

Abstract
Imidazolate-containing {Fe(NO)2}9 molecular squares have been synthesized by oxidative CO displacement from the reduced Fe(CO)2(NO)2 precursor. The structures of complex 1 [(Imidazole)Fe(NO)2]4, (Ford, Li, et al. Chem. Comm. 2005, 477–479), 2 [(2-Isopropylimidazole)Fe(NO)2]4, and 3 [(Benzimidazole)Fe(NO)2]4, as determined by X-ray diffraction analysis, find precise square planes of irons with imidazolates bridging the edges and nitrosyl ligands capping the irons at the corners. The orientation of the imidazolate ligands in each of the complexes results in variations of the overall structures, and molecular recognition features in the available cavities of 1 and 3. Computational studies show multiple low energy structural isomers and confirm that the isomers found in the crystallographic structures arise from intermolecular interactions. EPR and IR spectroscopic studies, and electrochemical results suggest that the tetramers remain intact in solution in the presence of weakly-coordinating (THF) and non-coordinating (CH2Cl2) solvents. Mössbauer spectroscopic data for a set of reference dinitrosyl iron complexes, reduced {Fe(NO)2}10 compounds A ((NHC-iPr)2Fe(NO)2), and C ((NHC-iPr)(CO)Fe(NO)2), and oxidized {Fe(NO)2}9 compounds B ([(NHC-iPr)2Fe(NO)2][BF4]), and D ((NHC-iPr)(SPh)Fe(NO)2) (NHC-iPr = 1,3-diisopropylimidazol-2-ylidene) demonstrate distinct differences of the isomer shifts and quadrupole splittings between the oxidized and reduced forms. The reduced compounds have smaller positive isomer shifts as compared to the oxidized compounds ascribed to the greater π-backbonding to the NO ligands. Mössbauer data for the tetrameric complexes 1, 2, and 3 demonstrate larger isomer shifts, most comparable to compound D; all four complexes contain cationic {Fe(NO)2}9 units bound to one anionic ligand and one neutral ligand. At RT the paramagnetic, S = ½ per iron, centers are not coupled.
Introduction
Prominent ligand donor sites in metalloproteins arise from thiolate sulfur and imidazole nitrogen in the side chain amino acid residues, cysteine and histidine, respectively. In both cases deprotonated forms produce anionic ligands that may serve as terminal or bridging ligands. The bridging capacity is important in the assembly of certain active sites as found in the cysteinyl thiolate-bridged nickel-iron hydrogenase or in the bovine erythrocyte superoxide dismutase containing a histidine imidazolate-bridged Cu-Zn active site, Figure 1.1,2 In the latter, copper may replace zinc resulting in a dicopper unit with noteworthy magnetic properties.3,4 In cases requiring valiant synthetic efforts to build in steric bulk within biomimetic ligands in order to prevent higher order aggregation and cluster formation, approaches to small molecule models of these active sites have been developed.4 In fact the clusters themselves may be of interest for unique structures and for molecular properties such as multiple redox events within a single molecule.
Figure 1.

Active site structures of a) [NiFe]-hydrogenase and b) bovine erythrocyte superoxide dismutaste demonstrating bridging cysteine or histidine (shown in blue) as found in metalloproteins.1,2
Dinitrosyl iron complexes (DNICs) comprise a special set of organometallic-like units, typically found in mononuclear tetrahedral geometry, with additional ligands of neutral L or anionic X types, or a combination: [L2Fe(NO)2]+/0, [X2Fe(NO)2]−/2−, and [LXFe(NO)2]0/−.5 The charges shown on the formulae reflect the possibility of two redox levels within the DNICs, in Enemark/Feltham electronic notations6 of {Fe(NO)2}9 or {Fe(NO)2}10, oxidized and reduced states, respectively. The EPR signal of g = 2.03 has been recognized as a characteristic signature of a DNIC, obviously only arising from oxidized, {Fe(NO)2}9 forms.7 Such DNICs have been detected in vivo,8 mainly arising from iron-sulfur cluster degradation in the presence of excess nitric oxide when released from some extrinsic NO-source,9 as a metabolite, or produced as a response to oxidative stress.10 Thiolato sulfurs, as protein-bound cysteine or in low molecular weight forms from free cysteine or glutathione are assumed to be the major ligands of in vivo DNICs.7 Nevertheless a range of natural donor ligands are known to support DNIC formation in cellular media.5c
That imidazoles, as neutral or anionic ligands, can also support DNIC formation, has been explored by ourselves and others.11–14 When deprotonated, the distal nitrogen functionality in the imidazolate ligands of [(Imid)2Fe(NO)2]−, can lead to aggregation through bridging, yielding interesting molecular squares comprised of four {Fe(NO)2}9 units bridged by imidazolates.11,13 The self-assembly of cationic {Fe(NO)2}9 into clusters is well known for thiolates in the ubiquitous Roussin’s red esters (RRE), (μ-SR)2[Fe(NO)2]2, for example.15 Of note is that the {Fe(NO)2}9 in the RRE complexes are spin coupled (the Fe···Fe distance is less than 3 Å). Liaw, et al., have designed a variation on the RRE as shown in Figure 2 that spaces the {Fe(NO)2}9 units at 4 Å apart. This diiron complex was found to have two non-coupled S = ½, {Fe(NO)2}9 centers.5c
Figure 2.

Structure and ball and stick rendition of a RRE in which the {Fe(NO)2}9 units are spaced 3.997 Å apart resulting in non-coupled S = ½, {Fe(NO)2}9 centers.5c
Magnetic susceptibility data demonstrate that each Fe within the imidazolate tetramers reported by Li, et al., [(Imid)Fe(NO)2]4 (complex 1)13 and by us, [(Imid-iPr)Fe(NO)2]4 (complex 2),11 with Fe···Fe distances 6 Å apart, can also be considered as non-coupled, S = ½ units at room temperature. This report will further explore the effect of sterically encumbered imidazoles on the structure and aggregation of these interesting molecular squares.
Experimental Section
General Methods and Materials
Solvents were reagent grade, further purified and degassed by a Bruker solvent purification system, and stored over molecular sieves. Reagents, including nitrosonium tetrafluoroborate and benzimidazole were purchased from Sigma-Aldrich Chemical Co. and were used as received. Standard Schlenk-line techniques (N2 atmosphere) and an Ar-filled glove box were used to maintain anaerobic conditions during preparation, isolation, and product storage. Fe(CO)2(NO)2,16 [Na-18-crown-6-ether][Fe(CO)3(NO)],17 [(Imid)Fe(NO)2]4, complex 1,13 and [(Imid-iPr)Fe(NO)2]4, complex 211 were prepared according to published procedures.
Physical Measurements
Infrared spectra were recorded on a Bruker Tensor 27 FTIR spectrometer in CaF2 solution cells of 0.1 mm pathlength. Solid state samples were recorded using the Pike MIRacle™ attachment from Pike Technologies for Attenuated Total Reflectance, ATR, Infrared Spectra. Mass spectrometry (ESI-MS) was performed by the Laboratory for Biological Mass Spectrometry at Texas A&M University. Nanoelectrospray ionization in positive mode used an Applied Biosystems QSTAR Pulsar (Concord, ON, Canada) equipped with a nanoelectrospray ion source. Solutions were flowed at 700 nL/min through a 50 μm ID fused-silica capillary that was tapered at the tip. Electrospray needle voltage was held at 1900 V.
Elemental analyses of crystalline samples were determined by Atlantic Microlab, Inc., Norcross, GA. EPR spectra were recorded in frozen THF using a Bruker ESP 300 equipped with an Oxford ER910 cryostat operating at 10 K, and in THF or CH2Cl2 solution at 298 K. 1H NMR spectra were recorded in d-THF using a Mercury 300 MHz NMR spectrometer.
Cyclic voltammograms were recorded on a BAS-100A electrochemical analyzer. All experiments were performed under an Ar blanket in CH2Cl2 solution containing 0.1 M [t-Bu4N][BF4] as electrolyte at 22°C, a 3.0 mm glassy carbon working electrode, a Ag/AgNO3 reference electrode, and a Pt coil counter electrode. All values have been internally referenced to the Cp2Fe/Cp2Fe+ couple set at zero.
Ambient Magnetism Measurements
Single crystals of 3 were used for Evans’ method magnetic susceptibility (d-THF) with values in the range of 1.45 to 2.05 BM.18
Mössbauer spectra were recorded with a MS4 WRC spectrometer (SEE Co., Edina, MN) with a 4.5 to 300 K closed-cycle refrigerated helium system. The spectra were collected at 5 K with a 700 G field applied parallel to the source of radiation. The spectra were analyzed with the WMOSS software package (also SEE Co., Edina, MN). Chemical shifts were calibrated relative to Fe metal foil at 298 K. Test compounds A ((NHC-iPr)2Fe(NO)2), B ([(NHC-iPr)2Fe(NO)2][BF4]), C ((NHC-iPr)(CO)Fe(NO)2), and D ((NHC-iPr)(SPh)Fe(NO)2) (NHC-iPr = 1,3-diisopropylimidazol-2-ylidene) were prepared according to published procedures.11
Preparation of [(Imid-benz)Fe(NO)2]4, Compound 3
Method A
In a 100 mL Schlenk flask, 0.75 g (1.64 mmol) [Na-18-crown-6-ether][Fe(CO)3(NO)] and 0.20 g (1.71 mmol) [NO]BF4 were dissolved in 15 mL CH2Cl2 and stirred for at least 10 min to produce Fe(CO)2(NO)2, which was vacuum transferred to a flask immersed in liquid N2. To this was added a solution of 0.39 g (3.3 mmol) benzimidazole, Imid-benz, in 15 mL THF, via cannula. Following overnight stirring at 22°, solvent was removed in vacuo from the brown solution, and 30 mL of CH2Cl2 was added to dissolve the red-brown residue, leaving behind the excess Imid-benz. Following filtration of the extract through Celite, solvent was removed in vacuo to afford 0.235 g (68%) of a brown solid, soluble in THF and CH2Cl2. X-ray quality crystals formed from slow diffusion of pentane into a CH2Cl2 solution of product at 0°C.
Method B
In a 50 mL Schlenk flask, 0.118 g (1.00 mmol) of Imid-benz was dissolved in 5 mL of THF and then a freshly prepared THF solution of Fe(CO)2(NO)2 (1.00 mmol) was added into the flask by cannula under a positive pressure of N2 at 22°C. The reaction mixture was stirred for 1 h after which IR monitoring indicated complete formation of (Imid-benz)(CO)Fe(NO)2. IR (THF): ν(CO) 1993 (m), ν(NO) 1746, 1701 cm−1.11 Dry air was bubbled into the solution with a color change within 10 s from greenish brown to dark brown. The solution was concentrated in vacuo, transferred to a degassed test tube, and layered with pentane to afford X-ray quality dark brown crystals at −35 °C. Isolation of the crystals yielded 0.147 g (63%) of analytically pure product. ν(NO) IR (THF): 1801 (m), 1736 (s) cm−1. Mass spectrometry: (Nano-(+)-ESI MS): The parent ion (Fe4C28H20N16O8, m/z = 931) isotope bundle was not observed in the mass spectrum. Base (100%) peak was centered at m/z 538 (Fe3C14H10N8O5, representing Fe3(NO)4O(Imid-benz)2) with other intense bundles centered at m/z 478.9 representing loss of 2 NO from the base peak, and at m/z 655.9 representing addition of one Imid-benz (+118 mass units) to the base peak. For a sample of single crystals: Anal. Calcd. for Fe4C28H26N16O8 (found): C, 36.1 (36.1); H, 2.17 (2.16); N, 23.7 (24.1).
X-ray Crystallography
Crystalline samples of 3 obtained by both routes described above (i.e., as isolated from CH2Cl2 and from THF) were coated in mineral oil, affixed to a Nylon loop, and placed under streaming N2 (110 K) in a single-crystal APEXii CCD diffractometer. X-ray diffraction data were collected by covering a hemisphere of space upon combination of three sets of exposures. The structures were solved by direct methods. H atoms were placed at idealized positions and refined with fixed isotropic displacement parameters and anisotropic displacement parameters were employed for all non-hydrogen atoms. The following programs were used: for data collection and cell refinement, APEX2;19 data reductions, SAINTPLUS, version 6.63;20 absorption correction, SADABS;21 structure solutions, SHELXS-97;22 structure refinement, SHELXL-97.23 Structure plots were generated in Mercury, version 2.3.24
Computational Details
Density functional theory calculations were performed at the BP8625,26/6-311G(d,p)27–30 level of theory appropriate for di-nitrosyl iron complexes containing N- and C-donor ligands.31 Calculations of complexes 1–3 focused on fundamental geometric features and orientation of the imidazolate ligands in the gas-phase computations vs. that experimentally found in the solid state, and electronic features as referenced to ν(NO) vibrational data were also determined. Three spin states, the quintet state (one unpaired electron on each Fe(NO)2 unit; fully ferromagnetic), the singlet state (fully antiferromagnetic, odd electron on each Fe), and the intermediary triplet state were included in the calculations.
Calculations were performed utilizing the Gaussian 09 software suite.32 Starting structures of both the X-ray crystallographic coordinates (for 1–3) as well as a highly symmetric “square-like” structure (for 3 only) were employed in the optimizations. Additionally, complex 3 was calculated at the crystallographic geometry of 1 (up, up, up, up) and complex 1 was calculated at the crystallographic geometry of 3 (up, down, up down) in order to compare electronic energies of isomers. While the singlet states of tetramers 2 and 3 optimized successfully by a typical unrestricted density functional theory approach, the singlet state of tetramer 1 converged with difficulty, so it was optimized using an initial guess generated by the broken-symmetry formalism described by Noodleman, et al.33 and recently described for similar dimeric copper systems.34
In difficult to converge cases, loose SCF parameters (SCF = 10−4) and smaller basis sets (6-31G) were utilized to produce a starting guess for subsequent calculations. All data presented here was terminally optimized at the BP86/6-311G(d,p) level of theory with a tight SCF convergence of 10−8. Subsequent frequency calculations were performed analytically as implemented into Gaussian 09.32
The vibrational frequencies of 1–3 were calculated both in the gas-phase and solution-phase using the polarizable continuum model (PCM) with CH2Cl2 parameters. The slight degree of asymmetry of the tetrameric units leads to multiple, near-degenerate vibrations, which have been averaged to two absorbances in order to make comparisons to the experimental data. Molecular orbitals were rendered in AGUI35 at an isosurface value of 0.03. All energies were recorded as electronic energies and converted from Hartrees to kcal/mol, and, in the case of energy comparisons of isomers, the lowest energy isomer has been set at 0 kcal/mol.
Results and Discussion
Synthesis and Composition
As described for the preparation of the [(Imid-iPr)Fe(NO)2]4 complex 2,11 addition of excess benzimidazole to freshly prepared Fe(CO)2(NO)2 in a mixture of THF/CH2Cl2 results in a color change from orange to red-brown and ultimately (the CO substitution is stepwise) an infrared spectrum representative of an oxidized DNIC (ν(NO) in CH2Cl2 = 1805(m), 1739(s)).11 Deliberate addition of dry air to the reaction mixture speeds up the reaction to completion within seconds. Water was detected as a byproduct under these conditions (Figure S1). Isolation and recrystallization from CH2Cl2/pentane and from THF/pentane yielded brown, X-ray quality crystals of complex 3, [(Imid-benz)Fe(NO)2]4; excellent elemental analysis was achieved from the high quality single crystals.
Complexes 1, 2, and 3 are paramagnetic at room temperature, with Evans’ method magnetic susceptibility measurements (d-THF solution) indicating one unpaired electron per iron in complex 3. The 1H NMR spectrum of complex 3 in d-THF (Figure S2) displayed broad signals centered around 7.97 and 6.59 ppm assigned to the Imid-benz ligand protons within the paramagnetic DNIC; free Imid-benz has signals at 8.14 (singlet), 7.6 (multiplet), and 7.24 (multiplet).
Mass spectral data were acquired by the nanoelectron spray ionization technique in the positive mode, however only for complex 2 was the parent ion observed (Fe4C24H36N16O8, isotope bundle centered at m/z = 900) and it was of very low intensity. The base (100%) peak was centered at m/z 538 (Fe3N9O5C12H18, representing Fe3(NO)5(Imid-iPr)2) with other intense bundles centered at m/z 478.8 representing loss of 2 NO from the base peak, and at m/z 647.9 representing addition of one iPr-imidazole (+110 mass units) to the base peak.11 Likewise for complex 3 the base (100%) peak was centered at a three-iron unit, m/z 538 (Fe3C14H10N8O5, representing Fe3(NO)3(NO2)(Imid-benz)2) (Figure S3). Other fragments are described in the experimental section.
Molecular Structures
Two x-ray structures of complex 3, as crystallized from THF and from CH2Cl2, were determined in order to assure that there was no solvent dependence on the state of aggregation or the orientation of the Imid-benz ligands in the tetramers. The X-ray diffraction analysis and the molecular structure of complex 3, [(Imid-benz)Fe(NO)2]4 are first reported here and compared to those of complexes 1, [(Imid)Fe(NO)2]4,13 and 2, [(Imid-iPr)Fe(NO)2]4.11 Two views of 3 are given as thermal ellipsoid plots in Figure 3; a full structure report is available in Supporting Information. As was found for complexes 1 and 2 (Figure 4), the structure of 3 consists of an almost precise square plane of irons, with nitrosyl ligands capping the irons at the corners and imidazolates bridging the edges. The contrasting structural features of complexes 1 – 3 are revealed from side views of the Fe4 plane, in Figures 4 and 5.
Figure 3.

a), b) Views of the thermal ellipsoid plot at 50% probability of [(Imid-benz)Fe(NO)2]4, complex 3. The labels correspond to the distance between opposite aryl C-C bonds at the widest point and at the closest point. Red: O, blue: N, black: C, orange: Fe, gray: H. c) Two molecules from the extended packing diagram to show the close contact of the benzyl groups of the closed portion to those of the open portion. d) Labeling scheme to demonstrate selected intermolecular C to C distances between benzyl groups of the closed portion to the open portion. Nitrosyl groups have been removed for clarity. C-C distances, Å: C8-C12 3.610; C8-C11 3.587; C11-C4 4.926; C11-C3 4.497; C11-C2 3.837; C10-C2 3.506; C10-C3 3.940.
Figure 4.

Ball and stick representations of the structures of complexes 1 – 3 as derived from X-ray diffraction analysis and with canting of the imidazoles emphasized by shaded planes. In each case, the view on the right is from a rotation of 90° relative to the left.
Figure 5.

Up and/or down orientations of imidazolate ligands of complexes 1 – 3 and an analogous Cu-containing molecular square.3 “Up” and “down” refers to the orientation of the ethenyl (HC=CH) group of the imidazolate with respect to the Fe4 plane. Imidazole substituents (in the case of complex 2 and 3) have been removed for clarity. Red: O, blue: N, black: C, orange: Fe, dark red: Cu.
Interestingly, the imidazolate units are oriented differently in the three tetramers in this series with respect to the position of the C2 and C1 units relative to the Fe4 square plane, Figure 5. A fourth structure shown in Figure 5 is that of a copper analogue, described more fully below.3 Associated with the imidazolate orientation is a canting of imidazole ligand planes shown best in the shaded planes of Figure 4. In complex 1, the methylene units of the imidazole are all positioned to the same side and oriented in towards one another (angle of intersection of the Fe4 plane and imidazole planes = 53°), closing off one side of the Fe4 plane. On the opposite side the ethylene units, designated as the all “up” configuration in Figure 5, render a bowl-like open side that in the complete molecular structure of Li, et al., includes an acetone solvent molecule of crystallization.13 For comparison to complex 3, extended packing structures of complexes 1 acetone and 2 are found in the S.I.
In complex 2, the C1 methylene carbons and the C2 ethylene units alternate up and down positions such that the canting of the planes renders both sides of the Fe4 square plane blocked by the inwardly pointing isopropyl groups, Figure 4b; as in 1, the ethylene units are outwardly oriented. Complex 3, Figures 4c and 3, shows another variation, in that the aryl groups attached to the ethylene unit are oriented outwardly and across from each other on one side of the Fe4 square plane (10.2 Å across from aryl C-C bonds at widest point) and inwardly on the other two (3.4 Å apart at the closest point). This configuration has the effect of sterically blocking only one side of the Fe4 plane and allows for highly efficient packing in the crystalline form as the closed side of one unit can nest into the open side of another, see Figure 3 and S.I. Note that the nested benzimidazolates have the benzyl groups arranged aryl edge to aryl plane (as they are in the solid state form of benzene) rather than π-stacked. While the closest intermolecular Fe···Fe distance is ~ 5.3 Å, the closest intermolecular C···C distance (from C10 of the nested benzimidazolate to C2 of the benzimidazolate of the open portion) is ~ 3.5 Å, Figure 3d. The separation of the nested benzyl groups measured by C11 to the methenyl carbon of the benzimidazolate of the open portion is ~ 3.6 Å.
A view down the center of the squares in the packing diagrams thus reveals a columnar cavity in complex 3; the centers of complexes 1 and 2 are blocked by the included acetone molecule or isopropyl groups, respectively. Graphics displaying these views are given in Figure 6.
Figure 6.

A view down the center of the squares demonstrating blocked cavities for complexes 1 and 2 and an open cavity for complex 3.
Metric parameters presented in Table 1 show similar ∠NNO-Fe-NNO in the range of 116.5 to 113.1° for 1 and 3; the Imid-iPr analogue 2 average is 110.5°. The ∠NImid-Fe-NImid angles vary somewhat with complex 3 having the smallest, avg. = 105°. Likely to accommodate the additional steric bulk of the isopropyl groups the C1 unit of the imidazole ring, complex 2 has a larger cavity as compared to 1 and 3: Fe to Fe average edge distances are 6.24 Å, 0.2 Å larger than that of 1 and 3; Fe to Fe cross distances are ca. 8.8 Å. The iron atoms are positioned at ca. 120° relative to the carbons adjacent to the N-donors in the imidazolates, thus the vectors that connect the iron atoms do not include the imid-nitrogen donors.
Table 1.
Selected bond distances (Å) and angles (°) for complexes 1, 2, and 3.
| 1 | 2 | 3 | |
|---|---|---|---|
| Bond Distances (Å) | |||
|
| |||
| Fe-NImid avg | 2.005(5) | 2.036(11) | 2.013(3) |
| N-O avg | 1.166(6) | 1.194(13) | 1.175(5) |
| Fe-NNO avg | 1.694(5) | 1.712(13) | 1.687(4) |
| Fe---Fe (adj) | 5.965 | 6.230 | 6.039 |
| 5.977 | 6.253 | 6.015 | |
| Fe---Fe (cross) | 8.697 | 8.755 | 8.278 |
| 8.183 | 8.898 | 8.761 | |
|
| |||
| Bond Angles (°) | |||
|
| |||
| NImid-Fe-NImid avg | 109.24(17) | 110.72(5) | 104.88(14) |
| NNO-Fe-NNO avg | 114.8(2) | 110.39(5) | 114.15(19) |
| Fe-N-O avg | 166.8(6) | 164.02(11) | 166.9(4) |
| Fe-NImid-CImid avg | 128.9(3) | 133.39(10) | 127.3(3) |
|
| |||
| Avg. Deviation of Fe from Fe4 Square Plane (Å) | |||
|
| |||
| 0.0094 | 0.0145 | 0.0219 | |
|
| |||
| Angle of intersection of Fe4 Square Plane With Imidazole Plane (°) | |||
|
| |||
| 129.8 | 107.8 | 117.5 | |
| 55.7 | 68.5 | 70.3 | |
These DNIC tetramer structures are similar to those of imid-azolate-bridged tetracopper squares.3,4 The example shown in Figure 5, finds the Cu(II) corners capped by the 1,4,7-triazacyclononane or tacn ligand.3 The overall charge of the cluster is 4+. Copper to copper distances along the edges are 5.89 and 5.99 Å, and the ∠NImid-Cu-NImid are in the range of 93°.3 This tetramer and other copper-containing analogues were synthesized as mimics of histidine-containing metalloproteins and examined for the ability of the bridging imidazolate to mediate antiferromagnetic interactions. Variable temperature magnetic susceptibility studies found antiferromagnetic interactions between adjacent Cu(II) centers at low temperatures, ascribed to a σ-exchange pathway through the imidazolate bridge,3,4 that optimized at Cu-NImid-CImid angles of ca. 132°.34 We note that the analogous Fe-NImid-CImid angles in complexes 1 – 3 are in the 127 – 133° range.
Computational Studies
The optimizations of 1–3, starting from crystallographic structure coordinates of the single tetramer, with the acetone molecule removed from the broad edge of the bowl in 1, were performed on the singlet, triplet, and quintet states of the molecules. The antiferromagnetic singlet state was found to be of lowest energy, with the triplet and quintet states only slightly higher in energy (for 1, 0.54 and 1.09 kcal/mol, for 2, 0.73 and 1.47 kcal/mol and for 3, 0.88 and 1.61 kcal/mol, respectively), indicating a very low energy barrier for spin flipping at room temperature, increasing with the larger imidazolate and a greater degree of molecular asymmetry. Energy diagrams for the isomers are described in Figures S11-13. Any coupling present in the tetramer is expected to be weak due to the large distances between the iron atoms. Consistent with this, room temperature Evans’ method magnetic susceptibility data indicates 1 unpaired electron per iron.
The computational methodology used reproduced the vibrational frequencies especially when solvation was included. The vibrational frequencies for 1calc. and 3calc. for the gas-phase and solution-phase are follows: 1calc.: 1824/1774 cm−1 (g.p.) and 1813/1739 cm−1 (CH2Cl2), similar to that found for 1exp., 1805 and 1737 in CH2Cl2. Similarly, the frequencies for 3calc. were found to be 1810/1767 cm−1 (g.p.) and 1797/1733 cm−1 (CH2Cl2), matching well with the 3exp. CH2Cl2 values of 1805 and 1739 cm−1. Additional results are found in Table S1.
The geometric parameters from optimized structures are not significantly different between singlet, triplet, and quintet states. Beginning from the orientations of the x-ray crystal structures, the calculated metric parameters of one unit of complexes 1, 2 and 3 substantially match experimental values (see Table S1). However, as shown in Figure 7 and Table S1, there are differences in the canting of the imidazolate planes, particularly for the benzimidazole of complex 3. The gas phase calculation of this tetramer finds the imidazolate planes ⊥ to the Fe4 plane and largely parallel to each other. This distinction suggests a role for non-covalent interactions or crystal packing in the solid state.
Figure 7.
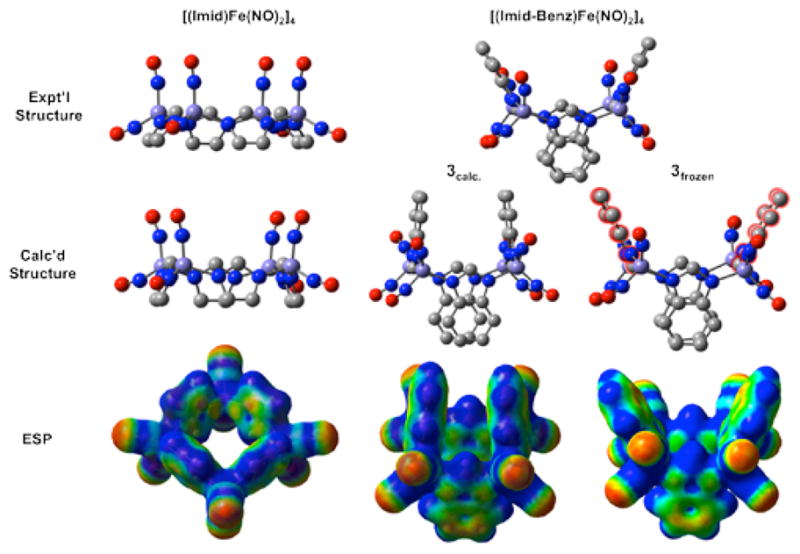
Experimental and computational structures and electrostatic potentials for 1 (left) and 3 (right), with the calculated coordinates shown taken from 3quint. Both 3calc. and 3frozen are shown, with the imidazolate groups held frozen in the latter shown outlined in red. Electrostatic potentials were generated at an isosurface value of 0.01.
An additional calculation of complex 3 was performed; a structure corresponding to a single tetramer in which bond distances, valence angles, and dihedral angles of the cross imid-azolate groups that correspond to the broad edge of the bowl were frozen to observe the effect on the cross imidazolate interactions corresponding to the narrow edge of the bowl (3frozen). In 3frozen, in which the arene rings of 3 opposite one another (the rings shown as 10.2 Å apart in Figure 3 and as outlined in red in Figure 7) were held frozen, the arene rings 3.4 Å apart were allowed to relax. In this conformation, i.e., with the observed canting as described in Figure 7 and in the experimental structure, the distance between the unfrozen rings expanded (see Table S1) and the calculated energy is 3.59 kcal higher than the optimized gas phase structure. It should be noted that the opposite frozen structure was calculated (i.e., the close arene rings held frozen with the distant arene rings allowed to relax), and a similar effect was found, with the arene rings 10.2 Å apart optimizing to a structure whereby they approximate a parallel orientation.
By computations we explored the interchange of imidazolate orientational isomers of 1 and 3 (1up/down and 3up/up), creating for 3 an “super-bowl” when in the (up, up, up, up) orientation, Figure 8. The difference in energy between the two isomers of 1 is 0.82 kcal/mol, favoring the all-up configuration that is found in the solid state structure. As an acetone molecule crystallized in the open bowl of complex 1, the energetic preference of this orientation even in its absence was unexpected. For gas-phase 3, the all-up orientation is favored over the (up, down, up, down) orientation, albeit by a mere 0.05 kcal/mol. This is in contrast with the orientation in the crystallographic structure, implying that intermolecular interactions are responsible for the observed conformation. Structural details of these additional computed isomers of 1 and 3 can be found in Figure 8 and Table S1.
Figure 8.

Orientational isomers of 1 (up/down, left) and 3 (up/up, right).
Electron Paramagnetic Resonance Spectral Data
The EPR spectra for complexes 1 – 3 (Figure 9) originate from paramagnetic {Fe(NO)2}9 units, all of which at 295 K display a broad isotropic signal at g = 2.03 (~ 2 mM THF solutions), characteristic of DNIC complexes. In frozen THF at 10 K, complex 2 shows a rhombic signal with g values of 2.055, 2.029, 2.012, whereas complex 3 shows a broad isotropic signal with a g value of 2.029 similar to the signal seen at room temperature. Under these conditions, 10 K in frozen THF, complex 1 also shows a broad isotropic signal with a g value of 2.021. In contrast, Li and coworkers report a nine-line spectrum with N-14 hyperfine coupling centered around a g value of 2.031 at 170 K in THF solution.13 The line width of Li’s signal is ~13 G, while the line width of the signal obtained for complex 1 in THF at 298 K is 14 G. An overlay of these spectra, Figure S7, suggests that the isotropic signal of 1 at room temperature is an unresolved envelope of the 9-line pattern seen for 1 at 170 K.
Figure 9.

EPR spectra of complex 1 in THF at 170 K (line width = 30 G), reproduced with permission from ref. 13, 298 K (frequency at 9.45 GHz, line width = 14 G), and in CH2Cl2 at 298 K (frequency at 9.45, line width = 14 G); complex 2 in THF at 10 K (frequency at 9.49 GHz, line width = 43 G), 298 K (frequency at 9.45 GHz, line width = 13 G), and in CH2Cl2 at 298 K (frequency at 9.45, line width = 12 G); and complex 3 in THF at 10 K (frequency at 9.49 GHz, line width = 19 G), 298 K (frequency at 9.45 GHz, line width = 18 G), and in CH2Cl2 at 298 K (frequency at 9.44 GHz, line width = 14 G).
On the basis of the nine-line EPR spectrum Li and coworkers suggested that complex 1 dissociated in THF solvent to produce a THF-bound monomer DNIC.13 In order to determine if there was any observable effect with a non-coordinating solvent on the signal shape or g value, EPR spectra of complexes 1 – 3 were also obtained in CH2Cl2 solution at 298 K. In all three cases, a broad isotropic signal is observed with a g value closely matching that of the EPR spectra taken in THF solution. No N-14 hyperfine was observed in spectra recorded in both the THF or CH2Cl2 solutions under various conditions: decreasing the concentration of the samples to ~ 0.5 mM or modifying the spectral parameters (increasing scan time, increasing or decreasing the power, or lowering the modulation amplitude). As the spectral line shapes and g values are similar in both THF and CH2Cl2, we assume that the tetramers remain intact in solution. The similarity of ν(NO) IR spectra in both solvents and in the solid state support this conclusion (see Figure S8).
Cyclic Voltammetry
The cyclic voltammograms of complexes 1, 2, and 3 were recorded in 2 mM CH2Cl2 solution with 100 mM [t-Bu4N][BF4] as the supporting electrolyte. Potentials were measured relative to a Ag/AgNO3 electrode using a glassy carbon working electrode and are referenced to Cp2Fe/Cp2Fe+. Multiple and overlapping redox events are observed for each complex. The cyclic voltammograms of complexes 1 – 3 in the cathodic region are given in Figure 10; the scans are reversed at different potentials for each complex in attempts to isolate individual redox events. The redox potentials for each complex are listed in Table 2.
Figure 10.

Scan reversals of the cyclic voltammograms to isolate successive waves of complexes a), 1; b), 2; c), 3 in 2 mM CH2Cl2 solution. All are referenced to Cp2Fe/Cp2Fe+.
Table 2.
Reduction and oxidation values observed for complexes 1 – 3.a
| Epc (V) | Epa (V) | Δ(Epa-Epc) (V) | Δ(Epc2-Epc1) (V) | |
|---|---|---|---|---|
| 1 | −1.20 | −1.03 | 0.17 | 0.19 |
| −1.39 | −1.29 | 0.10 | ||
| 2 | −1.29 | −1.09 | 0.20 | 0.38 |
| −1.67 | −1.41 | 0.26 | 0.31b | |
| −1.98 | ||||
| 3 | −1.12 | −0.99 | 0.13 | 0.16 |
| −1.28 | −1.21 | 0.07 |
In CH2Cl2 solution (0.1 M tBu4NBF4). All experiments were recorded using a glassy carbon working electrode and Pt counter electrode and referenced to Cp2Fe/Cp2Fe+ at a scan rate of 100 mV/s.
The potential difference refers to the difference between the second and third reductions.
For complexes 1 and 3, two reductive events are observed at −1.20 and −1.39 V and −1.12 and −1.28 V, respectively. Isolation of the successive waves suggest that the oxidation events observed upon scan reversal are associated with the corresponding reduction, i.e. for complex 3, the reductive event at −1.12 V is associated with the oxidative event at −0.99 V and the reductive event at −1.28 V is associated with the oxidative event at −1.21 V. The square wave voltammograms display broad peaks, rather than well-defined, separated peaks, suggesting that there are multiple overlapping events associated with the two main events of the cyclic voltammograms (Figure S9). For complex 2, three reduction events are observed at −1.29, −1.67, and −1.98 V. Again, isolation of the successive waves suggest that the oxidation events upon scan reversal at −1.09 and −1.41 V are associated with the events at −1.29 and −1.67 V, respectively. The square wave voltammogram is even broader, also suggesting the presence of several redox events in the −1.29 to − 2.0 V region. The patterns of the cyclic voltammograms are consistent with previously studied polymetallics that have multiple redox events.36,37
Assuming that the tetramers remain intact in CH2Cl2 solution during the electrochemical experiment, we might expect to observe four individual, {Fe(NO)2}9/10 redox events. The broad and poorly defined events in both the cyclic voltammograms and square wave voltammograms suggest that, if indeed four reduction events exist, they must occur at similar voltage values, resulting in signal overlap. Other neutral {Fe(NO)2}9 complexes, such as (NHC-iPr)(SPh)Fe(NO)211 and (Ar-nacnac)Fe(NO)2,38 show reversible, well-defined redox couples in THF at E1/2 = −1.33 and −1.34 V, respectively (Figure S10c; S10 a and b have CV’s of neutral reduced species for comparison). As imidazole is a weaker donor compared to N-heterocyclic carbenes, and imidazolates are weaker donors than thiolates and diketim-inates,11 we expect reduction of 1, 2, and 3 to be slightly easier (more positive), on the basis of typical donor/acceptor ability arguments. This is the case for all three complexes.
The small difference (< 0.2 V) between the first and second reduction potentials for complexes 1 and 3 (Table 2), compared with previously reported polymetallic complexes,36 suggest substantial localization of the reduction events. For complex 2, the slightly larger ΔE values (0.38 or 0.31 V) suggest somewhat greater delocalization of the reduction events; i.e., the first reduction seems to have a greater effect on the subsequent reductions.
Mössbauer Spectroscopic Studies
As frozen solution samples at ~ 50 mM in THF solvent, complexes 1, 2, and 3 exhibit sharp quadrupole doublets at 5 K (Figure 11) with isomer shifts of 0.270, 0.289, and 0.276, mm/s, respectively, and quadrupole splitting parameters of 0.700, 0.717, and 0.768 mm/s, respectively. For comparison, Mössbauer data were also obtained for a set of related, well-characterized, monomeric DNICs; these spectra and corresponding structures are given in Figure 12. Competing/complicating effects in hetero-ligated complexes requires cautious interpretation of Mössbauer data and careful comparisons to reference compounds. A classic and detailed study of neutral Fe(CO)2(NO)2, (PPh3)(CO)Fe(NO)2, and (PPh3)2Fe(NO)2 report small positive isomer shifts (ranging from +0.027 to +0.110 mm/s), more positive values corresponding to the better donors, poorer π-accepting phosphine derivatives. The quadrupole splitting values range from 0.332 to 0.687 mm/s.39 Salts of anionic [Fe(CO)3NO]− showed small and negative isomer shifts (ca. − 0.08 mm/s).39 In the current description of the redox states of DNICs, these classic organometallics contain reduced {Fe(NO)2}10 units, such that the electric field gradient at Fe is implied to be buffered by substantial electron delocalization.
Figure 11.

5 K Mössbauer spectra for tetrameric complexes 1, 2, and 3 in frozen THF solution in an applied field of 700 G.
Figure 12.

5 K Mössbauer spectra of test complexes A–D in frozen THF solution in an applied field of 700 G.
More recently, Mössbauer data been reported and interpreted for the reduced and oxidized [(Ar-nacnac)Fe(NO)2]−/0 complexes (Ar-nacnac = 2,6-diisopropylphenyl) prepared and characterized by the Lippard group.38,40 The isomer shifts for the reduced and oxidized forms are extremely close, 0.22 and 0.19 mm/s, respectively.38 As NO ligands are notoriously “non-innocent,” and the Ar-nacnac ligand is also potentially redox-active, the question of ligand-based versus metal-based redox process in the Lippard system was addressed by Ye and Neese in DFT studies.40 Their computations find that the 3d orbitals for iron in the reduced DNIC are energetically close to the NO π* orbitals resulting in strong π-backbonding and thus a smaller isomer shift than what might be expected for a metal-based reduction.
The compounds chosen here as references for the tetramers 1–3 contain spectator ligands with varying donor/acceptor properties (Figure 12).11 Test compounds A and C are reduced {Fe(NO)2}10 complexes. Complex A contains two strongly σ-donating NHC ligands bound to the Fe(NO)2 unit, whereas, for complex C one of the NHC ligands is replaced with CO, a weaker σ-donor, but a stronger π-backbonding ligand. Both the isomer shift and the quadrupole splitting are smaller in the latter, and are consistent with recorded data for (Ph3P)(CO)Fe(NO)2 and (Ph3P)2Fe(NO)2.39 Complex B, containing two strongly σ-donating NHCs, is the cationic, oxidized, {Fe(NO)2}9 analogue of complex A. Complex D, also in redox level {Fe(NO)2}9, is a neutral complex containing an anionic, strong σ-donor/strong π-donor, phenyl thiolate (SPh−), and a strongly σ-donating NHC. Note that the isomer shifts of the reduced {Fe(NO)2}10 complexes A and C, are distinctly lower than for B and D, the oxidized {Fe(NO)2}9 complexes. The analogous {Fe(NO)2}9 complex as a dithiolate, [(PhS)2Fe(NO)2]−, has Mössbauer parameters (δ = 0.18(2) mm/s and ΔEQ = 0.69(2) mm/s) similar to D.41,42 Although A and C contain Fe in a formally reduced state relative to B and D within the DNICs, the isomer shifts are found at lower values compared to those of B and D, suggesting greater Π-backbonding in the reduced complexes.
Test {Fe(NO)2}9 complexes B and D also demonstrate this trend, where B contains two NHC ligands and has an isomer shift of 0.114 mm/s, while D contains only one NHC ligand, and a strong π-donor in the PhS− ligand, but has a only slightly larger isomer shift of 0.151 mm/s.
The isomer shifts of tetramers 1, 2, and 3 are significantly larger than those of the test complexes, with the closest match coming from complex D, the complex containing the neutral, NHC ligand, and the anionic SPh−, overall rendering the oxidized Fe(NO)2 unit within the DNIC neutral. That is, the tetramers may be analyzed as containing cationic {Fe(NO)2}9 units, bound to one anionic and one neutral imidazole nitrogen. The larger shifts may be due to the fact that the σ-donating ability of the N-donors of the imidazole ligand, in a bridging mode, is less than that of the NHC and anionic thiolate. Note that the similar isomer shifts of the Ar-nacnac DNIC redox pair and that of the tetrameric complexes 1 – 3 suggest comparable electronic environments produced by N-donors, one of which is anionic on the {Fe(NO)2}9 unit.
Summary and Comments
Despite the presence of bridging imidazolates in biomolecules as well as a variety of inorganic complexes, to our knowledge, only one DNIC containing bridging imidazolate ligands, complex 1, was reported previous to this study.13 Tetrameric complexes 1, 2, and 3 form as molecular squares; the orientation of the imidazolate ligand with respect to the Fe4 plane in the crystal structures is influenced by both steric interactions of the imidazolate substituents and crystal-packing forces. The computationally optimized structures suggest that in gas phase or in solution there should be small energetic differences in the orientation of the imidazolates, and thus the imidazolate ligand plane should be flexible in the absence of crystal packing forces.
EPR spectroscopic studies find g values of ~ 2.03 for all three tetrameric complexes, however, N-14 hyperfine coupling is not observed. There are few examples of DNICs that exhibit hyper-fine coupling in their EPR spectra.13,43–45 Any correlation between DNIC structure and observation of hyperfine coupling to N-donor ligands or the NO ligands still remains unclear. Interestingly, the analogous copper based imidazolate molecular square also has an isotropic, temperature-independent EPR signal with no distinct N-14 hyperfine coupling. However, at low temperatures (below 100 K), a second spectral component with wide splittings is observed and assigned to the triplet state of the Cu tetramer.3
The Mössbauer data reported here, as one of the few examples thus far for DNICs in different redox levels, characterizes the oxidized {Fe(NO)2}9 complexes to have larger positive isomer shifts than the reduced {Fe(NO)2}10 analogues. While the nature of the spectator ligands varies within the four reference complexes as compared to the tetramers, the precise source of differences in quadrupole splitting parameters awaits more detailed interpretations through simulations. It is clear however that along with the ν(NO) IR data, Mössbauer spectroscopy could become a useful tool in detecting the presence of both oxidized and reduced DNICs in biological systems, and their response to ligand fields.
Biological Implications
Histidine-containing DNICs have been observed via EPR spectroscopy upon nitrosylation of several proteins containing histidine residues, including aconitase, mammalian ferritin, and the iron-quinone complex of photo-system II.46,47,48 From these studies, it is not clear whether the histidine residues were bound through the neutral N or the deprotonated N of the imidazole ring. Importantly, pH studies have demonstrated that deprotonation of the imidazole N-H occurs at a pKa of 14.5; however, when histidine is bound to Fe in the case of a 2Fe2S center, this pKa is lowered to 7.85.49,50 Thus, under physiological pH conditions, the possibility of histidine deprotonation leads to the prospect of aggregation of nitrosylated Fe in histidine-containing proteins as seen in the inorganic imidazolate-containing Fe(NO)2 aggregates discussed here, and as is known for copper(II) imidazolates. Whether the DNIC containing clusters could form in the cellular environment or what their function might be has yet to be discovered.
Supplementary Material
Acknowledgments
We appreciate financial support from the National Science Foundation (CHE-0910679 to MYD and CHE-0910552 to MBH) and the R. A. Welch Foundation (A-0924 to MYD and A-0648 to MBH). We sincerely thank Dr. Paul A. Lindahl for the use of his EPR and Mössbauer instruments (NIH grant GM46441) and especially Gregory P. Holmes-Hampton for his help in running the Mössbauer samples. We also thank Dr. Yohannes Rezenom for the mass spectrometry results.
Footnotes
Supporting Information. Additional spectroscopic and computational details, X-ray crystallographic data (CIF) from the structure determination and full list of metric parameters for complex 3. Complete Ref. 32. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]; b) Bagley KA, Duin EC, Roseboom W, Albracht SPJ, Woodruff WH. Biochemistry. 1995;34:5527–5535. doi: 10.1021/bi00016a026. [DOI] [PubMed] [Google Scholar]
- 2.a) Richardson JS, Thomas KA, Rubin BH, Richardson DC. Proc Nat Acad Sci USA. 1975;72:1349–1353. doi: 10.1073/pnas.72.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]
- 3.Chaudhuri P, Karpenstein I, Winter M, Lengen M, Butzlaff C, Bill E, Trautwein AX, Flörke U, Haupt HJ. Inorg Chem. 1993;32:888–894. [Google Scholar]
- 4.Kolks G, Lippard SJ, Waszczak JV, Lilienthal HR. J Am Chem Soc. 1982;104:717–725. [Google Scholar]
- 5.a) Hung M-C, Tsai M-C, Lee G-H, Liaw W-F. Inorg Chem. 2006;45:6041–6047. doi: 10.1021/ic0605120. [DOI] [PubMed] [Google Scholar]; b) Wang J-H, Chen C-H. In-org Chem. 2010;49:7644–7646. doi: 10.1021/ic101126v. [DOI] [PubMed] [Google Scholar]; c) Tsai M-L, Hsieh C-H, Liaw W-F. Inorg Chem. 2007;46:5110–5117. doi: 10.1021/ic0702567. [DOI] [PubMed] [Google Scholar]
- 6.Enemark JH, Feltham RD. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 7.Vanin AF. Nitric Oxide. 2009;21:1–13. doi: 10.1016/j.niox.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Vithayathil AJ, Ternberg JL, Commoner B. Nature. 1965;207:1246–1249. doi: 10.1038/2071246a0. [DOI] [PubMed] [Google Scholar]
- 9.Foster MW, Cowan JA. J Am Chem Soc. 1999;121:4093–4100. [Google Scholar]
- 10.Vanin AF. Methods Enzymol. 2008;436:445–461. doi: 10.1016/S0076-6879(08)36025-X. [DOI] [PubMed] [Google Scholar]
- 11.Hess JL, Hsieh CH, Reibenspies JH, Darensbourg MY. Inorg Chem. 2011;50:8541–8552. doi: 10.1021/ic201138f. [DOI] [PubMed] [Google Scholar]
- 12.Reginato N, McCrory CTC, Pervitsky D, Li L. J Am Chem Soc. 1999;121:10217–10218. [Google Scholar]
- 13.Wang X, Sundberg EB, Li L, Kantardjieff KA, Herron SR, Lim M, Ford PC. Chem Commun. 2005:477–479. doi: 10.1039/b412086h. [DOI] [PubMed] [Google Scholar]
- 14.Li L. Comments Inorg Chem. 2002;23:335–353. [Google Scholar]
- 15.Butler AR, Megson IL. Chem Rev. 2002;102:1155–1165. doi: 10.1021/cr000076d. [DOI] [PubMed] [Google Scholar]
- 16.McBride DW, Stafford SL, Stone FGA. Inorg Chem. 1962;1:386–388. [Google Scholar]
- 17.Connelly NG, Gardner C. J Chem Soc, Dalton Trans. 1976:1525–1527. [Google Scholar]
- 18.(a) Evans DF. J Chem Soc. 1959:2003–2005. [Google Scholar]; b) Grant DH. J Chem Educ. 1995;72:39–40. [Google Scholar]
- 19.APEX2, version 20097-0. Bruker AXS Inc; Madison, WI: 2007. [Google Scholar]
- 20.SAINTPLUS: Program for Reduction of Area Detector Data, version 6.63. Bruker AXS Inc; Madison, WI: 2007. [Google Scholar]
- 21.Sheldrick GM. SADABS: Program for Absorption Correction of Area Detector Frames. Bruker AXS Inc; Madison, WI: 2001. [Google Scholar]
- 22.Sheldrick GM. SHELXS-97: Program for Crystal Structure Solution. Universität Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 23.Sheldrick GM. SHELXL-97: Program for Crystal Structure Refinement. Universität Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 24.Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, van de Streek J. J Appl Crytsallogr. 2006;39:453–457. [Google Scholar]
- 25.Becke AD. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 26.Perdew JP. Phys Rev B: Condens Matter Mater Phys. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 27.Krishnan R, Binkley JS, Seeger R, Pople JA. J Chem Phys. 1980;72:650. [Google Scholar]
- 28.Wachters AJH. J Chem Phys. 1970;52:1033. [Google Scholar]
- 29.Hay PJ. J Chem Phys. 1977;66:4377–4384. [Google Scholar]
- 30.Raghavachari K, Trucks GW. J Chem Phys. 1989;91:1062–1065. [Google Scholar]
- 31.Brothers SM, Darensbourg MY, Hall MB. Inorg Chem. 2011;50:8532–8540. doi: 10.1021/ic201137t. [DOI] [PubMed] [Google Scholar]
- 32.Frisch MJ, et al. Gaussian 09, Revision A.1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 33.Noodleman L. J Chem Phys. 1981;74:5737. [Google Scholar]
- 34.Sun YM, Liu CB, Lin XJ, Bi SW. New J Chem. 2004;28:270. [Google Scholar]
- 35.AMPAC™ 9. Semichem, Inc; 12456 W 62nd Terrace - Suite D, Shawnee, KS 66216: 1992–2008. [Google Scholar]
- 36.Musie G, Farmer PJ, Tuntulani T, Reibenspies JH, Darensbourg MY. Inorg Chem. 1996;35:2176–2183. doi: 10.1021/ic9515968. [DOI] [PubMed] [Google Scholar]
- 37.Jeffery SP, Lee J, Darensbourg MY. Chem Commun. 2005:1122–1124. doi: 10.1039/b416898d. [DOI] [PubMed] [Google Scholar]
- 38.Tonzetich ZJ, Do LH, Lippard SJ. J Am Chem Soc. 2009;131:7964–7965. doi: 10.1021/ja9030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mazak RA, Collins RL. J Chem Phys. 1969;51:3220–3225. [Google Scholar]
- 40.Ye S, Neese F. J Am Chem Soc. 2010;132:3646–3647. doi: 10.1021/ja9091616. [DOI] [PubMed] [Google Scholar]
- 41.Harrop TC, Tonzetich ZJ, Reisner E, Lippard SP. J Am Chem Soc. 2008;130:15602–15610. doi: 10.1021/ja8054996. [DOI] [PubMed] [Google Scholar]
- 42.Sanina NA, Rakova OA, Aldoshin SM, Shilov GV, Shulga YM, Kulikov AV, Ovanesyan NS. Mendele-ev Commun. 2004:1–2. [Google Scholar]
- 43.Huang HW, Tsou CC, Kuo TS, Liaw WF. Inorg Chem. 2008;47:2196–2204. doi: 10.1021/ic702000y. [DOI] [PubMed] [Google Scholar]
- 44.Tsai ML, Liaw WF. Inorg Chem. 2006;45:6583–6585. doi: 10.1021/ic0608849. [DOI] [PubMed] [Google Scholar]
- 45.Chen CH, Ho YC, Lee GH. J Organomet Chem. 2009;694:3395–3400. [Google Scholar]
- 46.Kennedy MC, Antholine WE, Beinert H. J Biol Chem. 1997;272:20340–20347. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- 47.Lee M, Arosio P, Cozzi A, Chasteen MD. Biochemistry. 1994;33:3679–3687. doi: 10.1021/bi00178a026. [DOI] [PubMed] [Google Scholar]
- 48.Goussias C, Deligiannakis Y, Sanakis Y, Ioannidis N, Petrouleas V. Biochemistry. 2002;41:15212–15223. doi: 10.1021/bi026223e. [DOI] [PubMed] [Google Scholar]
- 49.Konkle ME, Elsenheimer KN, Hakala K, Robicheaux JC, Weintraub ST, Hunsicker-Wang LM. Biochemistry. 2010;49:7272–7281. doi: 10.1021/bi1007904. [DOI] [PubMed] [Google Scholar]
- 50.Hsueh K, Westler WM, Markley JL. J Am Chem Soc. 2010;132:7908–7918. doi: 10.1021/ja1026387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.