ST gene from strain WS-2773E. Oligonucleotides 4764 with 47120 and 47121 with 47112 were used to produce two template fragments, respectively, for overlap extension PCR with oligonucleotides 4764 and 47112. The resulting ΔestAE fragment was then ligated into pJCB12.
Plasmid pJCB12-ΔastA incorporates a defined 42-bp deletion covering the first 14 amino acids of the EAST1 coding sequence. Oligonucleotide pairs 4749 with 4750 and 4751 with 4752 were used to amplify DNA fragments upstream and downstream of the EAST1 gene. Oligonucleotides 4749 and 4752 were then used to fuse these PCR products by overlap extension PCR. The construct was cloned into pJCB12 to give pJCB12-ΔastA.
Plasmid pJCB12-ΔeltA incorporates a defined 658-bp deletion mutation of the LT subunit A gene (eltA) from strain WS-2773E. Using plasmid DNA from strain WS-2773E as template, two DNA fragments were amplified, one 5′ to eltA using oligonucleotides 4772 and 4773 and one including the 3′ end of eltA and most of eltB using oligonucleotides 4774 with 4746. The eltA-deletion fragment was generated in an overlap extension PCR using these two fragments as template and oligonucleotides 4772 with 4746 and was then ligated with pJCB12 using the SalI and SphI restriction sites incorporated into the primers.
The pJCB12 derivatives used to introduce deletion mutations into aroC, ompC, and ompF have been described previously (31).
Construction of suicide vector for insertion of CS1 operon at ΔompC locus.
The 5′ ompC DNA fragment was amplified from PTL003 genomic DNA using oligonucleotides 47229 and 47230, and the 3′ ompC DNA fragment was similarly amplified using oligonucleotides 47231 and 47232. These two fragments were then fused by overlap extension PCR using oligonucleotides 47229 and 47232, and the resulting fragment was digested with BamHI and SalI and ligated to pACYC184 similarly digested to produce pACYC-ΔompC. Part of the CS1 operon was then obtained from pACYC-CS1 on a PstI/XhoI DNA fragment, and this was ligated to PstI/XhoI-digested pACYC-ΔompC to generate pACYC-ΔompC::cooBA. The remainder of the CS1 operon was then obtained from pACYC-CS1 on an XhoI DNA fragment, and this was ligated to similarly digested pACYC-ΔompC::cooBA, thereby re-creating the entire CS1 operon flanked by ompC nucleotide sequences in the pACYC184 plasmid vector. The ΔompC::CS1 portion was then digested from this plasmid using SacI and SalI and ligated to similarly digested pJCB12 to create pJCB12-ΔompC::CS1.
Construction of suicide vector for insertion of Ptac-LTB construct at ΔompF locus.
A fragment from the ompF region was amplified from strain E1392/75-2A genomic DNA using oligonucleotides 47183 and 47257. The eltB gene was amplified from WS-2773E plasmid DNA using oligonucleotides 47236 and 47258, and this was joined to the above-described ompF fragment by overlap extension PCR using oligonucleotides 47183 and 47236. The resulting DNA fragment was ligated to pJCB12 using the incorporated BglII and SphI restriction sites to give plasmid pJCB-ompFLTB. A second DNA fragment was amplified from the ompF region using oligonucleotides 47184 and 47228. The Ptac-LTB region of plasmid pACYC-PtacLTB was amplified using oligonucleotides 47227 and 4746, and this fragment was joined to the second ompF fragment by overlap extension PCR using oligonucleotides 47184 and 4746. The resulting DNA fragment was digested with SalI and XbaI to yield two fragments, one of which includes the Ptac promoter fused to ompF. After ligation to similarly digested pJCB-ompFLTB, a plasmid incorporating the ompF-Ptac fragment next to the LTB-ompF fragment of pJCB-ompFLTB to give plasmid pJCB12-ΔompF::PtacLTB was identified.
The E. coli rrnB transcriptional terminator was amplified from pKK223-3 using oligonucleotides 47255 and 47256, both of which incorporate an SpeI site. This fragment was cloned into the SpeI site at the 3′ terminus of eltB in pJCB12-ΔompF::PtacLTB to give pJCB12-ΔompF::PtacLTBrrnB.
Construction of suicide vector for insertion of PltLTB construct at ΔompF locus.
The suicide vector pJCB12-ΔompF was digested with SalI/SphI and ligated with the SalI/SphI-digested PltLTB fragment from pLLTB to generate pJCB12-ΔompF::PltLTB, in which the LTB gene with its native promoter is flanked by nucleotide sequences that flank the ompF locus.
Introduction of genetic constructs into ETEC chromosome.
Plasmid pACYC-Tc was first transformed into recipient ETEC strains to permit their selection after conjugation using tetracycline. The pJCB12 derivatives described above were introduced into the recipient ETEC strain either by electroporation or, more usually, by conjugation from E. coli strain SM10λpir. ETEC strains that had integrated the suicide vector into the chromosome were selected by growth on tetracycline and chloramphenicol. Insertion at the chosen locus was confirmed by PCR. Transconjugants were then cultured in medium supplemented with sucrose to select derivatives in which the sacB gene (and therefore the suicide vector) had been lost. Desired recombinants were identified by PCR, and nucleotide sequence determinations confirmed that the deletion/insertion had occurred correctly. Upon completion of the genetic manipulations, pACYC-Tc was cured from the strains by introduction of plasmid pJCB12-ori15A, a pJCB12 derivative that carries the same replication origin as pACYC-Tc. Tetracycline-sensitive derivatives could then be cured of pJCB12-ori15A by growth on sucrose-supplemented media.
RESULTS
Characterization of ETEC clinical isolates WS-3504D and WS-2773E.
PCR and Southern blot analysis of strains WS-3504D (expressing CS2 and CS3) and WS-2773E (expressing CS5 and CS6) (Fig. 1) revealed that both possessed the estA gene coding for the STIb (STh) heat-stable toxin (11, 17, 30). Although both strains also possessed the LT genes, no LT was detectable in culture supernatants of WS-3504D using ELISA or functional assays (data not shown). Both strains harbored the astA gene coding for EAST1 (16). In addition, while WS-3504D was resistant to ampicillin, WS-2773E was sensitive to all antibiotics tested (see Materials and Methods).
Fig. 1.

Changes in plasmids between vaccine strains and their ancestral clinical isolates. Plasmid DNA from clinical isolates WS-3504D and WS-2773E, alongside that from vaccine candidate derives (ACAM2007 and ACAM2006, respectively), was electrophoresed through 0.6% agarose and stained with SYBR gold. ACAM2007 was further derivatized to yield ACAM2017 and ACAM2027; ACM2006 was further derivatized to yield ACAM2012 and ACAM2022. The approximate positions of size standards are shown down the left side, and plasmids are labeled with the Southern probes to which they hybridized. To the right of each gel picture are shown some example results of Southern hybridizations using the CS3, CS5, LT, and ST probes, as indicated. Southern hybridization of WS-3504D and ACAM2007 with the ST and EAST1 probes gave results identical to those obtained with the LT probe (data not shown). A faint signal for the ST gene is visible in ACAM2006 because the ST gene deletion in this strain is very short and lies within the bounds of the ST gene probe.
Deletion of toxin genes from ETEC isolates WS-3504D and WS-2773E.
In order to create a safe live attenuated vaccine strain, removal of the toxins is necessary. Strain WS-3504D was targeted first with pJCB12-ΔastA to generate a deletion in the EAST1 gene. When toxin-minus derivatives were analyzed by PCR using primers that annealed just outside the deleted sequence, no PCR product was obtained, suggesting that flanking DNA had been lost along with the EAST1 gene. Indeed, loss of the EAST1 gene was also accompanied by loss of the ST and LT genes, suggesting that these toxin genes may be linked on the same plasmid and lost by a large deletion. This was confirmed by Southern blot analysis (see later and Fig. 1). PCR analysis confirmed that the CS2 and CS3 loci remained.
In contrast, strain WS-2773E required a separate manipulation to delete each of the EAST1, ST, and LT genes. In WS-2773E, deletion of the ST gene repeatedly gave derivatives that had also lost genes from the CS5 and CS6 operons, suggesting that these CS and ST genetic loci are closely linked. Finally, a recombinant that had incorporated the defined ΔestA deletion by allelic exchange was obtained, and this was confirmed by nucleotide sequencing (data not shown).
In both strains, loss of the toxin genes was first identified by PCR, and then the absence of expressed toxins was confirmed by Southern hybridization, ELISA, the Y1 adrenal cell assay, and TEER. No toxins were detected by any of these assays (data not shown), and Southern hybridizations showed that the genes were no longer present in the plasmid DNA, where the toxins are usually encoded (Fig. 1).
Deletion of chromosomal loci from toxin-minus strains and removal of antibiotic resistance determinants.
The deletions of aroC, ompC, and ompF were sequentially introduced into both toxin-minus strains using the suicide vector method (see Materials and Methods). The structure of the aroC, ompC, and ompF chromosomal deletions in both strains was confirmed by nucleotide sequencing (data not shown). The resulting WS-2773E toxin-minus derivative with chromosomal deletions was named ACAM2006.
At this stage, the attenuated WS-3504D derivative retained the ampicillin resistance determinant. To ensure against any contribution to the spread of such antibiotic resistance, any live attenuated bacterial vaccine strains must be free of antibiotic resistance determinants. Therefore, removal of ampicillin resistance from the attenuated WS-3504D derivative was achieved by screening several thousand colonies for a spontaneous mutant which had lost this phenotype by replica plating from solid medium onto medium supplemented with ampicillin. An ampicillin-sensitive, attenuated WS-3504D derivative was obtained which was called ACAM2007. Both ACAM2006 and ACAM2007 were characterized at each stage of their construction to confirm retention of CS expression.
Characterization of changes in plasmids following generation of vaccine strains.
Plasmid DNA was isolated from ETEC strains WS-3504D and WS-2773E and their derivatives, ACAM2007 and ACAM2006, respectively, and Southern hybridization was performed with probes specific for the ST, LT, and EAST1 genes (data not shown) as well as with probes specific for the genes encoding CS3, CS5, and CS6 (data not shown). Figure 1 shows agarose gels of the large plasmids from these strains and which of these plasmids carry the ETEC virulence factors, as revealed by Southern hybridization. These data indicate that multiple virulence factors are often encoded on one plasmid. Thus, in WS-3504D, a plasmid of ∼120 kb codes for ST, LT, EAST1, and CS3. In its attenuated derivative, ACAM2007, this plasmid is reduced to about 100 kb and has lost the LT, ST, and EAST1 (data are shown only for LT) genes while retaining CS3 expression. In WS-2773E, LT is encoded on a plasmid separate from and smaller than that which codes for ST, CS5, and CS6. Loss of the LT gene is accompanied by a reduction in plasmid size of approximately 20 kb. The deletion of the ST gene in this strain was achieved by a small, precise deletion of the majority of the coding region of ST, such that the difference in plasmid sizes is not resolvable by gel electrophoresis. This was required, as all nonspecific deletions of the ST gene also resulted in the loss of CS6 expression. In strain WS-2773E, EAST1 was encoded by a gene located on the chromosome which was removed using the suicide vector pJCB12-ΔastA, which resulted in no change to the plasmids carried in the strain (data not shown). The results also show that loss of the toxins has frequently coincided with large deletions from the relevant plasmid, the only exception to this being in ACAM2006, where significant effort was required to identify a derivative with a defined ST deletion in order to retain expression of CS5 and CS6 (see above). The loss of another plasmid of ∼70 kb during construction of ACAM2007 is also apparent. The loss of ST was also observed to coincide with a large deletion from a large-molecular-size plasmid during construction of previously described vaccine candidate ACAM2010 (31). These results demonstrate the propensity of ETEC plasmids to undergo substantial rearrangements which can be isolated when cultures are placed under strong selective pressure.
Expression of CS1 from an operon inserted into chromosome of ACAM2007.
Rather than including 2 different strains in a final vaccine to express all of CS1, CS2, and CS3, ACAM2007 was modified to express CS1, in addition to the CS2 and CS3 that it expresses naturally. To achieve this and to keep further disruption of the chromosome to a minimum, the CS1 operon was inserted at the site of ompC deletion in ACAM2007. The ACAM2007 ompC::cooBACD (CS1) derivative was called ACAM2017. The nucleotide sequences across the ompC/CS operon junctions were determined and found to be as expected (data not shown).
When ACAM2017 was harvested from CFA agar, the amount of CS2 and CS3 expressed was similar to that in the parent strain, ACAM2007 (Fig. 2). In addition, CS1 was seen to be expressed in similar quantities to CS2, although apparently at somewhat lower levels than in ACAM2008, which expresses CS1 naturally (Fig. 2). This difference is not considered to be significant, as it represents in vitro expression levels, which are sensitive to many influences, such as culture density. It has been demonstrated in a clinical trial that specific immune responses to CS1 are generated following immunization with strain ACM2017 (5). There was no obvious difference between the growth rates of ACAM2007 and ACAM2017.
Fig. 2.
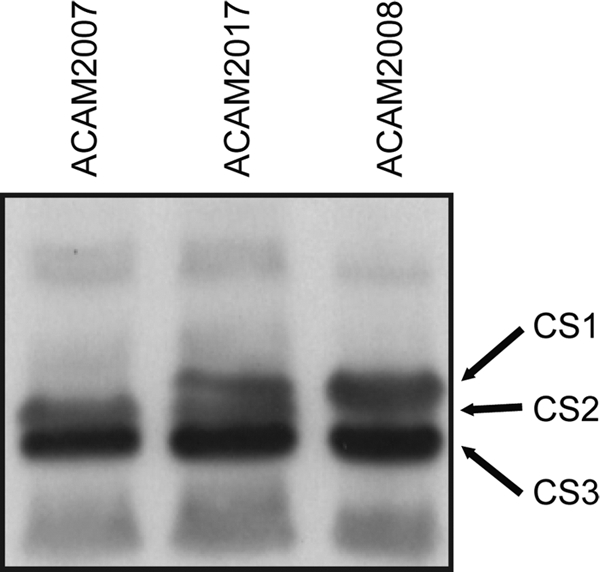
Western blot showing expression of CS1, CS2, and CS3 in ACAM2017. Following SDS-PAGE of heat-extracted CS protein and transfer to nitrocellulose, CSs were visualized using a mixture of three antisera, each of which was raised in rabbits against CS1, CS2, or CS3 detected with anti-rabbit Ig-HRP. The positions of CS1, CS2, and CS3 are indicated for ACAM2017 compared to those in strains expressing only CS2 and CS3 (ACAM2007) or CS1 and CS3 (ACAM2008).
Deletion of prophage from ACAM2006 to generate ACAM2012.
Screening of culture supernatants revealed that ACAM2006 was harboring a lysogenic bacteriophage. By isolating bacteriophage DNA from ACAM2006 culture supernatant, approximately 400 bp of nucleotide sequence data was generated from two different regions of the phage genome. This indicated that the bacteriophage was similar to lysogenic bacteriophage P2 and allowed the construction of a vector for deletion of a large amount of prophage DNA from the ACAM2006 chromosome to generate strain ACAM2012. Screening of ACAM2012 culture supernatants found no further evidence of bacteriophage.
Expression of LTB from a chromosomal location using the strong Ptac or native LT promoter.
The gene for the nontoxic LTB subunit was introduced into the chromosomes of three of the vaccine candidates to generate stable expression of the antigen in the absence of any selection mechanisms, which is required when expression is plasmid mediated. Since there is no way of predicting if natural, inducible expression levels or constitutive high levels of LTB expression will generate the best immune response, two different LTB constructs were generated, one in which the LTB gene was expressed from its native promoter (pLLTB) and one in which expression was from the strong tac promoter Ptac-LTB).
The LTB gene under expression from the Ptac promoter was inserted at the ompF deletion site in ACAM2017 to give ACAM2027 and into the analogous site in ACAM2012 to give ACAM2022. Likewise, the LTB gene under expression from the LT promoter was inserted at the ompF deletion site in ACAM2010 (35) to generate ACAM2025. The nucleotide sequences at the insertion sites were confirmed (data not shown). Levels of LTB expression in culture supernatants and in whole-cell lysates were determined by ELISA for ACAM2022 and ACAM2027. Both in culture supernatants and in whole-cell lysates, LTB was seen at levels that considerably exceeded those for clinical isolates. For example, ACAM2027 and ACAM2022 whole-cell lysates contained approximately 1,500 ng/ml LTB, whereas lysate from WS-2773E contained 36 ng/ml LTB. In Western blots, for ACAM2022 and ACAM2027, a band corresponding to LTB could clearly be seen, whereas no LTB band was visible for WS-2773E (Fig. 3), confirming higher levels of constitutive LTB expression in the ACAM strains than in the clinical isolate WS-2773E.
Fig. 3.
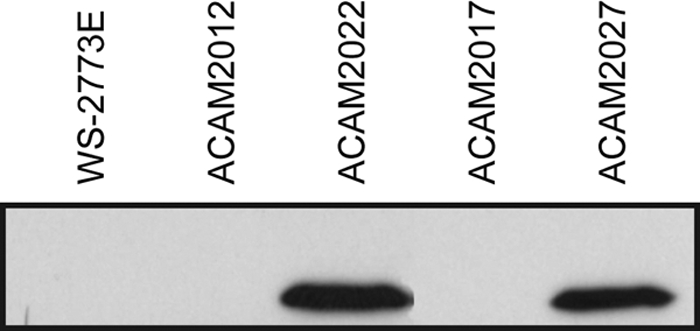
Western blot showing expression of LTB from vaccine candidate strains ACAM2022 and ACAM2027 and their precursors. Following SDS-PAGE of whole cells and transfer to nitrocellulose, LTB was visualized using rabbit anti-cholera toxin antisera detected with anti-rabbit Ig-HRP. LTB expression from ACAM2022 and ACAM2027 is shown in comparison with that of their precursors, ACAM2012 and ACAM2017, respectively, and with that of the ACAM2012 precursor, clinical isolate WS-2773E, which expresses native LT.
In ACAM2025, expression of the LTB fragment is significantly lower when grown in culture than in ACAM2022 and ACAM2027 (data not shown); however, higher levels of expression may be induced by activation of the natural promoter in vivo following vaccination.
DISCUSSION
This work describes the generation and characterization of six novel live attenuated ETEC vaccine strains, three of which have been chosen for inclusion in a multistrain vaccine to protect against ETEC diarrhea. From receipt at our laboratory, all strains were grown solely on certified media where the source of any animal-derived component was fully documented, and later, media were adapted to be completely free of animal-derived components to eliminate any potential risks from unknown animal zoonoses, such as occurred with bovine spongiform encephalopathy in the late 1990s to early 2000s.
All five vaccine candidate strains have had the known ETEC toxin genes (coding for ST, LT, and EAST1) removed and further defined attenuating deletion mutations made in aroC, ompC, and ompF. In addition to ST, LT, and EAST1, the major virulence factors of ETEC are CS antigens, which are generally fimbrial structures that mediate adherence to the intestinal epithelium. Immune responses against the toxins and CS antigens are likely to be important for protection against ETEC infection (14, 34). There are several known CS antigens, but natural ETEC strains express only one or two. Therefore, a vaccine must induce immune responses to several CS antigens if it is to protect effectively against most ETEC strains and will therefore need to consist of either several strains or fewer strains expressing additional CS antigens. Our vaccine candidate repertoire includes several strains coding for different CS antigens. Vaccine strain ACAM2025 expresses CFA/I; ACAM2012 and its derivative, ACAM2022, express CS5 and CS6; ACAM2007 expresses CS2 and CS3, and its derivatives, ACAM2017 and ACAM2027, in addition, express CS1. CS1 expression levels in these strains are comparable to those of CS2 and CS3 when grown on CS-inducing media, and the strains show no obvious signs that carriage of the additional CS operon is detrimental to growth. In a randomized, double-blind phase I clinical study (5), significant mucosal antibody secreting cell (ASC) and whole-gut lavage fluid immune responses against CS1 were induced by ACAM2017 but not by its parent strain, ACAM2007, which expresses only CS2 and CS3. Vaccine strains ACAM2022, ACAM2025, and ACAM2027 all express the LTB subunit and between them express CFA/I, CS1, CS2, CS3, CS5, and CS6. Precursors of ACAM2025 (ACAM2010) and ACAM2027 (ACAM2007 and ACAM2017) have already been demonstrated to be safe and immunogenic in human volunteers (5, 31), as has PTL003, which was similarly attenuated (15, 32).
A number of studies have shown that LT is expressed by 35 to 55% of ETEC strains, depending on location (19, 22, 28, 29, 35). Induction of an immune response to LT is therefore likely to provide limited protection against natural ETEC infection which should be significantly broadened by simultaneous immunization against the most common CFAs. Since it cannot be predicted which mode of LTB expression will generate the best immune response in human subjects, the LTB gene was expressed from the native promoter in ACAM2025 but from the strong tac promoter in ACAM2022 and ACAM2027. The level of expression of LTB under the control of the tac promoter is approximately 37-fold higher in attenuated vaccine strain ACAM2022 than in its parental wild-type strain WS-2773E when grown in vitro, and it is possible that such high levels of antigen present in attenuated vaccine strains will induce immune responses as high as or even higher than those induced by wild-type ETEC infection. This hypothesis is supported by the results of a phase I study (12). LTB is highly homologous to cholera toxin (CT), and the two toxins are immunologically cross-reactive. The recombinant CTB present in the commercial cholera vaccine Dukoral is reported to confer some protection against ETEC as a result of this immunological cross-reactivity. This raises the possibility that the LTB-expressing ETEC vaccine candidates described here may also provide some immune protection against cholera, a disease which, like ETEC, is endemic in some regions where sanitation and supplies of clean drinking water are inadequate as well as emerging as epidemics following natural or human-caused disasters.
The LTB gene constructs in ACAM2025, ACAM2027, and ACAM2022 and the CS1 operon in ACAM2017 and ACAM2027 were inserted into the chromosome. This has advantages over expression from plasmid vectors because the relevant genes or operons are less likely than a plasmid to be lost during manufacture of a vaccine formulation or after administration to vaccinees. In addition, there is no requirement for a selective marker such as antibiotic resistance, which would be needed if the operons were on a plasmid vector. The CS2 operon has been found on the chromosome in naturally occurring ETEC (20), indicating that the chromosome is an appropriate location from which to express CS operons. The colonization factors CS3, CS5, CS6, and CFA/I are expressed in the vaccine strains on native ETEC plasmids, which are large-molecular-size, low-copy-number, and stably inherited elements, or derivatives of them from which the toxin genes have been deleted. During many in vitro and in vivo passages, there was no evidence for loss of these plasmids (data not shown).
In this work, we chose to employ the technique of allelic exchange using suicide vectors to both delete and insert genes into the chromosome or plasmids of our strains. This method depends solely on the native homologous recombination systems and so is unlikely to catalyze any unforeseen recombination events between repeated nucleotide sequences in the genome. In addition, combining overlap extension PCR and the suicide vector method enables the generation of genetic constructs with absolute precision.
ACAM2025, ACAM2022, and ACAM2027, which between them express 6 different CS antigens (see above) and LTB, could provide protection against up to 90% of ETEC strains (35). These three strains have been combined into a vaccine formulation known as ACE527, which has recently been tested in a randomized, double-blind, placebo-controlled phase I safety and immunogenicity trial. ACE527 proved to be very well tolerated at a total dose up to 1011 CFU and induced strong immune responses to CFAs expressed on all three strains and to LTB (12), confirming the validity of the oral, attenuated multistrain vaccine approach.
ACKNOWLEDGMENTS
We are grateful to the United States Naval Medical Research Center, Silver Spring, MD, and Unit 3, Cairo, Egypt, for providing us with ETEC clinical isolates.
This work was funded by Acambis Research Ltd. and by a research grant from the Dual Use Science and Technology program of the U.S. Army Medical Research and Materiel Command, Fort Detrick, MD.
Footnotes
Published ahead of print on 12 October 2011.
REFERENCES
- 1. Amin T., Hirst T. R. 1994. Purification of the B-subunit oligomer of Escherichia coli heat-labile enterotoxin by heterologous expression and secretion in a marine vibrio. Protein Expr. Purif. 5:198–204 [DOI] [PubMed] [Google Scholar]
- 2. Anonymous. 2006. Future directions for research on enterotoxigenic Escherichia coli vaccines for developing countries. Wkly. Epidemiol. Rec. 81:97–104 [PubMed] [Google Scholar]
- 3. Chatfield S. N., et al. 1992. Use of the nirB promoter to direct the stable expression of heterologous antigens in Salmonella oral vaccine strains: development of a single-dose oral tetanus vaccine. Biotechnology (NY) 10:888–892 [DOI] [PubMed] [Google Scholar]
- 4. Cravioto A. 1980. Studies on the adhesive factors of pathogenic strains of E. coli isolated from man. University of London, London, United Kingdom [Google Scholar]
- 5. Daley A., et al. 2007. Genetically modified enterotoxigenic Escherichia coli vaccines induce mucosal immune responses without inflammation. Gut 56:1550–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Donta S. T., Moon H. W., Whipp S. C. 1974. Detection of heat-labile Escherichia coli enterotoxin with the use of adrenal cells in tissue culture. Science 183:334–336 [DOI] [PubMed] [Google Scholar]
- 7. Dower W. J., Miller J. F., Ragsdale C. W. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16:6127–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evans D. G., Evans D. J., Jr., Clegg S., Pauley J. A. 1979. Purification and characterization of the CFA/I antigen of enterotoxigenic Escherichia coli. Infect. Immun. 25:738–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frech S. A., et al. 2008. Use of a patch containing heat-labile toxin from Escherichia coli against travellers' diarrhea: a phase II, randomised, double-blind, placebo-controlled field trial. Lancet 371:2019–2025 [DOI] [PubMed] [Google Scholar]
- 10. Froehlich B. J., Karakashian A., Melsen L. R., Wakefield J. C., Scott J. R. 1994. CooC and CooD are required for assembly of CS1 pili. Mol. Microbiol. 12:387–401 [DOI] [PubMed] [Google Scholar]
- 11. Guzman-Verduzco L. M., Kupersztoch Y. M. 1989. Rectification of two Escherichia coli heat-stable enterotoxin allele sequences and lack of biological effect of changing the carboxy-terminal tyrosine to histidine. Infect. Immun. 57:645–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harro C., et al. 2011. A combination vaccine consisting of three live attenuated enterotoxigenic Escherichia coli strains expressing a range of colonization factors and heat-labile toxin subunit B is well tolerated and immunogenic in a placebo-controlled double-blind phase I trial in healthy adults. Clin. Vaccine Immunol. 18:2118–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lawes M., Maloy S. 1995. MudSacI, a transposon with strong selectable and counterselectable markers: use for rapid mapping of chromosomal mutations in Salmonella typhimurium. J. Bacteriol. 177:1383–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levine M. 1990. Vaccines against enterotoxigenic Escherichia coli infections, p. 649–660 In Levine M. M., Woodrow C. G. (ed.), New generation vaccines. Marcel Dekker, Inc., New York, NY [Google Scholar]
- 15. McKenzie R., et al. 2006. Comparative safety and immunogenicity of two attenuated enterotoxigenic Escherichia coli vaccine strains in healthy adults. Infect. Immun. 74:994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McVeigh A., et al. 2000. IS1414, an Escherichia coli insertion sequence with a heat-stable enterotoxin gene embedded in a transposase-like gene. Infect. Immun. 68:5710–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moseley S. L., Hardy J. W., Hug M. I., Echeverria P., Falkow S. 1983. Isolation and nucleotide sequence determination of a gene encoding a heat-stable enterotoxin of Escherichia coli. Infect. Immun. 39:1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nataro J. P., Kaper J. B. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oyofo B. A., et al. 2001. Toxins and colonization factor antigens of enterotoxigenic Escherichia coli among residents of Jakarta, Indonesia. Am. J. Trop. Med. Hyg. 65:120–124 [DOI] [PubMed] [Google Scholar]
- 20. Perez-Casal J., Swartley J. S., Scott J. R. 1990. Gene encoding the major subunit of CS1 pili of human enterotoxigenic Escherichia coli. Infect. Immun. 58:3594–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peruski L. F., Jr., et al. 1999. Phenotypic diversity of enterotoxigenic Escherichia coli strains from a community-based study of pediatric diarrhea in periurban Egypt. J. Clin. Microbiol. 37:2974–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qadri F., et al. 2000. Prevalence of toxin types and colonization factors in enterotoxigenic Escherichia coli isolated during a 2-year period from diarrheal patients in Bangladesh. J. Clin. Microbiol. 38:27–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qadri F., Svennerholm A. M., Faruque A. S., Sack R. B. 2005. Enterotoxigenic Escherichia coli in developing countries: epidemiology, microbiology, clinical features, treatment, and prevention. Clin. Microbiol. Rev. 18:465–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rappuoli R., Pizza M., Douce G., Dougan G. 1999. Structure and mucosal adjuvanticity of cholera and Escherichia coli heat-labile enterotoxins. Immunol. Today 20:493–500 [DOI] [PubMed] [Google Scholar]
- 25. Sambrook J., Russell D. W. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Savarino S. J., et al. 1996. Enteroaggregative Escherichia coli heat-stable enterotoxin is not restricted to enteroaggregative E. coli. J. Infect. Dis. 173:1019–1022 [DOI] [PubMed] [Google Scholar]
- 27. Scott J. R., Wakefield J. C., Russell P. W., Orndorff P. E., Froehlich B. J. 1992. CooB is required for assembly but not transport of CS1 pilin. Mol. Microbiol. 6:293–300 [DOI] [PubMed] [Google Scholar]
- 28. Shaheen H. I., et al. 2003. Phenotypic diversity of enterotoxigenic Escherichia coli (ETEC) isolated from cases of travelers' diarrhea in Kenya. Int. J. Infect. Dis. 7:35–38 [DOI] [PubMed] [Google Scholar]
- 29. Shaheen H. I., et al. 2004. Phenotypic profiles of enterotoxigenic Escherichia coli associated with early childhood diarrhea in rural Egypt. J. Clin. Microbiol. 42:5588–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stieglitz H., et al. 1988. Cloning, sequencing, and expression in Ficoll-generated minicells of an Escherichia coli heat-stable enterotoxin gene. Plasmid 20:42–53 [DOI] [PubMed] [Google Scholar]
- 31. Turner A. K., et al. 2006. Construction and phase I clinical evaluation of the safety and immunogenicity of a candidate enterotoxigenic Escherichia coli vaccine strain expressing colonization factor antigen CFA/I. Infect. Immun. 74:1062–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Turner A. K., Terry T. D., Sack D. A., Londono-Arcila P., Darsley M. J. 2001. Construction and characterization of genetically defined aro omp mutants of enterotoxigenic Escherichia coli and preliminary studies of safety and immunogenicity in humans. Infect. Immun. 69:4969–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walker R. I., Steele D., Aguado T. 2007. Analysis of strategies to successfully vaccinate infants in developing countries against enterotoxigenic E. coli (ETEC) disease. Vaccine 25:2545–2566 [DOI] [PubMed] [Google Scholar]
- 34. Wiedermann G., Kollaritsch H., Kundi M., Svennerholm A. M., Bjare U. 2000. Double-blind, randomized, placebo controlled pilot study evaluating efficacy and reactogenicity of an oral ETEC B-subunit-inactivated whole-cell vaccine against travelers' diarrhea (preliminary report). J. Travel Med. 7:27–29 [DOI] [PubMed] [Google Scholar]
- 35. Wolf M. K. 1997. Occurrence, distribution, and associations of O and H serogroups, colonization factor antigens, and toxins of enterotoxigenic Escherichia coli. Clin. Microbiol. Rev. 10:569–584 [DOI] [PMC free article] [PubMed] [Google Scholar]