

Abstract
The present study investigated the pharmacokinetics of meropenem and biapenem in bile and estimated their pharmacodynamic target attainment at the site. Meropenem (0.5 g) or biapenem (0.3 g) was administered to surgery patients (n = 8 for each drug). Venous blood samples and hepatobiliary tract bile samples were obtained at the end of infusion (0.5 h) and for up to 5 h thereafter. Drug concentrations in plasma and bile were analyzed pharmacokinetically and used for a Monte Carlo simulation to predict the probability of attaining the pharmacodynamic target (40% of the time above the MIC). Both drugs penetrated similarly into bile, with mean bile/plasma ratios of 0.24 to 0.25 (maximum drug concentration) and 0.30 to 0.38 (area under the drug concentration-time curve). The usual regimens of meropenem (0.5 g every 8 h [q8h]) and biapenem (0.3 g q8h) (0.5-h infusions) achieved similar target attainment probabilities in bile (≥90%) against Escherichia coli, Klebsiella pneumoniae, and Enterobacter cloacae isolates. However, against Pseudomonas aeruginosa isolates, meropenem at 1 g q8h and biapenem at 0.6 g q8h were required for values of 80.7% and 71.9%, respectively. The biliary pharmacodynamic-based breakpoint (the highest MIC at which the target attainment probability in bile was ≥90%) was 1 mg/liter for 0.5 g q8h and 2 mg/liter for 1 g q8h for meropenem and 0.5 mg/liter for 0.3 g q8h and 1 mg/liter for 0.6 g q8h for biapenem. These results help to define the clinical pharmacokinetics of the two carbapenems in bile while also helping to rationalize and optimize the dosing regimens for biliary tract infections based on site-specific pharmacodynamic target attainment.
INTRODUCTION
Meropenem and biapenem are therapeutic options for the treatment of biliary tract infections, including bacterial cholangitis and cholecystitis, and also for antibacterial prophylaxis in biliary tract surgery and endoscopic retrograde cholangiopancreatography (5, 6, 9, 15, 21).
Antibiotics act at the site of infection, and their ability to reach the site is a primary determinant of their efficacy. Therefore, it is important to understand the pharmacokinetics of meropenem and biapenem in bile in order to improve their efficacy for the treatment of biliary tract infections. However, little is known about how well both carbapenems penetrate into bile. There have been a few reports measuring meropenem concentrations in human bile (3, 7); however, these reports did not fully characterize its biliary pharmacokinetics. There have been no earlier reports on the biliary pharmacokinetics of biapenem, and other pharmacokinetic information is limited, because the drug is used clinically only in Asia and is under investigation in other countries (1).
Both carbapenems exert a time-dependent activity, and their antibacterial and therapeutic effects correlate with the exposure time that drug concentrations remain above the MIC for the bacterium (TMIC), particularly at the site of action. For both drugs, the TMIC target required for bactericidal effects is considered to be 40% of the dosing interval (27). Therefore, in order to define effective doses and to optimize the treatment regimen for biliary tract infections, it is important to evaluate the pharmacodynamic exposure in bile by predicting the probability that the TMIC target is attained at a certain MIC (the probability of target attainment [PTA]) and determining the expected population PTA for a specific population of bacteria (the cumulative fraction of response [CFR]) (17).
The objectives of the present study were to fully describe the human biliary pharmacokinetics of meropenem and biapenem and to estimate their site-specific pharmacodynamic target attainment. It is clinically significant to compare the pharmacokinetic and pharmacodynamic characteristics between the two carbapenems. In this study, a noncompartmental analysis was performed to estimate the rate and extent of drug penetration from the systemic circulation into bile. Population modeling was employed to estimate the compartmental pharmacokinetic parameters and their variability. The population model estimates were then used for a pharmacodynamic Monte Carlo simulation to predict the PTA values in bile for drug regimens and their CFRs against MIC distributions for clinical isolates of common bacteria that cause biliary tract infections, namely, Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, and Pseudomonas aeruginosa (5, 15).
MATERIALS AND METHODS
Study design and subject population.
This study was a prospective and open-label study of meropenem and biapenem conducted at Hiroshima University Hospital. The study protocol and informed consent form were in compliance with the Declaration of Helsinki and were reviewed and approved by the ethics committee of the institution.
Patients undergoing percutaneous transhepatic biliary drainage after hepatobiliary-pancreatic surgery were included in this study. The inclusion criteria were as follows: patients of both sexes older than 20 years, patients who were amenable to antibacterial prophylaxis for postoperative infections, and patients willing and able to provide their written informed consent. Any patients who were pregnant or who were hypersensitive to β-lactams were excluded.
Drug administration and sample collection.
Meropenem (0.5 g) or biapenem (0.3 g) was administered prophylactically by a 0.5-h intravenous infusion. Venous blood samples were obtained 0.5 h, 1 h, 1.5 h, 2.5 h, and 5.5 h after the start of the infusion. Intrahepatic bile samples were obtained from the hepatobiliary tract through a percutaneous transhepatic biliary drain 0.5 h, 0.75 h, 1 h, 1.25 h, 1.5 h, 2 h, 2.5 h, 3.5 h, 4.5 h, and 5.5 h after the start of the infusion. All samples were placed immediately in polypropylene tubes on ice and centrifuged at 3,000 × g for 10 min at 4°C. The plasma and supernatant bile were then removed, stabilized with an equal volume of 1-mol/liter 3-morpholino-propanesulfonic acid buffer (pH 7.0), and stored at −40°C within 0.5 h after sampling. All samples were assayed within 3 days of collection.
Meropenem assay.
The total concentrations of meropenem in plasma and bile were determined using high-performance liquid chromatography as reported previously (14). Plasma or bile samples (400 μl) were transferred to an ultrafiltration device (Nanosep 10K; Pall Corporation, Port Washington, NY). The device was centrifuged, and 20 μl of the filtered solution was injected onto a chromatograph with a reversed-phase column (Symmetry C18; 5 μm × 4.6 mm × 150 mm; Waters Corporation, Milford, MA) and detected by measuring the UV absorbance at 300 nm. A mixture of 10 mM sodium phosphate buffer (pH 7.4) and acetonitrile (100:10 [vol/vol]) was used as the mobile phase, at a flow rate of 1 ml/min. The lower limits of quantification were 0.05 mg/liter (plasma) and 0.1 mg/liter (bile), and both calibration curves were linear up to 100 mg/liter. For intra- and interday assays, the precision was 0.43% to 7.17% (plasma) or 0.69% to 10.8% (bile), and the accuracy was 97.7% to 106% (plasma) or 99.7% to 105% (bile).
Biapenem assay.
The total concentrations of biapenem in plasma and bile were determined using high-performance liquid chromatography as reported previously (14). The same assay methods as those for meropenem were used, except that the mobile phase was 100 mM sodium acetate buffer (pH 4.6) and acetonitrile (197:3 [vol/vol]). The lower limits of quantification were 0.04 mg/liter (plasma) and 0.1 mg/liter (bile), and both calibration curves were linear up to 100 mg/liter. For intra- and interday assays, the precision was 0.15% to 7.31% (plasma) or 1.51% to 9.19% (bile), and the accuracy was 7.3% to 118% (plasma) or 99.5% to 109% (bile).
Noncompartmental pharmacokinetic analysis.
For each drug, the area under the drug concentration-time curve (AUC) and the mean residence time (MRT) were calculated based on the trapezoidal rule, using the MULTI program (26). The AUC from 0 h to infinity (AUC0–∞) was estimated as the actual area (AUC0-5.5) plus the extrapolated area (AUC5.5–∞ = C5.5/λZ, where C5.5 is the drug concentration at 5.5 h and λZ is the terminal slope on a loge scale). The total clearance (CLtotal) was calculated as dose/AUC0–∞, and the volume of distribution at steady state (Vsteady state) was calculated as CLtotal × MRT. The Cmax was defined as the observed maximum drug concentration, and Tmax was the time to Cmax.
Population pharmacokinetic modeling.
For each drug, the concentration data for plasma (C1) and bile (C3) were fitted simultaneously to a multicompartment model with zero-order infusion (Rin) and first-order elimination and transfer processes (Fig. 1). In the modeling using the NONMEM program (version 7.1.0; Icon Development Solutions, Ellicott, MD), the fixed-effects parameters were volume of distribution of the central compartment (V1), volume of distribution of the peripheral compartment (V2), volume of distribution of the biliary compartment (V3), clearance from the central compartment (CL1), clearance from the biliary compartment (CL4), intercompartmental (central-peripheral) clearance (Q2), and intercompartmental (central-biliary) clearance (Q3). The interindividual variability was modeled exponentially, as follows: θi = θ × exp(ηi), where θi is the fixed-effects parameter for the ith subject, θ is the mean value for the fixed-effects parameter in the population, and η is a random interindividual variable which is normally distributed, with a mean of zero and a variance of ω2. The residual (intraindividual) variability was modeled with a proportional error model, as follows: Cobs, ij = Cpred, ij × (1 + εij), where Cobs, ij and Cpred, ij are the jth observed and predicted concentrations, respectively, for the ith subject and ε is a random intraindividual error which is normally distributed, with a mean of zero and a variance of σ2.
Fig. 1.
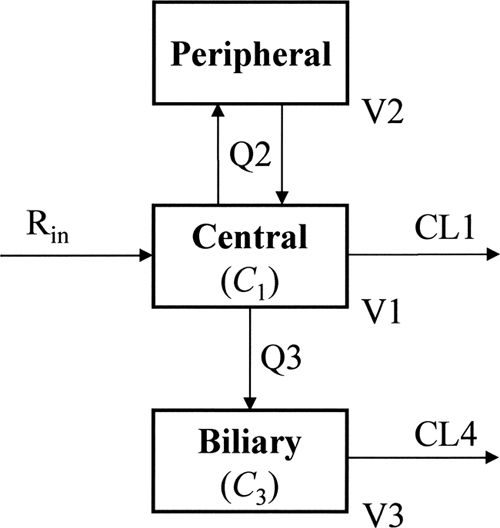
Multicompartment pharmacokinetic model for meropenem and biapenem. C1 and C3, drug concentrations in the central and biliary compartments (mg/liter); Rin, drug infusion rate (mg/h); V1, V2, and V3, volumes of distribution of the central, peripheral, and biliary compartments (liters); CL1 and CL4, clearances from the central and biliary compartments (liters/h); Q2 and Q3, intercompartmental clearances (liters/h).
A resampling technique called the bootstrap method was performed to check the validity and reliability of the population pharmacokinetic model that was developed. The program Wings for NONMEM (version 703; N. H. Holford, University of Auckland, Auckland, New Zealand) was used to create resampled new data sets. The 95% confidence intervals of the pharmacokinetic parameters estimated from 1,000 bootstrap replicates (2.5 to 97.5 percentiles of 1,000 estimates for V1, V2, V3, CL1, Q2, Q3, and CL4) were compared with the estimates from the developed model.
Pharmacodynamic Monte Carlo simulation.
A 10,000-subject Monte Carlo simulation was conducted to evaluate the pharmacodynamic exposure (TMIC) in plasma and bile. For each drug regimen (0.5-h infusion every 8 h [q8h]), the following process was iterated from the 1st to the 10,000th subject, using Crystal Ball software (version 2000; Oracle Corporation, Redwood Shores, CA) (12, 13, 20, 22). A set of fixed-effects parameters (θi values) (V1, V2, V3, CL1, Q2, Q3, and CL4) was randomly generated according to each mean estimate (θ) and interindividual variance (ω) of the population pharmacokinetic model that was developed. Using the set of seven θi values, the drug concentration-time curves (48 to 72 h after the start of the regimen) were created. For bile, the total concentration was employed because bile contains water, bile salts, mucus, and pigments but not protein; for plasma, the free fraction concentration was employed using protein binding values (2.4% for meropenem [10] and 3.0% for biapenem [11]). Next, the time point at which the drug concentration coincided with a specific MIC (0.031 to 64 mg/liter) was determined, and the percentages of a 24-h period that the drug concentrations remained above the MIC (free fraction %TMIC in plasma and %TMIC in bile) were finally calculated.
Determination of PTA and CFR.
Based on the pharmacodynamic Monte Carlo simulation, the PTA (%) in plasma and bile was determined as the fraction that achieved at least 40%TMIC (27) for each of 10,000 estimates. A probability of 90% was regarded as a clinically acceptable criterion, and the breakpoint (4, 12, 22) based on the biliary pharmacodynamic target attainment was defined as the highest MIC at which the PTA in bile was ≥90%.
The PTA at a specific MIC was then multiplied by the fraction of the clinical isolate population in each MIC category, and the sum of individual products was determined as the CFR (%). The MIC distribution data for meropenem and biapenem (Table 1) were derived from nationwide susceptibility surveillance data in Japan, including data from the study institution (25).
Table 1.
MIC distributions for meropenem and biapenem against clinical isolates of Escherichia coli (n = 141), Klebsiella pneumoniae (n = 129), Enterobacter cloacae (n = 92), and Pseudomonas aeruginosa (n = 322)
| Drug and bacterium | Fraction of isolates in each MIC category (mg/liter) |
MIC50 (mg/liter) | MIC90 (mg/liter) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ≤0.031 | 0.063 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 32 | ≥64 | |||
| Meropenem | ||||||||||||||
| E. coli | 0.979 | 0.021 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.031 | 0.031 |
| K. pneumoniae | 0.969 | 0.023 | 0.008 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.031 | 0.031 |
| E. cloacae | 0.359 | 0.402 | 0.152 | 0.054 | 0.022 | 0 | 0 | 0.011 | 0 | 0 | 0 | 0 | 0.063 | 0.125 |
| P. aeruginosa | 0 | 0.009 | 0.068 | 0.152 | 0.236 | 0.149 | 0.102 | 0.071 | 0.109 | 0.062 | 0.009 | 0.031 | 1 | 16 |
| Biapenem | ||||||||||||||
| E. coli | 0.447 | 0.482 | 0.057 | 0 | 0.014 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.063 | 0.063 |
| K. pneumoniae | 0.078 | 0.287 | 0.209 | 0.147 | 0.217 | 0.062 | 0 | 0 | 0 | 0 | 0 | 0 | 0.125 | 0.5 |
| E. cloacae | 0.043 | 0.217 | 0.370 | 0.250 | 0.120 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.125 | 0.5 |
| P. aeruginosa | 0 | 0.006 | 0.009 | 0.130 | 0.376 | 0.165 | 0.047 | 0.040 | 0.078 | 0.096 | 0.028 | 0.025 | 0.5 | 16 |
Statistical analysis.
The Mann-Whitney U test was performed using SPSS software (version 15.0J; IBM Japan, Tokyo, Japan) to determine differences between the meropenem and biapenem groups. P values of <0.05 were considered statistically significant.
RESULTS
Patients.
Eight patients were included in each group. For meropenem, the patient characteristics were as follows: five males and three females; age, 77.1 ± 5.2 years (mean ± standard deviation [SD]); body weight, 54.4 ± 11.5 kg; creatinine clearance, 71.9 ± 19.0 ml/min; and alanine aminotransferase level, 35.9 ± 18.0 IU/liter. For biapenem, the characteristics were as follows: four males and four females; age, 68.9 ± 13.1 years; body weight, 52.7 ± 13.7 kg; creatinine clearance, 78.9 ± 26.7 ml/min; and alanine aminotransferase level, 30.8 ± 13.0 IU/liter. The differences in demographic data between the two groups were not statistically significant.
Pharmacokinetics.
The noncompartmental pharmacokinetic parameters are summarized in Table 2. The proportions of the extrapolated AUC5.5–∞ to AUC0–∞ were all small (mean, 3.9% to 7.9%). For meropenem, the AUC0–∞ in bile was 21.07 ± 8.82 mg-h/liter, and its bile/plasma ratio was 0.38 ± 0.15. The Cmax in bile was 9.72 ± 3.54 mg/liter, and its bile/plasma ratio was 0.25 ± 0.10. For biapenem, AUC0–∞ and Cmax values were significantly lower than those for meropenem. However, the bile/plasma AUC0–∞ ratio, the bile/plasma Cmax ratio, and the Tmax in bile (mean, 1.31 h) were not significantly different between the two drugs.
Table 2.
Noncompartmental pharmacokinetic parameters for meropenem (0.5 g) and biapenem (0.3 g) after a 0.5-h infusion (mean ± SD; n = 8)
| Sample type and parameter | Value |
|
|---|---|---|
| Meropenem | Biapenem | |
| Plasma | ||
| AUC0–∞ (mg-h/liter) | 54.90 ± 7.49 | 38.93 ± 8.77 |
| AUC5.5–∞ (% of AUC0–∞) | 3.9 ± 1.7 | 6.4 ± 1.8 |
| Cmax (mg/liter) | 39.71 ± 7.84 | 20.84 ± 4.26 |
| Tmax (h) | 0.5 | 0.5 |
| CLtotal (liters/h) | 9.27 ± 1.38 | 8.01 ± 1.58 |
| Vsteady state (liters) | 13.47 ± 2.10 | 13.40 ± 2.16 |
| Bile | ||
| AUC0–∞ (mg-h/liter) | 21.07 ± 8.82 | 11.46 ± 3.75 |
| AUC5.5–∞ (% of AUC0–∞) | 6.2 ± 3.7 | 7.9 ± 2.8 |
| Cmax (mg/liter) | 9.72 ± 3.54 | 4.82 ± 1.21 |
| Tmax (h) | 1.31 ± 0.44 | 1.31 ± 0.22 |
| Both sample types | ||
| AUC0–∞, bile/AUC0–∞, plasma | 0.38 ± 0.15 | 0.30 ± 0.11 |
| Cmax, bile/Cmax, plasma | 0.25 ± 0.10 | 0.24 ± 0.08 |
The estimates of population pharmacokinetic parameters in the multicompartment model are listed in Table 3. Some parameter estimates of the interindividual variability (η) were finally fixed as zero in the NONMEM modeling because they were negligibly small (<0.0001); the other parameter estimates were all in the range of the 95% confidence intervals obtained using the bootstrap method. No high correlations (r > 0.7) were found among the seven population pharmacokinetic parameters for the two drugs. Figure 2 shows a good correspondence between the observed drug concentration (y axis) and the individual predicted concentration based on the population pharmacokinetic model parameters (x axis). The best-fit regression lines were as follows: y = 1.048x − 0.474 (r = 0.986) (plasma) and y = 1.193x − 0.914 (r = 0.980) (bile) for meropenem and y = 1.020x − 0.018 (r = 0.973) (plasma) and y = 1.187x − 0.466 (r = 0.982) (bile) for biapenem. For both drugs, the simulation curves drawn using the mean fixed-effects parameters fitted well with all of the mean measurements in plasma and bile (Fig. 3).
Table 3.
Estimates of population pharmacokinetic parameters for meropenem and biapenem in the multicompartment model (Fig. 1)
| Parameter | Meropenem |
Biapenem |
||
|---|---|---|---|---|
| Estimate | 95% Confidence interval (bootstrap method) | Estimate | 95% Confidence interval (bootstrap method) | |
| Fixed-effects parameters | ||||
| θV1 (liters) | 8.92 | 4.92–13.03 | 11.4 | 9.2–14.3 |
| θV2 (liters) | 6.00 | 4.30–7.46 | 4.80 | 1.86–7.30 |
| θV3 (liters) | 0.0404 | 0.0092–0.0504 | 0.0286 | 0.0027–0.0307 |
| θCL1 (liters/h) | 8.98 | 8.40–10.00 | 7.18 | 6.02–8.59 |
| θQ2 (liters/h) | 5.74 | 0.31–9.95 | 3.87 | 2.02–7.42 |
| θQ3 (liters/h) | 0.0201 | 0.0002–0.0279 | 0.0167 | 0.0033–0.0278 |
| θCL4 (liters/h) | 0.0422 | 0.0015–0.0531 | 0.0581 | 0.0013–0.0663 |
| Interindividual variability parameters | ||||
| ηV1 | 0.127 | 0.004–0.373 | 0.186 | 0.009–0.287 |
| ηV2 | 0 | None | 0.517 | 0.017–0.887 |
| ηV3 | 0.677 | 0.062–0.761 | 0.713 | 0.382–2.817 |
| ηCL1 | 0.114 | 0.001–0.132 | 0.202 | 0.002–0.308 |
| ηQ2 | 0 | None | 0 | None |
| ηQ3 | 0 | None | 0 | None |
| ηCL4 | 0.359 | 0.017–0.405 | 0 | None |
| Residual variability parameter | ||||
| ε | 0.0702 | 0.0047–0.0975 | 0.0738 | 0.0045–0.1042 |
Fig. 2.

Scatterplots of the observed concentrations of meropenem and biapenem in plasma (40 samples) and bile (80 samples) versus the individual predicted concentrations based on the population pharmacokinetic model parameters for each drug (Table 3). The lines represent unity (y = x).
Fig. 3.

Observed concentrations (mean ± SD; n = 8) and simulation curves for plasma and bile after 0.5-h infusions of meropenem (0.5 g) and biapenem (0.3 g). The simulation curves were generated using the mean fixed-effects parameters for each drug (Table 3).
Pharmacodynamic target attainment.
The PTA values for plasma (free fraction) and bile at specific MICs are shown in Fig. 4. For each drug regimen, the PTA value was lower for bile than for plasma. The highest MIC at which the PTA in bile was ≥90% was 1 mg/liter for 0.5 g q8h and 2 mg/liter for 1 g q8h (0.5-h infusions) for meropenem and 0.5 mg/liter for 0.3 g q8h and 1 mg/liter for 0.6 g q8h (0.5-h infusions) for biapenem.
Fig. 4.

Probabilities of target (40%TMIC) attainment in plasma (free fraction) and bile at specific MICs, using different meropenem and biapenem regimens. The gray lines represent 90% probability.
The CFR values in plasma (free fraction) and bile against four bacterial populations are given in Table 4. Although the CFR value in bile was lower than or equal to that in plasma for each drug regimen, meropenem at 0.5 g q8h and biapenem at 0.3 g q8h (0.5-h infusions) achieved 96.4% to 100% CFR values in bile against E. coli, K. pneumoniae, and E. cloacae isolate populations. However, against a P. aeruginosa isolate population, even meropenem at 1 g q8h and biapenem at 0.6 g q8h did not achieve a CFR value of ≥90% in bile.
Table 4.
Cumulative fractions of response in plasma (free fraction) and bile against clinical isolate populations, obtained using different meropenem and biapenem regimens (0.5-h infusions q8h)
| Bacterium | Cumulative fraction of response (%) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Meropenem at 0.5 g q8h |
Meropenem at 1 g q8h |
Biapenem at 0.3 g q8h |
Biapenem at 0.6 g q8h |
|||||
| Plasma | Bile | Plasma | Bile | Plasma | Bile | Plasma | Bile | |
| E. coli | 100 | 100 | 100 | 100 | 100 | 99.9 | 100 | 100 |
| K. pneumoniae | 100 | 100 | 100 | 100 | 99.6 | 96.4 | 100 | 98.5 |
| E. cloacae | 99.6 | 99.3 | 100 | 99.8 | 100 | 99.1 | 100 | 99.6 |
| P. aeruginosa | 76.4 | 71.1 | 85.3 | 80.7 | 74.6 | 62.2 | 81.5 | 71.9 |
DISCUSSION
The present study investigated the biliary pharmacokinetics of meropenem and biapenem in surgery patients and revealed that both carbapenems penetrated similarly into bile. This study also estimated the site-specific pharmacodynamic target attainment of the drugs and demonstrated that meropenem at 0.5 g q8h and biapenem at 0.3 g q8h (0.5-h infusions) achieved similar CFR values in bile (≥90%) against most of the tested bacterial populations. The biliary pharmacodynamic-based breakpoint was 1 mg/liter for 0.5 g q8h and 2 mg/liter for 1 g q8h for meropenem and 0.5 mg/liter for 0.3 g q8h and 1 mg/liter for 0.6 g q8h for biapenem.
Granai et al. (7) examined meropenem bile concentrations in patients with normal (unobstructed) bile ducts during endoscopic retrograde cholangiography, and based on a regression analysis, they showed the average concentration to be about 17 mg/liter at 1.5 h, 14 mg/liter at 2.5 h, and 11 mg/liter at 3.5 h after the start of 0.25- to 0.33-h infusions of 1 g meropenem. Condon et al. (3) also measured meropenem bile concentrations in patients during intra-abdominal surgery, and they showed the concentration (n = 3 or 4) to be 1.5 to 16.1 mg/liter at 0.5 to 1.5 h, 1.9 to 13.1 mg/liter at 1.5 to 3 h, and 1.3 to 18.9 mg/liter at 3 to 5 h after the start of a 0.5-h infusion of 1 g meropenem. An exact comparison between these earlier results and the current results was difficult because the dose and sampling times were different. Nevertheless, the half values of these concentrations (1 g meropenem) were in the range of the corresponding measurements (0.5 g meropenem) (Fig. 3): 1.07 to 2.32 mg/liter at 0.5 h, 2.94 to 11.79 mg/liter at 1.5 h, 1.16 to 10.01 mg/liter at 2.5 h, and 0.75 to 9.73 mg/liter at 3.5 h after the start of 0.5-h infusions.
Due to the fact that they did not observe the Cmax in bile, the earlier studies (3, 7) could not estimate the bile/plasma ratios of AUC0–∞ and Cmax for meropenem (commonly used as indices of drug penetration). However, because we used an adequate sampling scheme, the present study allowed us to observe the Cmax and to accurately estimate the AUC0–∞ with a small degree of extrapolation (Table 2, AUC5.5–∞). Meropenem and biapenem exhibited similar bile/plasma ratios for AUC0–∞ and Cmax (Table 2), although the absolute concentration values were lower for biapenem (0.3 g) than for meropenem (0.5 g), in concordance with the dose administered (Fig. 3).
The mean Tmax in bile for both drugs was also identically 1.31 h, that is, there was a 0.81-h delay after the Tmax in plasma. None of the earlier studies estimated the Tmax in bile for either meropenem or biapenem; however, Graziani et al. (8) showed that the Tmax of imipenem in bile (n = 12) was 1 to 2.5 h after the start of the 0.5-h infusion. These findings suggest that carbapenems with similar physical and chemical properties (such as lipophilicity and protein binding) exhibit similar pharmacokinetics in bile. As Graziani et al. speculated for imipenem (8), we believe that meropenem and biapenem are distributed to bile through liver cells by a simple diffusion mechanism rather than being concentrated by an active transport mechanism. The molecular weights (383.5 for meropenem and 350.4 for biapenem) are below the proposed threshold value of 450 to 500 which was reported to be needed when other β-lactams were taken up by active transport systems in liver cells (24).
In the NONMEM modeling, a multicompartment model (Fig. 1) was finally chosen because it described the concentration data better than the tested enterohepatic circulation models (16, 19), likely because neither meropenem nor biapenem is absorbed from the gastrointestinal tract (27). The mean noncompartmental pharmacokinetic parameters CLtotal (Table 2) (9.27 liters/h for meropenem and 8.01 liters/h for biapenem) and Vsteady state (13.47 liters for meropenem and 13.40 liters for biapenem) were consistent with the corresponding fixed-effects parameters for clearance (θCL1 + θCL4; 9.02 liters/h for meropenem and 7.24 liters/h for biapenem) (Table 3) and volume of distribution (θV1 + θV2 + θV3; 14.96 liters for meropenem and 16.23 liters for biapenem). The individual predicted concentrations in both plasma and bile were in good agreement with the observed drug concentrations (Fig. 2), and the simulation curves obtained using the mean fixed-effects parameters had a good fit with all of the mean measurements (Fig. 3). In addition, the estimates for θ, η, and ε were in the range of the 95% confidence intervals estimated from 1,000 bootstrap replicates. These results indicate the validity and reliability of the developed population pharmacokinetic models for both drugs, and therefore the models are considered to have a good predictive performance.
By using the developed model parameters, this study then predicted the concentrations of meropenem and biapenem to estimate the pharmacodynamic target attainment for the different dosing regimens. Since the total drug concentrations were lower in bile than in plasma (Fig. 3), the PTA values were lower for bile than for plasma (free fraction; 97.6% for meropenem and 97.0% for biapenem) (Fig. 4). This finding emphasizes that the free drug concentration in plasma does not always represent the drug level in a biological fluid. Therefore, it is important to directly use the drug level at a specific site of action in order to accurately estimate the site-specific pharmacodynamic target attainment. The results of the Monte Carlo simulation based on bile concentration (Table 4) suggest that 0.5-h infusions of meropenem at 0.5 g q8h and biapenem at 0.3 g q8h (the usual dosages) are similarly sufficient for the treatment of biliary tract infections due to E. coli, K. pneumoniae, and E. cloacae, which account for about 70% of the causative pathogens (21, 23). Since the Sanford guide recommends meropenem at 1 g q8h for the treatment of life-threatening conditions of cholangitis, cholecystitis, and biliary sepsis (6), the current results suggest that meropenem at 1 g q8h and biapenem at 0.6 g q8h should be recommended, especially when there is strong suspicion of a biliary tract infection with a carbapenem-resistant bacterium such as P. aeruginosa (Table 1) (MIC50 = 0.5 to 1 mg/liter and MIC90 = 16 mg/liter) and/or the MIC for the patient's causative pathogen is determined to be >1 mg/liter for meropenem and >0.5 mg/liter for biapenem. Nevertheless, the recommended regimens of meropenem (1 g q8h) and biapenem (0.6 g q8h) would still not always be sufficient for these therapeutic situations. Prolonging the infusion time of the regimens may be beneficial, although such prolongation may be unacceptable due to the increase in the patient's burden and the medical workload for the clinical staff (11, 13).
In an effort to provide an overview of the site-specific pharmacodynamic target attainment of meropenem and biapenem, the CFR was assessed against only four Gram-negative bacterial populations in Japan. However, the PTA versus MIC profiles (Fig. 4) should be applicable for other possible bacteria, regardless of the susceptibility pattern in other regions.
This study has some limitations. First, the drug concentrations were observed in intrahepatic bile from normal (unobstructed) hepatobiliary tracts of uninfected patients. However, patients with biliary tract infections often have obstructions of the bile ducts. Granai et al. (7) showed by a regression analysis that meropenem bile concentrations in the obstructed patient group were lower than those in the unobstructed patient group until 2.9 h postinfusion, and thereafter, this concentration relationship was reversed. A biliary obstruction therefore may affect the pharmacokinetics of meropenem and biapenem in bile. Second, infected patients often have renal dysfunction, which causes lower clearance, leading to the persistence of higher meropenem and biapenem concentrations in plasma (the TMIC will consequently be longer in bile) because both drugs are eliminated predominantly renally, with a 24-h urinary recovery of 60% to 70% (2, 18). The mean estimate for clearance from the central compartment was higher for the study patients (Table 3) (8.98 liters/h for meropenem and 7.18 liters/h for biapenem) than for infected patients with a creatinine clearance of 30 ml/min (4.75 liters/h for meropenem [10] and 4.24 liters/h for biapenem [11]). The creatinine clearance of the study patients was within a restricted range (71.9 ± 19.0 ml/min for meropenem and 78.9 ± 26.7 ml/min for biapenem), and there was only a small number of patients (n = 8 for each drug). Therefore, the pharmacokinetic and pharmacodynamic results of this study must be confirmed in a larger number of patients with various infections, with and without obstructed bile ducts.
In conclusion, intravenous meropenem and biapenem penetrated similarly into bile, with a mean bile/plasma Cmax ratio of 0.24 to 0.25 (a mean Tmax delay of 0.81 h after the end of the 0.5-h infusion) and a mean bile/plasma AUC0–∞ ratio of 0.30 to 0.38. The usual regimens of meropenem (0.5 g q8h) and biapenem (0.3 g q8h) (0.5-h infusions) achieved similar CFR values (≥90%) in bile against E. coli, K. pneumoniae, and E. cloacae isolate populations. However, against a P. aeruginosa isolate population, meropenem at 1 g q8h and biapenem at 0.6 g q8h were required for values of 80.7% and 71.9%, respectively. The biliary pharmacodynamic-based breakpoint was 1 mg/liter for 0.5 g q8h and 2 mg/liter for 1 g q8h for meropenem and 0.5 mg/liter for 0.3 g q8h and 1 mg/liter for 0.6 g q8h for biapenem. These results help to define the clinical pharmacokinetics of the two carbapenems in bile, while also helping to rationalize and optimize the dosing regimens for biliary tract infections based on the site-specific pharmacodynamic target attainment. Further studies are needed to both confirm the present findings and clarify their therapeutic significance.
ACKNOWLEDGMENT
No financial support was received for this work.
Footnotes
Published ahead of print on 26 September 2011.
REFERENCES
- 1. Bassetti M., Nicolini L., Esposito S., Righi E., Viscoli C. 2009. Current status of newer carbapenems. Curr. Med. Chem. 16: 564–575 [DOI] [PubMed] [Google Scholar]
- 2. Burman L. A., Nilsson-Ehle I., Hutchison M., Haworth S. J., Norrby S. R. 1991. Pharmacokinetics of meropenem and its metabolite ICI 213,689 in healthy subjects with known renal metabolism of imipenem. J. Antimicrob. Chemother. 27: 219–224 [DOI] [PubMed] [Google Scholar]
- 3. Condon R. E., et al. 1997. Penetration of meropenem in plasma and abdominal tissues from patients undergoing intraabdominal surgery. Clin. Infect. Dis. 24(Suppl. 2): S181–S183 [DOI] [PubMed] [Google Scholar]
- 4. Frei C. R., Wiederhold N. P., Burgess D. S. 2008. Antimicrobial breakpoints for gram-negative aerobic bacteria based on pharmacokinetic-pharmacodynamic models with Monte Carlo simulation. J. Antimicrob. Chemother. 61: 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. George J., Baillie J. 2005. Contemporary management of biliary tract infections. Curr. Infect. Dis. Rep. 7: 108–114 [DOI] [PubMed] [Google Scholar]
- 6. Gilbert D. N., Moellering R. C., Eliopoulos G. M., Chambers H. F., Saag M. S. 2010. The Sanford guide to antimicrobial therapy, 40th ed, p. 15 Antimicrobial Therapy, Sperryville, VA [Google Scholar]
- 7. Granai F., Smart H. L., Triger D. R. 1992. A study of the penetration of meropenem into bile using endoscopic retrograde cholangiography. J. Antimicrob. Chemother. 29: 711–718 [DOI] [PubMed] [Google Scholar]
- 8. Graziani A. L., Gibson G. A., MacGregor R. R. 1987. Biliary excretion of imipenem-cilastatin in hospitalized patients. Antimicrob. Agents Chemother. 31: 1718–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hara K., et al. 1999. Clinical evaluation of biapenem in various infectious diseases. Jpn. J. Antibiot. 52: 629–660 [PubMed] [Google Scholar]
- 10. Ikawa K., et al. 2010. Pharmacokinetic-pharmacodynamic target attainment analysis of meropenem in Japanese adult patients. J. Infect. Chemother. 16: 25–32 [DOI] [PubMed] [Google Scholar]
- 11. Ikawa K., et al. 2008. Pharmacokinetic-pharmacodynamic target attainment analysis of biapenem in adult patients: a dosing strategy. Chemotherapy 54: 386–394 [DOI] [PubMed] [Google Scholar]
- 12. Ikawa K., et al. 2009. Pharmacokinetics and pharmacodynamics of biapenem in human cerebrospinal fluid. Int. J. Antimicrob. Agents 34: 184–185 [DOI] [PubMed] [Google Scholar]
- 13. Ikawa K., et al. 2009. Penetrability of intravenous biapenem into the peritoneal fluid of laparotomy patients and the peritoneal pharmacodynamics against Gram-negative bacteria. J. Appl. Res. 9: 107–112 [Google Scholar]
- 14. Kameda K., et al. 2010. HPLC method for measuring meropenem and biapenem concentrations in human peritoneal fluid and bile: application to comparative pharmacokinetic investigations. J. Chromatogr. Sci. 48: 406–411 [DOI] [PubMed] [Google Scholar]
- 15. Karachalios G., Charalabopoulos K. 2002. Biliary excretion of antimicrobial drugs. Chemotherapy 48: 280–297 [DOI] [PubMed] [Google Scholar]
- 16. Lehr T., et al. 2009. A quantitative enterohepatic circulation model: development and evaluation with tesofensine and meloxicam. Clin. Pharmacokinet. 48: 529–542 [DOI] [PubMed] [Google Scholar]
- 17. Mouton J. W., Dudley M. N., Cars O., Derendorf H., Drusano G. L. 2005. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J. Antimicrob. Chemother. 55: 601–607 [DOI] [PubMed] [Google Scholar]
- 18. Nakashima M., et al. 1993. Phase 1 study of L-627, biapenem, a new parenteral carbapenem antibiotic. Int. J. Clin. Pharmacol. Ther. Toxicol. 31: 70–76 [PubMed] [Google Scholar]
- 19. Roberts M. S., Magnusson B. M., Burczynski F. J., Weiss M. 2002. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin. Pharmacokinet. 41: 751–790 [DOI] [PubMed] [Google Scholar]
- 20. Soga Y., et al. 2010. Peritoneal pharmacokinetics and pharmacodynamic target attainment of meropenem in patients undergoing abdominal surgery. J. Chemother. 22: 98–102 [DOI] [PubMed] [Google Scholar]
- 21. Tanaka A., et al. 2007. Antimicrobial therapy for acute cholangitis: Tokyo guidelines. J. Hepatobiliary Pancreat. Surg. 14: 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsumura R., et al. 2008. The pharmacokinetics and pharmacodynamics of meropenem in the cerebrospinal fluid of neurosurgical patients. J. Chemother. 20: 615–621 [DOI] [PubMed] [Google Scholar]
- 23. Westphal J. F., Brogard J. M. 1999. Biliary tract infections: a guide to drug treatment. Drugs 57: 81–91 [DOI] [PubMed] [Google Scholar]
- 24. Westphal J. F., et al. 1997. Assessment of biliary excretion of piperacillin-tazobactam in humans. Antimicrob. Agents Chemother. 41: 1636–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamaguchi K., et al. 2007. Nationwide surveillance of parenteral antibiotics containing meropenem activities against clinically isolated strains in 2006. Jpn. J. Antibiot. 60: 344–377 [PubMed] [Google Scholar]
- 26. Yamaoka K., Tanigawara Y., Nakagawa T., Uno T. 1981. A pharmacokinetic analysis program (multi) for microcomputer. J. Pharmacobiodyn. 4: 879–885 [DOI] [PubMed] [Google Scholar]
- 27. Zhanel G. G., et al. 2007. Comparative review of the carbapenems. Drugs 67: 1027–1052 [DOI] [PubMed] [Google Scholar]