

Abstract
Apoptosis and inhibition of host gene expression are often associated with virus infections. Many viral polypeptides modulate apoptosis by direct interaction with highly conserved apoptotic pathways. Some viruses induce apoptosis during late stages of the infection cycle, while others inhibit apoptosis to facilitate replication or maintain persistent infection. In previous work, we showed that Chilo iridescent virus (CIV) or CIV virion protein extract induces apoptosis in spruce budworm and cotton boll weevil cell cultures. Here, we characterize the product of a CIV gene (iridovirus serine/threonine kinase; istk) with signature sequences for S/T kinase and ATP binding. ISTK appears to belong to the superfamily, vaccinia-related kinases (VRKs). The istk gene was expressed in Pichia pastoris vectors. Purified ISTK (48 kDa) exhibited S/T kinase activity. Treatment with ISTK induced apoptosis in budworm cells. A 35-kDa cleavage product of ISTK retaining key signature sequences was identified during purification. Pichia-expressed 35-kDa polypeptide, designated iridoptin, induced apoptosis and inhibition of host protein synthesis in budworm and boll weevil cells. A mutation in the ATP-binding site eliminated both kinase and apoptosis activity of iridoptin, suggesting that kinase activity is essential for induction of apoptosis. Analysis with custom antibody confirmed that ISTK is a structural component of CIV particles. This is the first demonstration of a viral kinase inducing apoptosis in any virus-host system and the first identification of a factor inducing apoptosis or host protein shutoff for the family Iridoviridae.
INTRODUCTION
Apoptosis and host protein shutoff are often associated with virus infection. Several viral gene products regulate apoptosis by interacting directly with components of apoptotic pathways. Viruses have evolved ways to modulate apoptosis. Some viruses encode proteins that induce apoptosis late in the infection cycle, enabling dissemination of progeny particles. Viral proteins may also inhibit apoptosis. This results in either prolonging the survival of infected cells to maximize production of progeny virus or facilitating viral persistence. Host organisms have evolved ways to utilize apoptosis as a defense against viruses (16). Many viruses also inhibit protein synthesis in their hosts. Depending on the viral system, host shutoff may be advantageous to the virus toward freeing metabolic precursors and host synthetic machinery. Host shutoff may also reduce host defenses and thus enhance viral pathogenesis (38).
The family Iridoviridae (isometric cytoplasmic DNA viruses) (39) consists of several genera in vertebrate and invertebrate hosts. Recently, a member of the genus Iridovirus was associated with honey bee colony collapse disorder (5). Some members of the genus Ranavirus have been linked to global amphibian decline (11, 24). The mechanisms underlying these effects are not known, but it is intriguing that several members of the Iridoviridae induce or inhibit apoptosis (8, 9, 14, 18–20, 28, 34, 40). A Bcl-2-like protein and an inhibitor of apoptosis (IAP) block apoptosis in the Iridoviridae (21, 29). However, the genes or proteins responsible for induction of apoptosis have remained elusive. Earlier work in our laboratory showed that a virion protein extract from Chilo iridescent virus (CIV) induces apoptosis and host protein shutoff; viral gene expression is not necessary for apoptosis induction by this extract (9, 34). Kinase activity was detected in the CIV particle by Monnier and Devauchelle (33) and in CIV virion protein extract (CVPE) in our laboratory (34). Work with an iridovirus from vertebrates suggested that phosphorylation of eukaryotic initiation factor 2 (eIF2) by a viral component is probably responsible for inhibition of host gene expression in infected cells (7). However, the specific viral factor responsible for the induction of apoptosis or host protein shutoff in the Iridoviridae has not been identified.
In our search for viral genes with potential utility as plant-incorporated protectants against insect pests, we focused on insect iridovirus genes that shut down host protein synthesis or induce apoptosis. We partially sequenced the CIV genome and identified an open reading frame (ORF) with similarities to the B1R gene of vaccinia virus. Vaccinia B1R kinase was earlier suspected as having a role in host protein shutoff (2, 30) but has since been shown to allow cell survival toward completion of the viral replication cycle (36). Transcription work in our laboratory showed that the CIV B1R-like ORF was expressed as an early gene during viral replication (13). This ORF had signature sequences for a predicted serine/threonine kinase, and we designated the putative gene iridovirus serine/threonine kinase (istk). It was of interest to determine if the CIV istk gene was involved in apoptosis or host protein shutoff in insect cells. The complete genomic sequence of CIV was described by Jakob et al. (23).
We cloned and expressed the istk gene from CIV in the Pichia pastoris expression system. In this report, we show that the ISTK product induces apoptosis in insect cells upon external application and is a component of the CIV particle. The ISTK polypeptide appeared to be unstable under some laboratory conditions. We identified a 35-kDa cleavage product of ISTK. The 35-kDa polypeptide expressed in the Pichia system was designated iridoptin and was shown to be a potent inducer of apoptosis. In addition to inducing apoptosis, iridoptin shuts off host protein synthesis in insect cells, and both ISTK and iridoptin have kinase activity. Mutant iridoptin lacking kinase activity does not induce apoptosis. This is the first report showing that a viral protein kinase induces apoptosis and the first identification of a protein from the family Iridoviridae associated with apoptosis induction or host protein shutoff.
MATERIALS AND METHODS
Virus rearing and purification.
CIV was raised in larvae of the greater wax moth, Galleria mellonella, and purified by sucrose gradient centrifugation as described previously (17).
Virus was extracted by maceration in Tris-NaCl buffer (50 mM Tris-HCl, 150 μM NaCl, pH 7.4) using a Waring blender. The slurry was filtered through cheesecloth into Sorvall GSA centrifuge tubes to remove large particulate material. The supernatant was then transferred into Sorvall SS-34 tubes and centrifuged at 17,000 rpm in a Sorvall SS34 rotor for 30 min at 4°C. The pellet was resuspended overnight in Tris-NaCl buffer and again centrifuged in SS-34 tubes. After overnight resuspension of the second pellet, the virus was layered onto 10 to 60% sucrose (wt/vol; in Tris-NaCl) gradients and centrifuged for 2 h at 36,000 rpm in a Beckman SW27 or Sorvall Surespin 630 rotor at 4°C. The major CIV band was harvested, and bands representing empty particles or aggregates were avoided. Sucrose was removed by dilution and further differential centrifugation. The resuspended pellet was run on a second set of sucrose gradients, and virus bands were processed as described above. The final virus suspension was filtered through a series of 0.45- and 0.22-μm-pore-size Millipore GV filters. Purified virus was examined by transmission electron microscopy after staining with phosphotungstic acid; preparations were homogenous for intact CIV particles and devoid of aggregates or observable impurities.
Preparation of CIV virion protein extract.
CIV virion protein extract was prepared from purified virus using a modification of the method described by Cerutti and Deauvechelle (6). CIV was purified as described above (17), pelleted, and resuspended in CHAPS [3-(3-cholamidopropyl) dimethylammonio-1-propanesulfonate] extraction buffer (10 mM Tris-HCl, 10 mM CHAPS, and 1 M KCl, pH 7.4). CHAPS concentration was optimized based on yields of kinase activity levels, which were 80% higher with 10 mM CHAPS than with 1 mM CHAPS, with diminishing returns from 10 mM to 50 mM CHAPS (C. W. Henderson and S. L. Bilimoria, unpublished data). The suspension was then incubated at 30°C for 15 min, layered on a 6-ml cushion of 20% sucrose in Beckman SW-41 tubes, and centrifuged for 2 h at 36,000 rpm and 4°C. The supernatant above the cushion was collected and subjected to four cycles of ultrafiltration (YM-10 membrane; Amicon, Billerica, MA) against Tris-NaCl buffer (50 mM Tris-HCl, 150 μM NaCl, pH 7.4). This process removed residual sucrose and allowed concentration of the viral protein extract to a final volume of approximately 1 ml. The concentrate was filtered through 0.22-μm-pore-size Millipore GV filters and stored at −80°C. Mock extracts were prepared from uninfected G. mellonella larvae.
Cell cultures and virus.
IPRI-CF-124T (CF) cells (4) from the spruce budworm Choristoneura fumiferana and BRL-AG-3A (AG) cells (37) from the boll weevil Anthonomus grandis were cultured in Corning 25-cm2 flasks using Hink's TNM-FH medium supplemented with 10% fetal bovine serum (HyClone Laboratories) and incubated at 28°C. CF and AG cells were typically subcultured at 6-day intervals at a ratio of 1:10 (17). Chilo iridescent virus was obtained from James Kalmakoff (Dunedin, New Zealand) and reared in G. mellonella larvae as described previously (17).
Polyacrylamide gel electrophoresis and immunoblot analysis.
SDS-PAGE analysis was carried out using a standard protocol and as described previously (34). Protein concentration was determined by Bradford assay. Bands were detected by Coomassie blue staining, silver staining, or Western analysis using appropriate antibodies. For Western analysis, samples were wet transferred to 0.45-μm-pore-size nitrocellulose membranes; alternatively, iBlot Dry Blotting System (Invitrogen, CA) and 0.2-μm-pore-size polyvinylidene difluoride (PVDF) membranes were utilized. The membranes were blocked for 1 h with 5% (wt/vol) bovine serum albumin (BSA) in phosphate-buffered saline with 0.05% Tween 20 (PBS-T). Upon washing with PBS-T, the blots were incubated overnight at 4°C with primary mouse anti-His6 monoclonal antibody (Invitrogen, CA) diluted in PBS-T or with rabbit antiserum (1:500) against a custom-synthesized iridoptin polypeptide, I64-C77 (GenScript USA, Inc.). Alkaline phosphatase-labeled anti-mouse or anti-rabbit IgG (Invitrogen, CA) was added as appropriate after excess primary antibody was washed off. The blots were developed with nitroblue tetrazolium/5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP; Invitrogen) per the manufacturer's protocol.
PCR amplification of istk DNA sequence.
PCR primers were designed to amplify a 1.2-kbp region from the CIV genome that corresponds to the istk gene encoding the 411-amino-acid iridovirus serine-threonine kinase (ISTK). The forward and reverse primers synthesized for amplification were 5′ ATG GAT CTT AAA GAC GAA TTT ATT 3′ and 5′ GTT AAA AAT GTT ATG TAA TAG TTG ATA 3′, respectively. PCRs were performed in a final volume of 25 μl using 2.5 μl of 10× reaction buffer, 1.5 mM MgCl2, 200 μM each deoxynucleoside triphosphate (dNTP), 50 ng/ml template DNA, 1 μmol each of forward and reverse primer, and 2 units of Takara Taq polymerase. After an initial denaturing step at 94°C for 5 min, 30 cycles of denaturation, annealing, and elongation were performed successively at 94°C, 55°C, and 72°C, respectively, for 2 min each.
Cloning and expression in Pichia.
The CIV istk gene was PCR amplified, and the product was cloned in TOPO pCR 2.1 vector (Invitrogen, CA). The TOPO construct was used to transform Escherichia coli strain DH5α. Ampicillin (100 μg/ml) was used for selection in E. coli, and recombinants were detected by blue/white colony screening using 20 μg/ml 5-bromo-4-chloro-3-indolyl-β-d- galactopyranoside (X-Gal) and 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Selected clones were identified and sequenced using M13 reverse primers (Center for Biotechnology and Genomics, Texas Tech University [TTU]) to determine the orientation of the insert. The gene was then cloned into pPICZ C (Invitrogen, CA) and sequenced using AOX1 forward and reverse primers to confirm that the gene was in frame with the His6 tag at the C terminus. The selected clones were introduced into P. pastoris strain X33 and used for induction studies. Expression in Pichia was induced with methanol (1%, vol/vol), and fractions were collected every 24 h for 5 days. Pichia cells were lysed using breaking buffer (2% [vol/vol] Triton X-100, 1% [wt/vol] SDS, 100 mM NaCl, 10 mM Tris-Cl, pH 8.0, 1 mM EDTA, pH 8.0, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μM pepstatin A, and 0.6 μM leupeptin) and 0.5-mm glass beads. The iridoptin gene sequence was also cloned and expressed in Pichia using the procedure described above. Iridoptin utilized in the experiment shown in Fig. 3A was expressed in E. coli Rosetta(DE3)/pLysS cells (EMD Biosciences) per the manufacturer's protocol.
Fig. 3.

Immunoblot analysis of CIV polypeptides with custom antibody to the synthetic peptide I64-C77 from the iridoptin sequence. (A) CIV, 5 μg of purified CIV; IRI, 4 μg of iridoptin. (B) CIV, 6 μg of purified CIV; CVPE, 10 μg of viral protein extract. The numbers represent molecular mass in kDa. See Materials and Methods for details.
ProBond purification of expressed product.
Chromatography on resin columns was performed under native conditions using a ProBond Protein Purification System (Invitrogen, CA) per the manufacturer's protocol. The column was first rinsed with deionized/distilled water and then with four column volumes of binding buffer (0.5 M NaCl and 10 mM imidazole). Yeast cells were lysed as described above. The lysate was centrifuged at 3,000 × g for 10 min at 4°C, and the supernatant was collected. The clear cell lysate was then added to the resin and allowed to mix gently at 4°C for ∼30 min. The column was then treated with wash buffer (0.5 M NaCl and 20 mM imidazole) to remove nonspecifically bound proteins. The His-tagged protein was released with elution buffer (0.5 M NaCl and 250 mM imidazole). Eluant was collected as 1-ml fractions.
C-terminal sequencing.
C-terminal sequencing was conducted by the Macromolecular Structure, Sequencing and Synthesis Facility located in the Department of Biochemistry at Michigan State University using an Applied Biosystems Sequenator. The C-terminal residues of polypeptides were successively cleaved by carboxypeptidases yielding individual amino acids, which were then identified by high-performance liquid chromatography (HPLC).
MALDI-TOF MS.
Matrix-assisted laser desorption-ionization time of flight mass spectrometry (MALDI-TOF MS) was used to confirm identity of the 35-kDa polypeptide expressed in P. pastoris. Sequencing of the 35-kDa polypeptide was conducted by the Core Facility, Institute for Cellular and Molecular Biology, The University of Texas at Austin, using a MALDI-TOF mass spectrometer (PerSeptive Biosystems Voyager).
Synthesis of kinase-dead iridoptin.
Kinase-dead mutants were generated using an in vivo Pichia expression system. Iridoptin was point mutated from a 153 lysine (AAA) to a glutamine (CAA) codon using megaprimer PCR. CAA was chosen per codon usage for Pichia. Primer sequences were the following: forward, 5′-AGTATAGGTACCATGGATCTTAAAGACGAATTTAT-3′; reverse, 5′-CATACATCTAGAAAACCATTATGTGCTTTTTTAG-3′. The reverse primer which introduced the mutation was 5′-CGTCTTGGTAGTTACCAT-3′ (mutated base in italics). The mutated iridoptin sequence was then subcloned in frame with a C-terminal His6 tag into the pPICZ B Pichia expression vector using KpnI and XbaI restriction enzymes. Mutation at the desired sequence in iridoptin was confirmed by DNA sequencing. The expression vector was then transferred to Pichia (strain X33). Expression of mutant iridoptin was induced in Pichia and purified using His tag purification columns as described for normal iridoptin.
Kinase assays. (i) Calbiochem protein kinase assay kit.
A Calbiochem protein kinase assay kit was used to detect kinases belonging to the protein kinase C family, which include tyrosine and serine/threonine kinases. Purified (electroeluted) ISTK product, induced lysate, uninduced lysate, and CVPE were assayed for the presence of kinase activity. These kinases use ATP as the phosphate source to phosphorylate the synthetic peptide, which is detected by enzyme-linked immunosorbent assay (ELISA) using biotinylated monoclonal antibody specific to phosphorylated serine residues. Purified ISTK (10 ng) was incubated with ATP (30 μM) in kinase buffer (25 mM HEPES [pH 7], 1 mM MnCl2, and 5 mM MgCl2) for 15 min at 30°C. After a 15-min incubation at 30°C, the plate was treated three times with wash buffer (0.05% Tween in PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.5) and incubated with peroxidase-conjugated streptavidin (Calbiochem). Color was developed by addition of chromogen o-phenylenediamine to 3 μM, and the reaction was halted with stop solution (20% H3PO4). Absorbance was read at 492 nm with a Perkin-Elmer Victor microplate reader.
(ii) In vitro kinase assay of iridoptin.
Enzyme (iridoptin or mutant iridoptin) was added to protamine sulfate (1 mg/ml) as a substrate and [γ-32P]ATP in a final reaction volume of 25 μl in kinase reaction buffer (25 mM Tris-HCl, pH 8.0, 8 mM MgCl2, 100 μm cold ATP). The kinase reaction was carried out at 28°C for 30 min and run on SDS-PAGE gels.
Blebbing and TUNEL assays.
Blebbing and terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays were performed as described earlier (34). Cells were treated with iridoptin, actinomycin D, heat-inactivated iridoptin, or Rinaldini's balanced salt solution (RBSS; 0.5 M NaCl, 0.2 mM KCl, 5 mM sodium citrate, 0.04 mM NaH2PO4, 10 mM NaHCO3, 0.1% glucose) and observed for apoptotic blebbing by phase-contrast microscopy. Regression lines for dose-response analysis were fitted, and R2 values were calculated using Microsoft Excel 2003 software. In the TUNEL assay, dinucleotide terminal transferase was used to transfer biotin-labeled nucleotides to the ends of fragmented DNA. Horseradish peroxidase-labeled streptavidin was then bound to the biotin molecules. DNA fragments were then detected using peroxidase substrate (hydrogen peroxide) and chromogen (diaminobenzidine [DAB]).
Host protein shutoff.
An assay for translation inhibition was carried out using [35S]methionine precursor as described by Paul et al. (34). Bands were analyzed using Typhoon 9410 (Molecular Dynamics) phosphorimager software and plotted using Microsoft Excel 2003. Regression lines for dose-response analysis were fitted, and R2 values were calculated using Microsoft Excel 2003 software.
RESULTS
PCR, cloning, and temporal expression of the istk gene.
Partial sequencing of the CIV genome revealed an ORF for an iridovirus serine/threonine kinase (istk); this ORF was identical to ORF 389L described by Jakob et al. (23). Over the ∼100- to 400-amino-acid (aa) region, the derived ISTK polypeptide has 28% identity with the B1R product of vaccinia virus (28, 36) and is similar to other vaccinia-related kinases (VRKs) (25) from Tribolium castaneum (33% identity), Nasonia vitripennis (32% identity), Apis mellifera (29% identity), and several other mammalian and invertebrate species. The first 100 aa in ISTK were not found in any other VRK, and comparison of this 100-aa sequence with protein sequence databases did not detect significant similarities with other polypeptides or conserved domains. A high level of identity with other polypeptides was found in the conserved domain region containing the ATP-binding site and S/T kinase motif. PCR amplification of the istk gene sequence yielded a 1.2-kbp DNA fragment that corresponded to the predicted size for this gene. The gene was overexpressed in the Pichia system. Western analysis of lysates from induced yeast cells using His6 antibody detected the predicted 48-kDa product and a 17-kDa band (Fig. 1A, lane L).
Fig. 1.

(A) Western analysis of ISTK with anti-His6 antibody. ISTK was expressed in P. pastoris and analyzed as described in Materials and Methods. Lane M, molecular mass markers (kDa); lane L, Pichia lysate showing ISTK (48 kDa) and a 17-kDa polypeptide; lane P, purified eluate from nickel-affinity column after imidazole treatment; lane I, silver stain of the eluate in lane P showing a 35-kDa band. (B) Amino acid sequence of ISTK. The motifs for ATP binding and S/T kinase are underlined. The ISTK cleavage site resulting in the 35-kDa product (bold type) during downstream processing of ISTK is shown (▾). (C) Downstream processing of ISTK and derivation of active 35-kDa polypeptide from the ISTK gene product.
Kinase activity of ISTK.
Purified ISTK showed levels of kinase activity that were three times that in CIV virion protein extract (CVPE) and 28 times that in induced Pichia lysates expressing ISTK on a per microgram basis (Table 1 ). To demonstrate that the purified ISTK product belongs to the serine/threonine (S/T) class of kinases, inhibition studies with the antibiotic staurosporine were conducted. Staurosporine specifically inhibits S/T kinases. The kinase activity of ISTK was drastically inhibited by staurosporine. Heat inactivation resulted in a 4-fold decrease in kinase activity (Table 1).
Table 1.
Activity of ISTK and iridoptin
| Assay type and sample | Activity |
|---|---|
| ISTK protein kinase assay (OD490/μg of protein)a | |
| Purified ISTK | 1.97 ± 0.12 |
| ISTK + staurosporine (40 μg/ml) | 0.01 ± 0.0005 |
| CVPE | 0.62 ± 0.07 |
| Induced lysate | 0.07 ± 0.009 |
| Uninduced lysate | 0.01 ± 0.005 |
| Buffer | 0.0 ± 0.005 |
| In vitro iridoptin kinase assay (cpm/μg of total protein)b | |
| BSA | 111 ± 63 |
| Iridoptin | 1,004 ± 154 |
| Δ Iridoptin | 236 ± 19 |
| CVPE | 923 ± 49 |
Kinase assays for ISTK were carried out using a Calbiochem protein kinase assay kit. Results are means of three replicates. OD490, optical density at 490 nm.
Kinase assays for iridoptin were carried out as described in Materials and Methods. [γ-32P]ATP was used as the label, and protamine was used as the substrate. Samples were spotted onto phosphocellulose paper, and radioactivity was counted after excess label was rinsed off. Δ Iridoptin, heat-inactivated iridoptin. Results are means of duplicates.
ISTK induces apoptosis.
Induction of apoptosis by ISTK was confirmed using a TUNEL assay, which detects DNA fragmentation during apoptosis. IPRI-CF-124T (CF) cells were treated and processed for the TUNEL assay after incubation at 28°C for 18 h. Positive controls (actinomycin D; 4 μg/ml) induced nuclear diaminobenzidine (DAB) signal indicative of apoptosis in 95% of the CF cell population. ISTK treatment (7 μg/ml) resulted in nuclear DAB signals in 58% (standard deviation [SD], 4.2%) of the cell population. Cells treated with buffer were used as negative controls and showed nuclear DAB signal in 2% of the cell population (SD, 1%).
Generation of cleaved 35-kDa product from ISTK.
Lysates from Pichia constructs induced for ISTK expression revealed the predicted 48-kDa band and an additional 17-kDa product by Western analysis with anti-His6 antibody (Fig. 1A, lane L). This lysate was then put through nickel columns, and polypeptides with His6 were eluted with imidazole. Western analysis of this eluant with anti-His6 antibody did not detect any ISTK (Fig. 1A, lane P). Interestingly, silver staining of SDS-PAGE gels loaded with the eluant revealed a 35-kDa band (Fig. 1A, lane I). Nickel column eluate from yeast lysates with a β-galactosidase control construct yielded the predicted 116-kDa polypeptide and not the 35-kDa product visualized after expression of the istk construct. These data suggest that the 35-kDa band is a cleavage product of the ISTK gene.
C-terminal sequencing and MALDI-TOF analysis of the 35-kDa cleavage product.
To determine the exact cleavage point for the derivation of the 35-kDa polypeptide from CIV ISTK, we sequenced the C terminus following carboxypeptidase Y digest. The data revealed that the C-terminal residues of the 35-kDa polypeptide were Asn-Gly. This amino acid pair was detected at four points in the DNA-derived ISTK sequence (Fig. 1B). MALDI-TOF analysis of the cleaved 35-kDa polypeptide revealed signatures for both S/T kinase and the ATP-binding site, indicating derivation through cleavage at the Gly-Thr site (G303-T304) (Fig. 1B). Figure 1C summarizes the cleavage pathway for the derivation of the 35-kDa polypeptide from ISTK (48 kDa).
Cloning and expression of recombinant 35-kDa polypeptide.
C-terminal analysis determined the ISTK cleavage point yielding the 35-kDa polypeptide and therefore the truncated DNA sequence coding for the derived polypeptide. This sequence was utilized to clone and express the 35-kDa polypeptide. SDS-PAGE analysis of induced Pichia lysates showed that Pichia constructs containing this sequence yielded a 35-kDa band (Fig. 2A, lane P). Western analysis with anti-His6 antibody also revealed a 35-kDa band in induced yeast lysates and upon purification on nickel columns (Fig. 2B, lanes L and P, respectively). MALDI-TOF analysis of the Pichia-expressed product confirmed identity of the 35-kDa polypeptide (data not shown). The Pichia-expressed 35-kDa polypeptide is designated iridoptin.
Fig. 2.
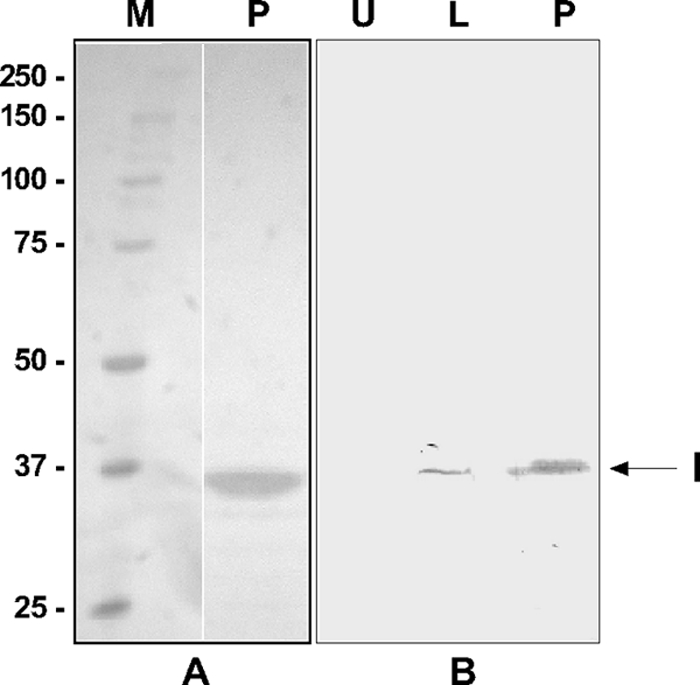
SDS-PAGE analysis of His-tagged iridoptin expressed in the Pichia system and purified on Ni-affinity columns. (A) Silver stain analysis. Lane M, molecular mass (kDa) markers; lane P, iridoptin purified on ProBond columns. Note the 35-kDa band. (B) Western analysis with anti-His6 antibody. Lane U, uninduced yeast lysate; lane L, induced lysate showing 35-kDa iridoptin band (I); lane P, iridoptin purified on ProBond affinity columns.
ISTK and the 35-kDa polypeptide are components of CIV particles and virion protein extracts.
Custom antibody to a 14-amino-acid synthetic peptide from iridoptin (I64-C77) was utilized to show that ISTK is present in purified CIV particles. This antibody is highly reactive to purified iridoptin and detected a 48-kDa band as well as a minor 100-kDa band in CIV particles (Fig. 3A). The antibody preparation was also utilized to detect ISTK and its derivatives in CIV virion protein extracts, where Western analysis revealed the 48-kDa ISTK polypeptide as well as 35-kDa and 37-kDa polypeptides (Fig. 3B).
Induction of apoptotic blebbing by iridoptin.
CF cells were treated in triplicate with iridoptin, actinomycin D, heat-inactivated iridoptin, or Rinaldini's balanced salt solution (RBSS) and observed for apoptotic blebbing (Fig. 4). CF cells treated with iridoptin (10 μg/ml) showed blebbing in 92% (SD, 4.7%) of the population (Fig. 4A). Positive controls (actinomycin D, 4 μg/ml) yielded 98% blebbing (SD, 1.1%) (Fig. 4B). Cells treated with heat-inactivated iridoptin (10 μg/ml at 65°C for 30 min) showed 5% blebbing (Fig. 4C). Mock treatment of cells with RBSS yielded 7% blebbing (Fig. 4D). Triplicate dose-response analyses of iridoptin in CF cells indicated that the amount required to induce blebbing in 50% of the cell population was 0.1 μg/ml (Fig. 5A). Blebbing was also observed in AG cells treated with iridoptin. Iridoptin at 20 μg/ml induced 87% (SD, 2.3%) blebbing in AG cell populations. Actinomycin D (4 μg/ml) induced 99% (SD, 1.5%) blebbing. Heat-inactivated iridoptin (20 μg/ml at 65°C for 30 min) induced 7% (SD, 1.7%) blebbing. RBSS induced negligible blebbing (2.7%; SD, 1.9%). Triplicate dose-response analyses of iridoptin in AG cells indicated that the amount required to induce blebbing in 50% of the cell population was 0.5 μg/ml (Fig. 5B).
Fig. 4.

Iridoptin induces apoptotic blebbing in spruce budworm (CF) cells. Cells in Nunc 60-well trays (5.6 × 103 cells per well in 7 μl of medium) were treated with iridoptin (10 μg/ml) (A), actinomycin D (4 μg/ml) (B), heat-inactivated iridoptin (10 μg/ml) (C), or RBSS (D). The cells were then incubated at 28°C for 24 h and observed for blebbing by phase-contrast microscopy. Scale bar, 10 μm.
Fig. 5.

Dose-response analysis of iridoptin-induced apoptosis in budworm and boll weevil cells. (A) Budworm cells (CF) were treated with dilutions of iridoptin starting at 20 μg/ml (final concentration) and incubated at 28°C for 24 h. Cells were observed for apoptotic blebs with a phase-contrast microscope. Percent blebbing was determined from approximately 250 cells per field. Linear regression line and R2 value were generated using Microsoft Excel 2003. The linear regression line was generated with the following equation: y = 24x + 75, where x is the log of final iridoptin concentration in μg/ml and y is the percentage of cell population with blebs. The R2 value for the fitted line was 0.95. The highest dilution of toxin-produced blebbing in 50% of the cell population was approximately 0.1 μg/ml. (B) Boll weevil (AG) cells. The line was generated by the following equation: y = 17.5x + 55, where x is the log of final iridoptin concentration in μg/ml and y is the percentage of cell population with blebs. The R2 value for the fitted line was 0.94. The final concentration of iridoptin required to induce blebbing in 50% of the cell population was 0.5 μg/ml. Assays for both cell lines were performed in triplicate.
Iridoptin-induced apoptosis detected with TUNEL assay.
Fig. 6 shows that AG cells treated with iridoptin (10 μg/ml at 28°C for 18 h) exhibited nuclear diaminobenzidine (DAB) signal indicative of apoptosis. Nearly 94% (SD, 2.1%) of the cell population stained positive. Positive actinomycin D controls (4 μg/ml) exhibited nuclear DAB signal in approximately 96% (SD, 0.8%) of the population. Cells mock-treated with RBSS or treated with heat-inactivated iridoptin (65°C for 30 min) (data not shown) produced negligible DAB signal. Similar TUNEL assay results were obtained with CF cells (data not shown).
Fig. 6.

Confirmation of iridoptin-induced apoptosis in boll weevil (AG) cells by TUNEL assay. AG cells were grown on glass slides in CellStar six-well plates (5.6 × 103 cells per well in 2.5 ml of medium). Cells were treated as described in Materials and Methods and viewed by phase-contrast microscopy. Brown staining of the nuclei indicated positive DAB signal. Assays were performed in duplicate. Treatments were as follows: 10 μg/ml iridoptin (A), 20 μg/ml iridoptin (B), 4 μg/ml actinomycin D (C), and mock treatment (D). Magnification, ×200. Insets show detail of nuclear DAB signal (magnification, ×400).
Host protein shutoff by iridoptin.
Pulse-labeling experiments with [35S]methionine showed that treatment with iridoptin inhibited host protein synthesis in CF and AG cells. Inhibition by iridoptin was 93% of positive controls for CF cells (Fig. 7A) (63% with iridoptin at 7 μg/ml versus 68% with actinomycin D at 4 μg/ml) and 61% of positive controls for AG cells (Fig. 7B). Inhibition was greater than 60% by 3 h posttreatment. Heat-inactivated iridoptin caused only 12 to 15% host protein shutoff, a 70 to 80% reduction. Controls consisting of cells treated with RBSS did not induce host protein shutoff (data not shown). Dose-response analysis of iridoptin in AG cells indicated that the amounts required to induce 50% and 90% inhibition of protein synthesis were 10 μg/ml and 23 μg/ml, respectively (Fig. 7C).
Fig. 7.

Iridoptin induces inhibition of protein synthesis in insect cells. Cells were pulsed with [35S]methionine 3 h after treatment with iridoptin (7 μg/ml), actinomycin D (ActD; 4 μg/ml), or heat-inactivated iridoptin (ΔIridoptin; 7 μg/ml). Polypeptides were separated on 10% SDS-polyacrylamide gels, and radioactive protein was visualized with a Typhoon 9410 (Molecular Dynamics) phosphorimager. Inhibition values are percentages of transmittance against mock lanes. (A) Budworm (CF) cells. (B) Boll weevil (AG) cells. (C) Dose-response analysis of iridoptin-induced host shutoff in boll weevil (AG) cells. A linear regression line was generated using Microsoft Excel 2003 and the following equation: y = 98.4x − 44.9, where x is the log of final iridoptin concentration in μg/ml and y is percent inhibition of host protein synthesis. The R2 value for the fitted line was 0.99. The final concentrations of iridoptin required to inhibit 50% and 90% host protein synthesis were 10 μg/ml and 23 μg/ml, respectively. Data are means of triplicate determinations.
Kinase-dead iridoptin does not induce apoptotic blebbing.
Kinase-dead iridoptin was generated in the Pichia system as described in Materials and Methods. CF cells cultured in 96-well plates were treated with either mutant iridoptin or normal iridoptin. Additionally, cells were either mock treated with RBSS (negative control) or treated with actinomycin D (positive control). Figure 8A shows wild-type and mutant iridoptin bands. Figures 8B and D show that kinase activity in mutant iridoptin was extremely low compared to that of normal iridoptin. Figure 8C shows a control for protamine loading. Figure 8E shows that mutant iridoptin induced blebbing in 5% of the cell population, compared to 70% for normal iridoptin. Actinomycin D-treated cells had 90% blebbing. Mock-treated cells did not show significant blebbing. These results are consistent with the data obtained for kinase activity of iridoptin (Fig. 8B and D).
Fig. 8.

Kinase activity and apoptosis induction by wild-type and mutant iridoptin. (A) Wild-type (WT) and mutant (Mut) iridoptin were separately purified from induced Pichia lysates using nickel affinity columns (ProBond), separated by SDS-PAGE, and stained with Coomassie blue. A single 35-kDa band was observed for each preparation. (B) Kinase activity. Assays were carried out as described in Materials and Methods. [γ-32P]ATP was used as the label, and protamine (5 kDa) was used as the substrate. (C) Substrate loading controls. (D) Band intensity mean integrated pixel density units of [32P]protamine substrate in plate B quantified with NIH ImageJ software. (E) CF cells treated with 9 μg/ml of iridoptin or kinase-dead mutant (m; K153Q) iridoptin and observed for apoptotic blebbing at 24 h. ActD, actinomycin D; Mock, RBSS. Percentages are means of three fields each comprising 150 cells.
DISCUSSION
We show that a virion-associated serine/threonine kinase from Chilo iridescent virus (CIV S/T kinase; ISTK) induces apoptosis in two insect cell lines. ISTK is a member of the superfamily, vaccinia-related kinases (VRKs) (26) but has an additional and unique 100-aa N-terminal region of unknown function (per NCBI, October 2011). We previously reported apoptosis-inducing and insecticidal activity by CIV and CVPEs (3, 34). During purification, ISTK was cleaved to a 35-kDa polypeptide, which we expressed using the Pichia pastoris system. The Pichia-expressed 35-kDa polypeptide was designated iridoptin.
Our data suggest that ISTK is a component of the CIV particle. Immunoblots using antibody to iridoptin indicated that ISTK is associated with virions twice purified on sucrose gradients. ISTK as well as 35-kDa and 37-kDa polypeptides was detected in Western analyses of CVPE using custom peptide antibody to I64-C77 of the ISTK sequence. However, we have not established whether iridoptin is simply a product of our purification protocol or whether ISTK is cleaved to iridoptin or a similar polypeptide during viral infection or storage. MALDI-TOF analysis suggested that cleavage of ISTK to iridoptin probably occurs via aminopeptidase N (APN). APNs have been shown to function as receptors for coronaviruses and other viruses of humans (31, 42) in addition to Bacillus thuringiensis toxin in the lepidopteran midgut (12, 27). Detailed cleavage studies on the occurrence and potential role of ISTK derivatives during natural infection and storage are needed.
A CHAPS-based protocol (6, 34) for CVPE was applied to freshly washed, purified virus and optimized for kinase activity yields (34; Henderson and Bilimoria, unpublished). These conditions suggest that ISTK is strongly associated with hydrophobic regions of the virus particle. Moreover, in a recent proteomic analysis of Wiseana iridescent virus (WIV, or invertebrate iridescent virus 9 [IIV-9]), a serine-threonine kinase (ORF 023L) was associated with viral particles (41).
Both ISTK and iridoptin are potent inducers of apoptosis and have high protein kinase activity. Kinase-dead mutants of iridoptin failed to induce apoptosis, suggesting that kinase activity is necessary for apoptosis induction. Whereas ISTK and iridoptin induced apoptosis in the absence of other virion components, our data do not preclude the role of additional factors in CVPE or virions toward apoptosis induction.
Protein kinases play major roles in various aspects of viral life cycles, including infectivity, uncoating, transcription, and replication (22). Essbauer and Ahne (14) found that apoptosis in epizootic hematopoietic necrosis virus (genus Ranavirus, family Iridoviridae) involves activation of protein kinases, especially double-stranded RNA-dependent protein kinase (PKR). PKR inhibits translation initiation through phosphorylation of the alpha subunit of initiation factor eIF2α and is an important element in the mode of action for interferon. However, iridoptin probably activates the apoptotic pathway by a different mechanism since insects lack interferon. Viral infections activate stress-related, mitogen-activated protein kinase pathways, leading to increased phosphorylation of cellular transcription factors and resulting in the induction of apoptosis (16). Previous work showed that CIV virions and CVPE require endocytosis and the Jun N-terminal kinase (JNK) signaling pathway to induce apoptosis in insect cells (9). Ongoing research in our laboratory suggests that inhibition of endocytosis with bafilomycin A1 (1 μM) for 30 min prior to iridoptin treatment (10 μg/ml) reduces apoptosis induction in CF cells from 90% to 8%. Pretreatment of CF cells with a JNK inhibitor (SP600125; 25 nM) lowered iridoptin-induced apoptotic blebbing from 90% to 10% (N. S. Chitnis and S. L. Bilimoria, unpublished data). These results suggest that iridoptin induces apoptosis in CF cells via endocytosis and the JNK pathway, as described for CVPE and CIV by Chitnis et al. (9). The role of JNK in apoptosis induction by ISTK, CVPE, and CIV suggests a feasible role for ISTK-induced apoptosis during the viral infection cycle. VRKs have been shown to play essential roles in regulating cell signaling, apoptosis, cellular stress response, nuclear envelope dynamics, and chromatin modifications (26).
Several mechanisms for direct inhibition of host gene expression have been described for viruses of eukaryotes; PKR or inactivation of host translational factors, such as eIF2, is utilized by a number of RNA viruses (38). Herpesviruses utilize the virion-associated, viral host shutoff (VHS) protein to inhibit protein synthesis (25). A number of competing mechanisms, including the virion surface tubule protein, have been proposed for host shutoff in poxvirus infections (1, 15, 32, 35).
Our data allow for a cautious analysis of the relationship between the apoptotic and host shutoff effects of iridoptin. Paul et al. (34) showed that when CVPE is utilized as inducer, apoptosis but not host shutoff is inhibited by caspase inhibitors. This could be due to the presence of factors other than ISTK or iridoptin in CVPE. However, distinct or multiple mechanisms for apoptosis and host shutoff are not precluded. The 50% effective concentration (EC50) of CVPE for host shutoff in CF cells is 700 times higher than that for apoptotic blebbing (34). Whereas we did not test the host shutoff effect of ISTK, the EC50 ratio of shutoff over apoptosis induction for iridoptin was 20 in AG cells and ∼70 in CF cells. These data suggest that the mechanisms for apoptosis and host shutoff may be distinct.
In considering the significance of ISTK or iridoptin at the cellular level, it should be noted that these polypeptides induce apoptosis without requiring viral gene expression. Excessively high levels of CIV induce massive apoptosis in cell culture within several hours of viral adsorption. Viral levels sufficient to infect 95% of the cell population in the first cycle of replication readily induce apoptosis when viral gene expression is blocked with cycloheximide (9). Expression of the immediate-early iap gene in CIV inhibits or delays apoptosis (21). Insects probably adapted to undergo apoptosis upon infection with CIV in order to control viral spread and pathogenesis in the host. However, it is likely that CIV adapted to this limitation with early IAP expression so as to inhibit apoptosis toward premature termination of the viral cycle.
The role of apoptosis in insect virus infections at the organismal level has been examined with baculovirus models. Recent research shows that apoptosis is a powerful response to baculovirus infections in larvae, where it reduces viral replication, infectivity, and the ability of the virus to disseminate (10). Thus, further research on the significance of ISTK or iridoptin in iridovirus infections of larvae is warranted.
This report documents the first demonstration of a viral kinase inducing apoptosis in any virus-host system and the first identification of a factor inducing apoptosis or host protein shutoff for the family Iridoviridae. This system provides a significant model for basic studies on the mechanism of apoptosis and host shutoff in the family Iridoviridae. Moreover, iridoptin has insecticidal activity (S. L. Bilimoria, U.S. patent application 2009/0069239A1). Chilo iridescent virus is hampered as a biopesticide by its low infectivity and transmission rate (39). However, the application of iridoptin as an insecticide or plant-incorporated protectant is feasible.
ACKNOWLEDGEMENTS
This work was supported in part by grants to S.L.B. from the Texas Advanced Research Program (003644-0148), Texas Advanced Technology Program (003644-0046), Research Enhancement Fund (Texas Higher Education Coordinating Board/Texas Tech University), and Research Development Fund (THECB/TTU). Support was also provided by the Office of the Provost and the Department of Biological Sciences at TTU. C.W.H. was supported by an ARCS Foundation Scholarship. P.V.T. was supported by a Howard Hughes Undergraduate Fellowship. N.S.C., E.R.P., and S.M.D. were supported by TTU Summer Research Awards. S.G. was supported by a Preston and Ima Smith Graduate Scholarship (TTU), and K.S.V. was supported by a Study Abroad Competitive Scholarship (TTU International Affairs). A.R.M. was supported by a TTU Honors College Undergraduate Research Fellowship.
We thank Susan San Francisco for services provided by the Center for Biotechnology and Genomics. We also thank Hua-jian Yao and Jonathan Townsend for technical assistance.
Footnotes
Published ahead of print on 12 October 2011.
REFERENCES
- 1. Bablanian R., Banerjee A. K. 1986. Poly(riboadenylic acid) preferentially inhibits in vitro translation of cellular mRNAs compared with vaccinia virus mRNAs: possible role in vaccinia virus cytopathology. Proc. Natl. Acad. Sci. U. S. A. 83:1290–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Banham A. H., Leader D. P., Smith G. L. 1993. Phosphorylation of ribosomal proteins by the vaccinia virus B1R protein kinase. FEBS Lett. 321:27–31 [DOI] [PubMed] [Google Scholar]
- 3. Bilimoria S. L. March 2001. Use of viral proteins for controlling the cotton boll weevil and other insect pests. U.S. patent 6200561
- 4. Bilimoria S. L., Sohi S. S. 1977. Development of an attached strain from a continuous insect cell line. In Vitro 13:461–466 [DOI] [PubMed] [Google Scholar]
- 5. Bromenshenk J. J., et al. 2010. Iridovirus and microsporidian linked to honey bee colony decline. PLoS One 5:e13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cerutti M., Devauchelle G. 1979. Cell fusion induced by invertebrate virus. Arch. Virol. 61:149–155 [DOI] [PubMed] [Google Scholar]
- 7. Chinchar V. G., Dholakia J. N. 1989. Frog virus 3-induced translational shut-off: activation of an eIF-2 kinase in virus-infected cells. Virus Res. 14:207–223 [DOI] [PubMed] [Google Scholar]
- 8. Chinchar V. G., Bryan L., Wang J., Long S., Chinchar G. D. 2003. Induction of apoptosis in frog virus 3-infected cells. Virology 306:303–312 [DOI] [PubMed] [Google Scholar]
- 9. Chitnis N. S., D’Costa S. M., Paul E. R., Bilimoria S. L. 2008. Modulation of iridovirus-induced apoptosis by endocytosis, early expression, JNK, and apical caspase. Virology 370:333–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clem R. J. 2005. The role of apoptosis in defense against baculovirus infection in insects. Curr. Top. Microbiol. Immunol. 289:113–129 [DOI] [PubMed] [Google Scholar]
- 11. Collins J. P., Storfer A. 2003. Global amphibian declines: sorting the hypotheses. Divers. Distrib. 9:89–98 [Google Scholar]
- 12. Crava C. M., et al. 2010. Study of the aminopeptidase N gene family in the lepidopterans Ostrinia nubilalis (Hübner) and Bombyx mori (L.): sequences, mapping and expression. Insect Biochem. Mol. Biol. 40:506–515 [DOI] [PubMed] [Google Scholar]
- 13. D’Costa S., Yao H., Bilimoria S. L. 2004. Transcriptional mapping in Chilo iridescent virus infections. Arch. Virol. 149:723–742 [DOI] [PubMed] [Google Scholar]
- 14. Essbauer S., Ahne W. 2002. The epizootic haematopoietic necrosis virus (Iridoviridae) induces apoptosis in vitro. J. Vet. Med. B Infect. Dis. Vet. Public Health 49:25–30 [DOI] [PubMed] [Google Scholar]
- 15. Gierman T. M., Frederickson R. M., Sonenberg N., Pickup D. J. 1992. The eukaryotic translation initiation factor 4E is not modified during the course of vaccinia virus replication. Virology 188:934–937 [DOI] [PubMed] [Google Scholar]
- 16. Hay S., Kannourakis G. 2002. A time to kill: viral manipulation of the cell death program. J. Gen. Virol. 83:1547–1564 [DOI] [PubMed] [Google Scholar]
- 17. Henderson C. W., Johnson C., Lodhi S. A., Bilimoria S. L. 2001. Replication of Chilo iridescent virus in the cotton boll weevil, Anthonomus grandis, and development of an infectivity assay. Arch. Virol. 146:767–775 [DOI] [PubMed] [Google Scholar]
- 18. Hu G. B., Cong R. S., Fan T. J., Mei X. G. 2004. Induction of apoptosis in a flounder gill cell line by lymphocystis disease virus infection. J. Fish Dis. 27:657–662 [DOI] [PubMed] [Google Scholar]
- 19. Huang Y. H., Huang X. H., Gui J. F., Zhang Q. Y. 2007. Mitochondrion-mediated apoptosis induced by Rana grylio virus infection in fish cells. Apoptosis 12:1569–1577 [DOI] [PubMed] [Google Scholar]
- 20. Imajoh M., Sugiura H., Oshima S. 2004. Morphological changes contribute to apoptotic cell death and are affected by caspase-3 and caspase-6 inhibitors during red sea bream iridovirus permissive replication. Virology 322:220–230 [DOI] [PubMed] [Google Scholar]
- 21. Ince I. A., et al. 2008. Open reading frame 193R of Chilo iridescent virus encodes a functional inhibitor of apoptosis (IAP). Virology 376:124–131 [DOI] [PubMed] [Google Scholar]
- 22. Jacob T., Van den Broeke C., Favoreel H. W. 2011. Viral serine/threonine kinases. J. Virol. 85:1158–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jakob N. J., Muller K., Bahr U., Darai G. 2001. Analysis of the first complete DNA sequence of an invertebrate iridovirus: coding strategy of the genome of Chilo iridescent virus. Virology 286:182–196 [DOI] [PubMed] [Google Scholar]
- 24. Jancovich J. K., et al. 2005. Evidence for emergence of an amphibian iridoviral disease because of human-enhanced spread. Mol. Ecol. 14:213–224 [DOI] [PubMed] [Google Scholar]
- 25. Karr B. M., Read G. S. 1999. The virion host shutoff function of herpes simplex virus degrades the 5′ end of a target mRNA before the 3′ end. Virology 264:195–204 [DOI] [PubMed] [Google Scholar]
- 26. Klerkx E. P., Lazo P. A., Askjaer P. 2009. Emerging biological functions of the vaccinia-related kinase (VRK) family. Histol. Histopathol. 24:749–759 [DOI] [PubMed] [Google Scholar]
- 27. Knight P. J., Crickmore N., Ellar D. J. 1994. The receptor for Bacillus thuringiensis CrylA(c) delta-endotoxin in the brush border membrane of the lepidopteran Manduca sexta is aminopeptidase N. Mol. Microbiol. 11:429–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lai Y. S., et al. 2008. Characterization of apoptosis induced by grouper iridovirus in two newly established cell lines from barramundi, Lates calcarifer (Bloch). J. Fish Dis. 31:825–834 [DOI] [PubMed] [Google Scholar]
- 29. Lin P. W., et al. 2008. Iridovirus Bcl-2 protein inhibits apoptosis in the early stage of viral infection. Apoptosis 13:165–176 [DOI] [PubMed] [Google Scholar]
- 30. Lin S., Chen W., Broyles S. S. 1992. The vaccinia virus B1R gene product is a serine/threonine protein kinase. J. Virol. 66:2717–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luan Y., Xu W. 2007. The structure and main functions of aminopeptidase N. Curr. Med. Chem. 14:639–647 [DOI] [PubMed] [Google Scholar]
- 32. Mbuy G. N., Morris R. E., Bubel H. C. 1982. Inhibition of cellular protein synthesis by vaccinia virus surface tubules. Virology 116:137–147 [DOI] [PubMed] [Google Scholar]
- 33. Monnier C., Devauchelle G. 1980. Enzyme activities associated with an invertebrate iridovirus: protein kinase activity associated with iridescent virus type 6 (Chilo iridescent virus). J. Virol. 35:444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paul E. R., et al. 2007. Induction of apoptosis by iridovirus virion protein extract. Arch. Virol. 152:1353–1364 [DOI] [PubMed] [Google Scholar]
- 35. Rice A. P., Roberts B. E. 1983. Vaccinia virus induces cellular mRNA degradation. J. Virol. 47:529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Santos C. R., Blanco S., Sevilla A., Lazo P. A. 2006. Vaccinia virus B1R kinase interacts with JIP1 and modulates c-Jun-dependent signaling. J. Virol. 80:7667–7675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stiles B., McDonald I. C., Gerst J. W., Adams T. S., Newman S. M. 1992. Initiation and characterization of five embryonic cell lines from the cotton boll weevil Anthonomus grandis in a commercial serum-free medium. In Vitro Cell. Dev. Biol. 28:355–363 [Google Scholar]
- 38. Thompson S. R., Sarnow P. 2000. Regulation of host cell translation by viruses and effects on cell function. Curr. Opin. Microbiol. 3:366–370 [DOI] [PubMed] [Google Scholar]
- 39. Williams T., Barbosa-Solomieu V., Chinchar V. G. 2005. A decade of advances in iridovirus research. Adv. Virus Res. 65:173–248 [DOI] [PubMed] [Google Scholar]
- 40. Williams T., Chitnis N. S., Bilimoria S. L. 2009. Invertebrate iridovirus modulation of apoptosis. Virol. Sin. 24:295–304 [Google Scholar]
- 41. Wong C. K., Young V. L., Kleffman T., Ward V. K. 2011. Genomic and proteomic analysis of invertebrate iridovirus type 9. J. Virol. 85:7900–7911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yeager C. L., et al. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422 [DOI] [PMC free article] [PubMed] [Google Scholar]