

Abstract
An analytically feasible, deterministic model for the spread of drug resistance among human malaria parasites, which incorporates all characteristics of the complex malaria-transmission cycle was introduced by Schneider and Kim (Theor. Popul Biol, 2010). The model accounts for the fact that only a fraction of infected hosts receive drug treatment and that hosts can be co-infected by differently many parasites. Furthermore, the model also incorporates host heterogeneity. Antimalarial-drug resistance is assumed to be caused by a single locus with two alleles—a sensitive one and a resistance one. The most important result for this model is that an analytical solution for the frequencies of a linked neutral biallelic locus exists. However, the exact solution does not admit an explicit form, and cannot straightforwardly be interpreted in terms of the model parameters. Here, we establish simple approximations for the equilibrium frequency at the neutral locus. Under the assumption that the resistant allele is initially rare—the biologically most relevant assumption in this context—and that recombination is weak, the approximations become similar to the approximations in the standard hitchhiking model. However, there are crucial differences. In particular, because of the high degree of selfing among malaria parasites in their sexual phase, a genome-wide reduction of relative heterozygosity occurs if selection is sufficiently strong. It turns out that the approximations are accurate even if the recombination rates are not small and the resistant allele is initially not very rare. The main advantage of our approximations is that they are easy to interpret in terms of model parameters. Moreover, they allow to make predictions of the size of the valley of reduced heterozygosity around the selected locus for given model parameters. Reversely, for a given reduction of heterozygosity, it is possible to identify the corresponding parameters. Moreover, we will show that incorporating host heterogeneity leads to an increased hitchhiking effect.
Keywords: Selective sweep, Co-infections, Drug concentration, Relative heterozygosity, Host heterogeneity, Malaria, Plasmodium falciparum
1 Introduction
Human malaria is an infectious disease caused by parasites belonging to the genus Plasmodium, which is endemic in most tropical and subtropical regions in the world. Infections with Plasmodium falciparium, the most virulent form of human malaria, result worldwide in one to 3 million deaths per year (cf. WHO 2000; Korenromp et al. 2005). Malaria control is highly dependent on drug treatments that kill parasites in infected hosts. However, attempts to control malaria have been thwarted by rapid evolution of antimalarial-drug resistance, a fact that has been described to be a public health disaster (cf. Marsh 1998).
The limited repertoire of safe, effective, and affordable antimalarial drugs has made research on the emergence and dispersion of resistance a global health priority. Mathematical models that can use input from genetic data to investigate the dynamics of mutations associated with drug resistance are urgently needed for designing drug-deployment policies that can increase the lifespan of the available drugs. This requires a detailed understanding of population genetic processes that lead to the emergence and dispersion of drug resistance. Unfortunately, the highly complex nature of the malaria-transmission cycle as well as complex demographic and environmental factors aggravate the efforts to elaborate theoretical models.
Malaria parasites undergo a complex transmission cycle with sexual phases in the mosquito vector and asexual phases in the infected host (cf. e.g. Daily 2006; Prugnolle et al. 2009). A human host is inoculated with sporozoites by the bite of an infected Anopheles mosquito during its blood meal. In the human host the sporozoites migrate first to the liver where they differentiate into hepatic merozoites. These are released into the blood stream where some of the hepatic merozoites form gametocytes. The haploid gametocytes are extracted by a mosquito during its blood meal and immediately reproduce sexually in the mosquito’s gut. Consequently, they undergo recombination. Completing the transmission cycle, this step is followed by the production of haploid sporozoites in the mosquitos’s salivary glands from which they can be inoculated into a human host.
Another source of complication is that many environmental and clinical factors differ significantly across the worldwide distribution of this parasite. The transmission rate and hence the number of secondary infections varies from very low rates in parts of South America, over intermediate rates in Southeast Asia, to high rates in Africa. On the other hand the level of host-acquired immunity is much higher (which means a high number of asymptomatic infections) in most of the affected areas in Africa than in other parts of the world. Such variation in host-acquired immunity affects drug use.
Among others, two important variables that summarize the demographic and clinical setting of the particular geographic area are the (average) number of parasites (m) co-infecting a given host (the average multiplicity of infection), which is determined by transmission intensity, and the proportion (α) of infected hosts that are drug treated. This parameter depends mainly on how many hosts acquired immunity.
Several studies built population genetic models that demonstrated profound effects of m and α on the rate of drug-resistance evolution (Hastings and Mackinnon 1998; Mackinnon and Hastings 1998; Hastings 2003,2006). However, it is still unclear which mechanisms are important to the spread of resistance, i.e., intra-host dynamics, drug half life, multiple drug treatment, migration, multiple infections, recombination, mutation etc.
Over the last 2 decades substantial advances regarding the genetic basis of antimalarial-drug resistance have been made. It is known that specific point mutations in the dhfr and dhps regions underlie resistance to pyrimethamine and sulfadoxine (cf. Cowman et al. 1988; Triglia and Cowman 1994; Brooks et al. 1994), that point mutations in mitrochandrial DNA underlie resistance to atovaquone (cf. Schwobel et al. 2003), and that mutations in the pfcrt gene are causing resistance against Chloroquine (CQ).
The spread of mutations causing drug resistance leads to a valley of reduced genetic variation at linked neutral regions. This removal of pre-existing variation occurs because recombination cannot effectively break the initial association with the neutral background in which the mutant first occurred, a process known as genetic hitchhiking or a selective sweep (Maynard Smith and Haigh 1974; Stephan et al. 1992; Barton 2000). For instance, Nair et al. (2003) observed a severe reduction of variation at microsatellite loci spanning over a 100 kb region surrounding the dhfr gene in a Southeast Asian population of P. falciparum. The extent of this pattern depends on how fast the favored allele increases to high frequency while meiotic recombination is constantly eroding the association between the favored allele and the surrounding chromosome segment (Kim and Stephan 2002). Selective sweeps have been initially studied for randomly mating populations of constant size, and homogenous constant selection pressures (e.g., Maynard Smith and Haigh 1974), to which we refer as the ‘standard model’, or standard ‘hitchhiking’.
In malaria biology, detection of selective sweeps mainly contributed to confirming the location of drug resistant mutations and elucidating their mutational origins (Wootton et al. 2002; Nash et al. 2005; Mita et al. 2007; Nair et al. 2007; McCollum et al. 2008), while fewer studies attempted to relate the span of selective sweeps with the strength of drug selection (Nair et al. 2003). Recently, Schneider and Kim (2010) introduced an analytical feasible model for the spread of antimalarial-drug resistance, which allows to study genetic hitchhiking. The model covers the important characteristics of the transmission cycle, incorporates host heterogeneity, i.e., different classes of treated and untreated hosts, and accounts for the fact that hosts can be infected by differently many parasites. Moreover, the model allows simple conditions for the spread of resistance and its speed in terms of the fitness parameters and α. Hence, it is useful to find ‘optimal’ treatment strategies to prevent or slow down the spread of resistance (for more discussion see Schneider and Kim 2010).
Studies based on known recombination rates and the result of the standard model of selective sweeps concluded that the observed patterns of selective sweeps are compatible with the predictions (Nair et al. 2003). However, Schneider and Kim (2010) discuss in detail why the application of the standard selective-sweep model to a malaria-parasite population is highly problematic. In particular, the high degree of selfing among malaria parasites will lead to a genome-wide reduction of heterozygosity if selection is sufficiently strong (cf. also Hedrick 1980).
In the absence of reliable public-health records, a retrospective analysis of the mechanisms and parameters underlying the spread of antimalarial drug resistance may be achieved indirectly through the patterns of selective sweeps. However, the analytical solution for the valley of reduced heterozygosity in the model of Schneider and Kim (2010) is not explicit, which limits their applicability. Moreover, it is not at all straightforward to interpret these results based on the model parameters.
In this article we derive accurate approximations to the exact, analytical solutions of Schneider and Kim (2010). The approximations under the assumption that the resistant allele is initially rare and that recombination is weak are similar to but different from the usual approximation for standard hitchhiking. Unlike in standard hitchhiking, our approximations are accurate even if the recombination rates are large and the resistant allele is initially not very rare. Moreover, using the approximations, we will show that incorporating host heterogeneity will result in an increased hitchhiking effect compared to our basic model, with corresponding selection parameters, which accounts just for one class of treated and one class of untreated hosts. The simple form of the approximations will allow us to easily interpret the effect of genetic hitchhiking in terms of the model parameters. Such approximations are extremely useful to achieve applicability of the results of Schneider and Kim (2010) to real data. Moreover, we show that the approximate solutions can be easily applied to identify the range of parameters that give rise to given levels of reduced heterozygosity in a given range of recombination distances. Finally, we discuss the differences compared with standard hitchhiking and give an outlook how our results can be applied to real data.
2 The model
We consider two biallelic loci. The first locus is subject to selection with a sensitive allele AS and a resistant allele AR segregating. The second locus is selectively neutral with the alleles N1 and N2 segregating. We will use the notation and parametrization summarized in Table 1.
Table 1.
Summary of notation
| Haplotypes | ASN1 | ASN2 | ARN1 | ARN2 | ||||
|---|---|---|---|---|---|---|---|---|
| Frequency | p1 | p2 | p3 | p4 | ||||
| Fitness in |
|
|
|
|
||||
| Untreated hosts | 1 | 1 | 1 − s | 1 − s | ||||
| Fitness in |
|
|
|
|
||||
| Treated hosts | 1 − dS | 1 − dS | 1 − dR | 1 − dR | ||||
| Sensitive/resistant | Sen. | Sen. | Res. | Res. |
The tables shows the notation of the four haplotypes, their frequencies, fitnesses in treated and untreated hosts, the parametrization of fitnesses that we are going to use for the illustrations in the following sections, and whether the haplotypes are sensitive or resistant. Here, s reflects metabolic costs of the resistant allele, while dS and dR indicate how efficiently the drug wipes out the sensitive and resistant parasites, respectively
Let p denote the frequency of the resistant allele AR. We have p = p3 + p4. Furthermore, we denote the frequency of the neutral allele N1 among the sensitive and resistant haplotypes by R and Q, respectively. We therefore have and .
Moreover, we denote the recombination rate between the two loci by r, and the vector of haplotype frequencies by p = (p1, … , p4). We assume that each host is infected randomly and independently by exactly m haploids (parasite strains). (We assume m to be a fixed parameter until Sect. 4.2, where we assume that m follows a fixed frequency-distribution).
Hosts acquire parasite strains according to their frequencies in the mosquito phase. Hence, the configuration of infections in hosts is multinomially distributed with parameters m and p1, … , p4. We assume that all haploids in a newly infected host have equal frequencies. Hence, the relative frequency of a haploid drawn from the parasite population among mosquitos is . Let us denote a multi-index by m = (m1, … , m4), and the sum over its components by . The probability that a host is infected by mi copies of haplotype i (i = 1, … , n, |m| = m) is given by
| (1) |
where denotes the respective multinomial coefficient and, as usual, .
After a host is infected the parasites reproduce clonally in the host. An infected host either receives drug treatment or is untreated. We assume that a fixed proportion α of infected hosts in the population is treated, whereas the remaining hosts are untreated. Thus, the probability for a host to be treated is simply α. The rate of reproduction of the haplotypes is different in treated and untreated hosts. The absolute fitness of a parasite strain is the expected number of its descendants in the host at the time of the mosquito visit. The absolute frequency (fitness) of haplotype i in an untreated host before a mosquito takes its blood meal is denoted by , whereas the absolute frequency of haplotype i in a treated host before a mosquito takes its blood meal is denoted by . [In the following, wherever it is appropriate, we use the superscript to (.) to resemble the superscripts (U) and (T).] Some of these haploids form gametocytes in male or female expressions. The frequencies of those are assumed to be proportional to the number of respective haplotypes. Furthermore, we impose that male and female gametocytes occur at the same frequencies. We assume that the number of different gametocytes taken by a mosquito during its blood meal from an infected host is proportional to its frequency in the host. Let γ denote the proportional constant, which is assumed to be the same for each mosquito. Hence, if the absolute frequencies of parasites in an infected host are , the absolute frequencies of parasites absorbed during the blood meal are . Note that this takes drug efficiency into account, because a mosquito will absorb a smaller number of parasites from a host in which drugs efficiently eliminated parasites.
In the mosquitos’ guts recombination occurs immediately after the blood meal during the phase in which meiosis occurs. In the gut of a mosquito, which has taken its blood meal from a host initially infected with mi haplotypes i (i = 1, … , 4), the probability that a male k-gametocyte fertilizes a female l-gametocyte is
where
is the frequency of parasite haploids in the mosquito’s gut. The above probability is the relative frequency of a male k-gametocyte times that of a female l-gametocyte. Therefore, the absolute frequency of pairings of a male k-gametocyte and a female l-gametocyte is, the probability of such a fertilization times the absolute numbers of parasites in the gut, i.e.,
| (2) |
The probability that a fertilization of a male k-gametocyte to a female l-gametocyte produces a haplotype i is denoted by R(kl → i). Therefore, the absolute frequencies of haplotype i in the population of mosquitos that took their blood meal from treated and untreated hosts are respectively
where the sum runs over all multi-indices with |m| = m. Therefore, the relative frequencies of haplotypes in the mosquito population become
| (3a) |
where
| (3b) |
For what follows, let us define the average fitness of the resistant allele among treated and untreated hosts by
| (4a) |
and that of the sensitive by
| (4b) |
If p(t) denotes the frequency of the resistant allele at time t, it changes according to , i.e., according to the standard haploid one-locus selection model (cf. Schneider and Kim 2010). Thus, the resistant allele will sweep through the population if and only if λ > μ, otherwise it will get lost (or remain constant in frequency). Hence, in the following we will always assume λ > μ without further mentioning. Note that the spread of the resistant allele is independent from m. Moreover, it is obvious to calculate the time until a given level of resistance is reached (cf. Result 2 in Schneider and Kim 2010). Because of the relatively simple form of the dynamics of the resistant allele it is possible to derive simple conditions for the spread of resistance and its speed. Example 1 in Schneider and Kim (2010) directly links the fitness parameters to the spread of fixation. Furthermore, assuming that the fitnesses in treated hosts are functions of the administered drug concentration, Example 2 in Schneider and Kim (2010) illustrates the impact of the drug concentration and α on the spread of resistance and its speed. Assuming that the fitness of resistant parasites decays slower than that of the sensitive ones as a function of the drug concentration they found that resistance spreads most quickly for intermediate drug concentrations and large α.
Anyhow, the focus of this article is genetic hitchhiking. The following relations were derived in Schneider and Kim (2010).
| (5a) |
where
| (5b) |
| (5c) |
and
| (5d) |
It was further shown in Schneider and Kim (2010) that
| (6a) |
and
| (6b) |
From the relations (5) and (6) the equilibrium frequency of the neutral allele N1 was calculated to be
| (7) |
where p0, Q0, and R0 are the respective initial frequencies,
and
| (8) |
It was mentioned by Schneider and Kim (2010) that, for practical purposes, it suffices to sum (7) until the time of quasi-fixation, for which they provided an explicit formula. However, it is not at all obvious how Q̂(m) is influenced by the various model parameters. In particular, when studying genetic hitchhiking using (7) it seems infeasible to predict the width of the valley of reduced heterozygosity in terms of the recombinational distance. Hence, simple but accurate approximations for (7) that permit a simple interpretation in terms of the model parameters are highly desirable.
3 Approximations
We shall now calculate simple approximations for (7). First, we treat the special case m = 2, and later deduce the general case from it.
3.1 Equilibrium frequencies at the neutral locus for m = 2
The equilibrium frequency of the neutral allele N1 is given by
where
| (9) |
is independent of the trajectory pt of the resistant allele and in particular of p0, and
| (10) |
In the following we will derive upper and lower bounds as well as approximations for Q̂ which exhibit a much simpler form. This permits us to explore the effects of the various parameters on Q̂.
First, note that Λl is monotone decreasing in l. To see this note that Λl < Λl−1 is equivalent to
This can be simplified to
By dividing through λl−1 μl−1 this simplifies to
Moreover, note that
| (11) |
Hence, by using
| (12) |
we obtain
Furthermore, because λ > μ we have
| (13) |
and consequently Λ̃ ≥ Λ̅.
Let us define
| (14) |
Note that by relabeling the alleles, we can assume without loss of generality that R0 > Q0. Hence, (14) is monotone increasing in a. Thus, we can formulate our first result.
Result 1
Let Q(a) be defined as in (14). Then, Q(a) is monotone increasing in a. Moreover, let Λ̲, Λ̅ and Λ̃ be defined as in (11), (12), and (13), respectively. Then
are upper bounds for Q̂, and
is a lower bound for Q̂.
Although the expressions of this bounds are simpler than that of Q̂ they are still not very explicit. Hence, we shall derive upper and lower bounds for Q̅ and Q̲, or more generally for Q(a).
First, note that ϑ < λ. This is because it is equivalent to
which obviously holds because
Essentially the same calculation shows ϑ < μ. Therefore, we have and . This combined with (11) and (13) implies ,since . Hence, in the following we will always assume .
Notice that Q̃ has exactly the same form as in the standard haploid hitchhiking model derived by Maynard Smith and Haigh (1974; eq. 8), with r replaced by and p0 replaced by . Thus, we can interpret this as studying standard hitchhiking with an ‘effective’ recombination rate , which is smaller than r, and an ‘effective’ initial frequency of , which is smaller than the actual initial frequency. The adjusted recombination rate leads to a more severe hitchhiking effect, whereas the adjusted initial frequency leads to a less pronounced hitchhiking effect. Combination of this factors lead to a more severe hitchhiking effect (cf. Schneider and Kim 2010).
Next, we need two definitions. For a ∈ ℝ and k ∈ ℕ+ the upper Pochhammer symbol is defined by
The hypergeometric function is defined by
where r, s ∈ ℕ, z, a1, …, ar ∈ ℝ, b1, … , .
With the above definition we are already able to formulate our first theorem.
Theorem 1
Assume for l ∈ ℕ, and let csc denote the cosecant of x. If , the function
| (15) |
is a lower bound for Q(a). If ,
| (16) |
is an upper bound for Q(a).
Proof
Consider the function
| (17) |
for x > −1 with a ∈ (0, 1), and let
| (18) |
By calculating the derivative, which is given by
and obviously always negative, we recognize that g is monotone decreasing as a function in x. Thus, we obtain the estimate
| (19) |
Since, we have
it follows from (19) that
| (20) |
is a lower bound for Q(a), and that
| (21) |
is an upper bound for Q(a).
We will now manipulate the above equations. First, rewrite g as
| (22) |
where
| (23) |
Assume first that cexη < 1. Hence, g is a geometric series, can be rewritten as
| (24) |
and converges absolutely for all x satisfying cexη < 1. If cexη > 1, we have and can expand (22) into the power series
which converges absolutely for all x satisfying cexη > 1.
Let us further define . The assumption guarantees x0 > 0. The absolute convergence of the above series enables us to integrate by summands, i.e., for x < x0 we have
| (25a) |
| (25b) |
| (25c) |
Note that we have used the fact that for all k ∈ ℕ in (25b), which is guaranteed by the assumption for all k ∈ ℕ.
Similarly, for x > x0 we have
| (26a) |
| (26b) |
| (26c) |
| (26d) |
It should be mentioned that we did not need the assumption for all k ∈ ℕ in (26).
Next, assume i.e., x0 > 0. Hence, we have
Since , the Leibnitz criterium implies that
| (27) |
converges, i.e., −∞ < G1(x0) < ∞. Furthermore, by a similar argument we obtain −∞ < G2(x0) < ∞. We also see that
Hence, for x > x0 this implies that the series representation (26d) of G2(x) is absolute convergent. Therefore, we obtain
| (28) |
and
| (29) |
Similarly, we see that G1(x) is absolute convergent for x0 < x. Therefore, we have
| (30) |
We obtain
| (31) |
If , we have x0 > − 1. The above calculations are still valid and yield
| (32) |
Let us now simplify (31) and (32). Note that
By using the the well known series
we arrive at
Because for k ∈ ℕ we have . Because, we assume a ∈ [0, 1] and λ > μ we also have for k ∈ ℤ−. Hence, we have . Therefore, we obtain
and
Combining the above with (20) and (21) and the definitions of ξ, η, and c immediately yields (15) and (16).
The assumptions , or hold whenever one aims to study the hitchhiking effect of a mutation that is initially rare. If the initial frequency of the mutation is not rare because one is interesting in studying mutations from standing genetic variation that become beneficial, for instance, because of a change in the environment, i.e., if one wants to study soft selective sweeps (cf. Hermisson and Pennings 2005), Theorem 1 is not applicable. The study of soft selective sweeps is relevant and hence we shall also treat the cases and .
Remark 1
The proof of Theorem 1 reveals that we have to replace (15) by
| (33) |
if . Moreover, we have to replace (16) by
| (34) |
if . Notably, (33) and (34) also hold if for k ∈ ℕ. Furthermore, (33) and (34) are the continuations of (15) and (16) as functions in p0.
Theorem 1 assumes for all k ∈ ℕ. If this assumption is violated we have to make the following adjustments.
Remark 2
Assume l ∈ ℕ. If ,
| (35) |
is a lower bound for . If ,
| (36) |
is an upper bound for . Moreover, (35) and (36) are the continuations of (15) and (16) in the limit . If , (35) has to be replaced by (33). If , (36) has to be replaced by (34).
The proof can be found in Appendix A.1.
Let
| (37) |
and analogously
| (38) |
We immediately obtain the following corollary.
Corollary 1
We have Ψ̃ ≥ Ψ̅ ≥ Q̅ ≥ Q̂ ≥ ψ̲, hence Ψ̃ and Ψ̅ are upper bounds for Q̂, and ψ̲ is a lower bound for Q̂. Moreover, we have Ψ̃ ≥ Q̃ ≥ ψ̃, Ψ̅ ≥ Q̅ ≥ ψ̅, and Ψ̲ ≥ Q̲ ≥ ψ̲.
Figure 1 illustrates Q̂ and its bounds Q̲ and Q̅, as well as their bounds Ψ̅, Ψ̲, ψ̅, and ψ̲ for various parameters. It becomes obvious that the upper bound Q̅ is very close to Q̂, whereas the lower bound Q̲ greatly underestimates Q̂.
Fig. 1.
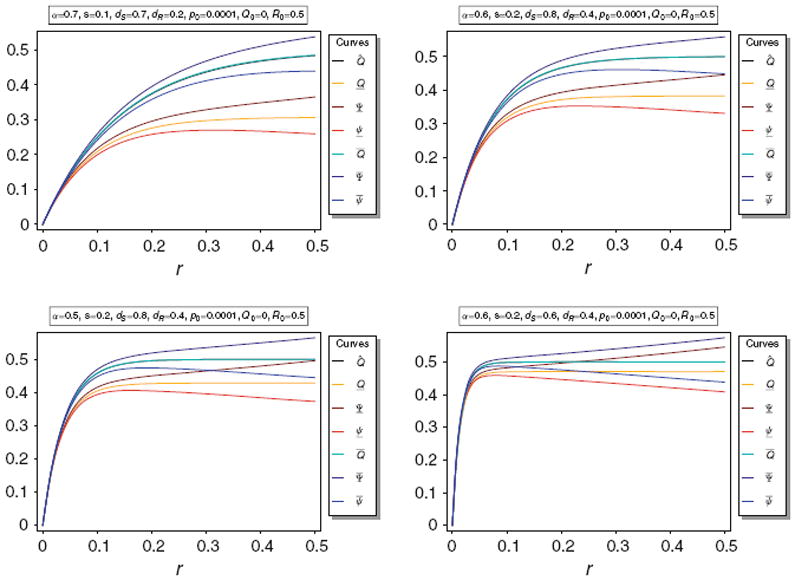
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds for Q̂ as a function of r for different parameter combinations. The panels show Q̂ along with its upper and lower bounds Q̅ and Q̲, as well as their respective upper and lower bounds Ψ̅ and ψ̅, and Ψ̲ and ψ̲. The parameters for the various plot panels are specified in the boxes above the panels. In all panels Q̂ and Q̅ are almost identical
It turns out that Q̃ and Q̅ are almost identical unless p0 is large. The same holds for for Ψ̃ and Ψ̅. This is illustrated in Fig. 2.
Fig. 2.
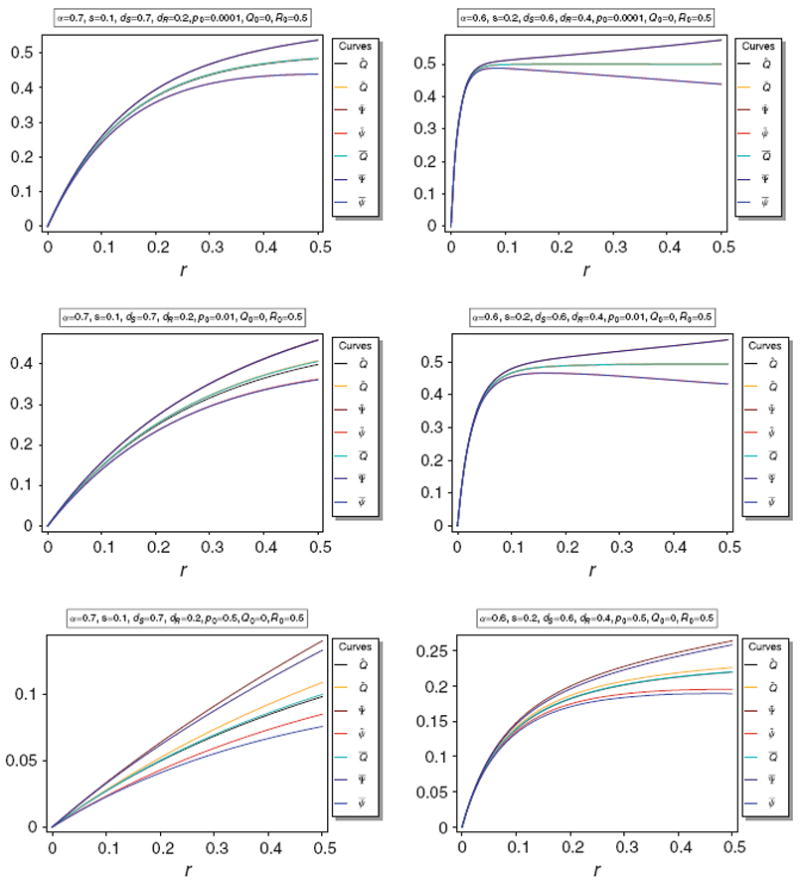
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds of Q̂ as a function of r for different parameter combinations. The panels show Q̂ along with its upper bounds Q̅ and Q̃, as well as their respective upper and lower bounds Ψ̅ and ψ̅, and Ψ̲ and ψ̲. The parameters for the various plot panels are specified in the boxes above the panels
Although, Ψ (a) and ψ(a) have closed expressions, these expressions involve the hypergeometric function. In the following we shall derive approximations for Ψ(a) and ψ(a) that make no use of the hypergeometric function. Our first step is the following theorem.
Theorem 2
For let
| (39) |
and, for let
| (40) |
If , ϕ(a) ≈ ψ(a) and if , Φ(a) ≈ Ψ (a).
The proof can be found in Appendix A.1.
If , or we have to modify the definitions of ϕ(a), or Φ(a), respectively.
Remark 3
For let
| (41) |
and, for let
| (42) |
If , ϕ(a) ≈ ψ(a) and if , Φ(a) ≈ Ψ(a). Moreover, (41) and (42) are the continuations of (39) and (40) regarded as functions in p0.
The proof can be found in Appendix A.1.
From Theorem 2, Remark 3, and the definitions of Λ̲, Λ̅, and Λ̃ we immediately obtain the following corollary.
Corollary 2
Let Ψ, ψ, Φ and ϕ be defined as in Theorems 1 and 2, and Remarks 1–3. Furthermore, let
| (43) |
and
| (44) |
Then Φ̃ ≈ Ψ̃, Φ̅ ≈ Ψ̅, Φ̲ ≈ Ψ̲, ϕ̃ ≈ ψ̃, ϕ̅ ≈ ψ̅, and ϕ̲ ≈ ψ̲.
The approximations provided in Corollary 2 are very accurate unless p0 becomes too large. This is illustrated in Fig. 3.
Fig. 3.
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds of Q̂ as a function of r for different parameter combinations. The panels show Q̂ along with its upper bounds Q̅ and Q̃, as well as their respective upper and lower bounds Ψ̅ and ψ̅, and Ψ̃ and ψ̃, along with their approximations Φ̅ and ϕ̅, and Φ̃ and ϕ̃, respectively. The parameters for the various plot panels are specified in the boxes above the panels
Note that we derived the upper and lower bounds for Q(a) by using the estimate (19). However, we can use (19) for the standard estimate
| (45) |
Hence, the estimates summarized in the next corollary follow immediately.
Corollary 3
Estimates for Q̂ are given by
| (46) |
and
| (47) |
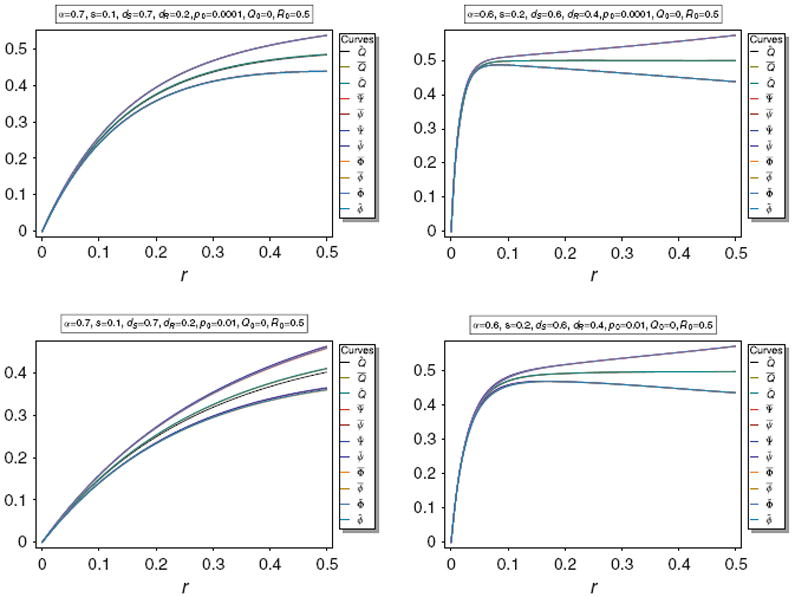
The approximations of Corollary 3 are illustrated in Fig. 4. It is obvious that the above approximations are very accurate for various parameters.
Fig. 4.
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds of Q̂ as a function of r for different parameter combinations. The panels show Q̂ along with its upper bounds Q̅ and Q̃, as well as its approximations Q̅*, Q̃*, Q̅°, and Q̃°. The parameters are the same as in Fig. 3
Although we already arrived at relatively simple approximations for Q̂, they are still difficult to interpret in terms of the involved parameters. In the following we will concentrate on the case in which the initial frequency p0 of the resistant allele is small. This is the most relevant case for studying genetic hitchhiking.
3.2 Approximations for rare mutations
So far in our derivations we did not assume that the initial frequency p0 of the resistant allele is small. However, since this is the biologically most relevant situations we shall derive further approximations for the equilibrium frequencies of the neutral alleles under the assumption that the mutation is initially rare, i.e., p0 ≈ 0 and 1 − p0 ≈ 1. Since we have Λ̃ ≈ Λ̅ under this assumption we focus on estimates based on Λ̃.
For p0 ≈ 0 we obtain the following theorem.
Theorem 3
Let
| (48) |
For p0 ≈ 0 we have Q°(a) ≈ A(a).
The proof can be found in Appendix A.1.
In accordance to out previous notation we set
| (49) |
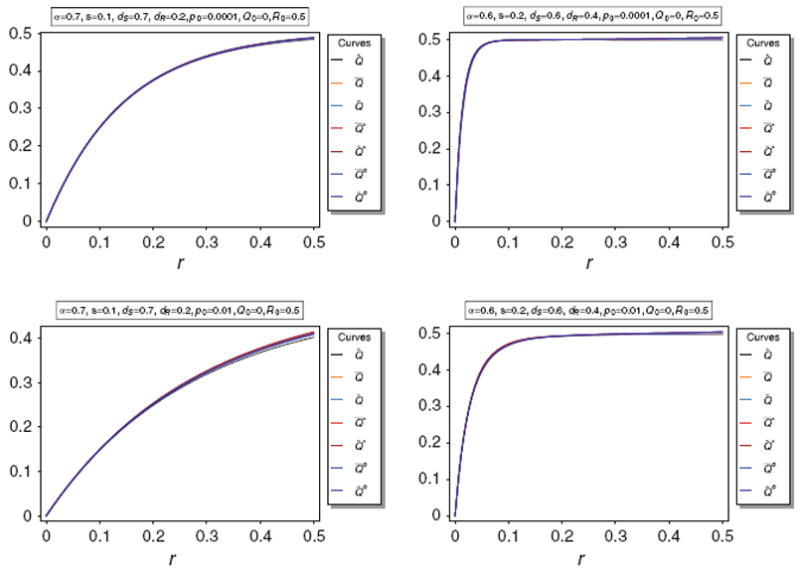
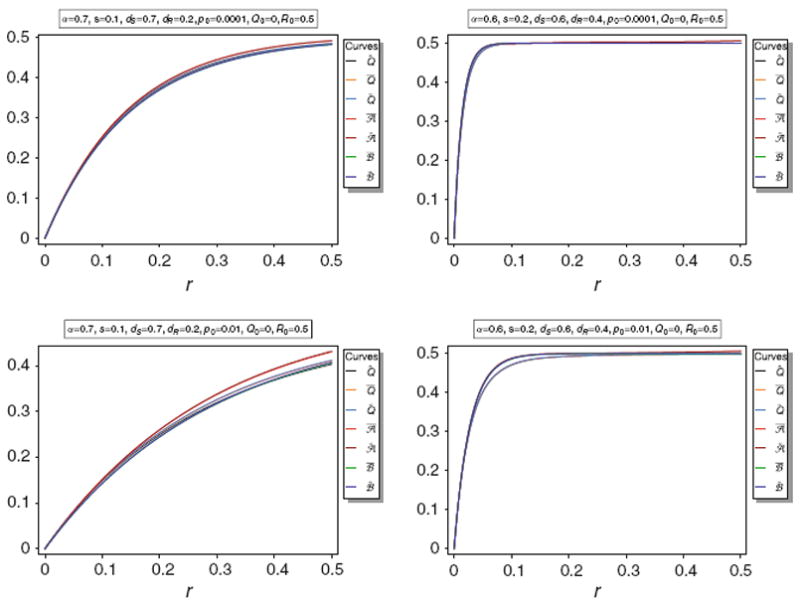
These approximations are illustrated in Fig. 5.
Fig. 5.
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds of Q̂ as a function of r for different parameter combinations. The panels show Q̂ along with its upper bounds Q̅ and Q̃, as well as the approximations A̅, Ã, B̅, B̃. The parameters are the same as in Fig. 3
Now, we shall additionally assume that a ≈ 1.
Theorem 4
Let a(x) = 1 − rx with and
| (50) |
For p0 ≈ 0 and rx ≈ 0 we have Q°(a(x)) ≈ B(x).
Proof
We have log a(x) ≈ −rx. Hence,
If rx ≈ 0, we can neglect and obtain
Moreover, because p0 ≈ 0 we have . Thus,
Note that we have and . Let us define
| (51) |
We obtain the following corollary.
Corollary 4
We have
| (52) |
For p0 ≈ 0 and , we have Q̃° ≈ B̃.
Figure 5 illustrates the approximations A̅, Ã, B̅, B̃. It is easily seen that the approximations are very accurate unless p0 is too large (p0 ≳ 0.1). Anyway, the approximations are still acceptably accurate for p0 = 0.1.
By expanding into a Taylor series around x = 0, and by setting we obtain
| (53) |
Hence, for r ≈ 0 we can further approximate this by neglecting terms of order O(r2) or O(r3) and higher, and obtain
| (54) |
and
| (55) |
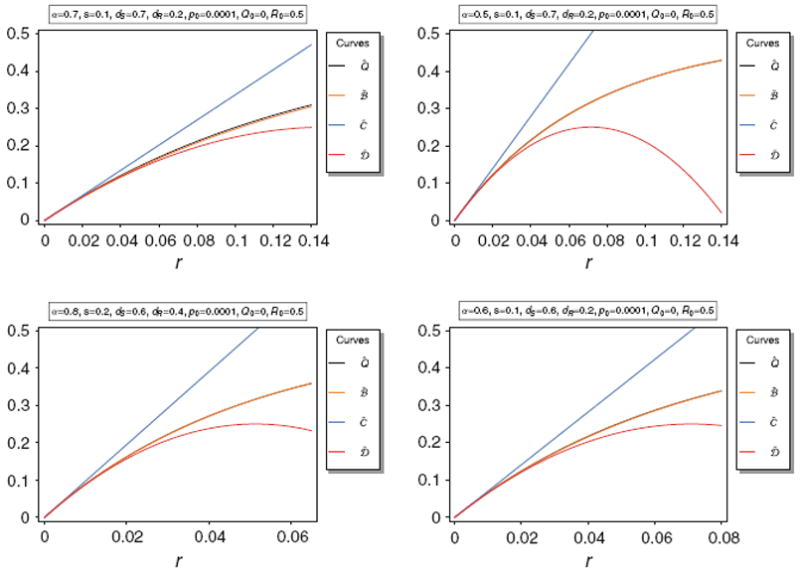
Figure 6 shows Q̂ along with its approximations C̃ and D̃. It is obvious that these approximations are only accurate for very small r. Not surprisingly D̃ is a much better approximation than the linear approximation C̃.
Fig. 6.
Equilibrium frequency Q̂ of the neutral allele N1 and various bounds of Q̂ as a function of r for various parameter combinations. The panels show Q̂ along with its upper bounds Q̅ and Q̃, as well as the approximations C̃ and D̃.
3.3 The hitchhiking effect for general m
Since the sensitive allele goes extinct in the population, the equilibrium frequency of the neutral allele N1 is given by (7), i.e., by
The above formula has the same structure as in the case m = 2. If we set m = 1 there is no recombination and hence the initial proportions of the neutral allele among genotypes with the resistant allele remain constant over time. Hence, we have .
We shall now derive approximations for Q̂(m). As easily seen from (7), we can apply our approximations from the case m = 2 if we make the approximation and ϑpτ,m ≈ b. We first approximate ϑpτ,m by a constant. From (5b) we obtain
Let
| (56) |
and
| (57) |
Hence, we have
where the last equality follows from the binomial formula. Similarly, we obtain ϑpτ,m ≥ ϑ̲(m).
Therefore, we obtain
Using the same approximation as in the case m = 2 yields
Hence, by replacing ϑ and a by ϑ̅(m) and Λ̅(m), or ϑ̲(m) and Λ̲(m), respectively, we obtain the upper and lower bounds
and
respectively.
It turns out that the upper and lower bounds are very inaccurate estimates for Q̂(m). However, it is possible to find relative accurate approximations for Q̂(m), at least if p0 is small, which is the most important case for our current purpose. If p0 ≈ 0, we have , so that we obtain
| (58) |
From (5c) we see that ϑ̃(m) = θ0,m Moreover, define
| (59) |
Hence, by approximating ϑpτ,m by ϑ̃(m) and Λm,l by Λ̃(m) we obtain
| (60) |
This approximation turns out to be sufficiently accurate unless p0 is too large.
Clearly, Q̃(m) can be further approximated as in the case m = 2. However, note that for m > 2 the assumption that p0 is small is always implicitly made because otherwise the approximation (58) for ϑpτ,m will be inaccurate. Hence, only those approximations that were derived under the assumption of p0 ≈ 0 are meaningful if m > 2, i.e., those of Result 4 and Corollary 4. We can therefore summarize:
Result 2
Let m ≥ 2 and
| (61) |
Then we have Q̂(m) ≈ B̃(m).
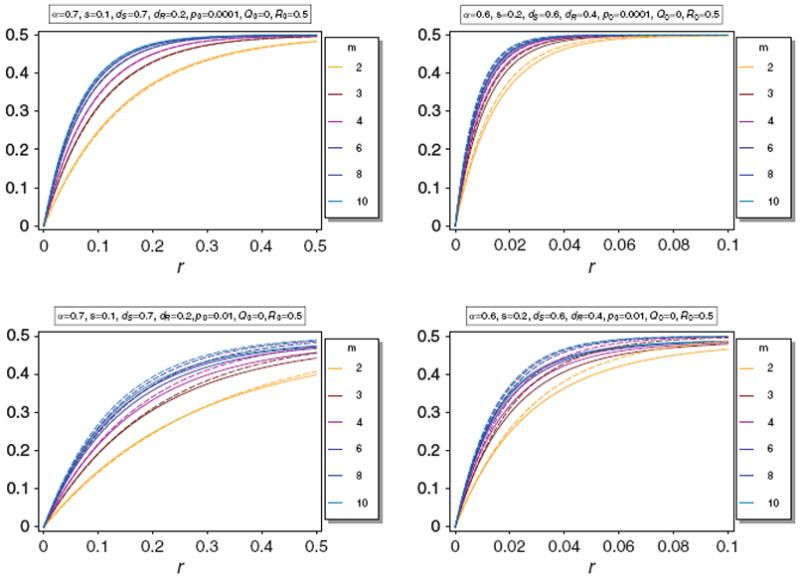
Note, that the approximations become worse for large m. However, as noted in Schneider and Kim (2010), the differences in Q̂(m) for different values of m become small for large m. Since, in reality m should be bounded by a maximum possible value it should be sufficient to assume m < 10. For these values Result 2 is still accurate. This is illustrated in Fig. 7. Furthermore, note that Result 2 becomes delicate for large m, because it is based on the approximation (58), which becomes very inaccurate as m → ∞.
Fig. 7.
Equilibrium frequency Q̂ (m) (thin lines) of the neutral allele N1 versus the approximation B(m) (thick dashed lines) for various values of m. The parameters are as in Fig. 3
4 The general model
Schneider and Kim (2010) also presented two generalizations of their model. First, they argued that the differentiation into treated and untreated hosts is oversimplified, and that host heterogeneity should be taken into account. Host heterogeneity can reflect for instance different levels of drug concentration, drug decay, different levels of host-acquired immunity, different immune responses etc. Second, they argued that it is oversimplified to assume that each host is infected by the same number of parasites, i.e., that m is a fixed parameter, and showed how the model can be generalized to the case in which m follows a given frequency distribution.
We shall briefly summarize the two generalizations, and show how our results are generalized.
4.1 Host heterogeneity
Assume again a fixed proportion α of hosts is treated. We divide the treated and untreated host into various different discrete “treated classes” and “untreated classes”. Let the proportion of infected hosts that fall into class j (j ∈ ℕ) be αj. Let U ⊆ ℕ and T = ℕ\U be the sets of treated and untreated classes, respectively. Hence, we have and . Let us denote the fitnesses of parasite haplotypes carrying the sensitive and resistant allele in hosts that fall into class j by and , and and , respectively.
Note that in the original formulation of the model we have , and . Hence, it can be regarded as the approximation that all untreated hosts are subsumed in just one class, in which is the mean fitness of the parasites among them and all treated hosts are subsumed in just one class, in which is the mean fitness of the parasites among them.
In the model accounting for host heterogeneity the equilibrium frequency of the neutral allele N1 is still given by (7), however the parameters λ, μ, are given by
| (62a) |
and
| (62b) |
whereas ϑp,m and Λm,t are still given by (5b) and (8), however in (5b) θk,m has to be replaced by
| (63) |
Hence, in all approximations ϑ̃(m) has to be replaced by
| (64) |
We summarize:
Result 3
Let m ≥ 2. The equilibrium frequency of the neutral allele N1, Q̂(m), is approximately given by
| (65) |
where λ,μ, and ϑ̃(m) are given by (62a), (62b), and (64), respectively.
It was mentioned in Schneider and Kim (2010) that accounting for host heterogeneity results in a more pronounced hitchhiking compared to the basic model if the values of λ and μ coincide, or more precisely if , and . The reason is that under this assumption ϑ̃(m) given by (64) is smaller than if it is given by (58), and (for fixed m) the respective approximations for the hitchhiking effect corresponds to standard hitchhiking with recombination rate and initial frequency . We will prove this in the Sect. 5.
4.2 Number of co-infections
Now, assume that the number m of parasites that infect a host follows some probability distribution over the population. Let κm denote the probability that a host is infected by m ≥ 1 parasites. Naturally, we have .
As shown in Schneider and Kim (2010), the equilibrium frequency of the neutral allele N1 is given by
| (66a) |
with
| (67a) |
where ϑpτ,m and Λm,t are given by (5b) and (8) (eventually with λ, μ, ϑp,m, Λm,t, and θk,m adjusted as in Sect. 4.1 to incorporate host heterogeneity). Hence, the equilibrium frequency of the neutral allele N1 can be approximated following the calculations of Sect. 3. We can summarize this in the following result.
Result 4
The equilibrium frequency of the neutral allele N1, Q̂(κ) is approximately given by
| (68) |
with , where λ, μ, and ϑ̃(m) are given by (4), and (58), respectively. If one wants to incorporate host heterogeneity, λ, μ, and ϑ̃(m) are given by (62), and (64), respectively.
5 Equilibrium heterozygosity and the hitchhiking effect
From now on we assume that m follows a probability distribution as in Sect. 4.2 and we account for host heterogeneity as in Sect. 4.1 unless otherwise mentioned.
Remember that the equilibrium heterozygozity is given by
| (69) |
We have seen in the last section that the equilibrium frequency, and its upper and lower bounds and approximations are given by expressions of the form Q0 + (R0 − Q0) A(r), where A is a function of the recombination rate that has to be chosen appropriately. For brevity we will suppress the dependence of Q̂(κ) on r unless necessary. Let us regard R0, i.e., the initial frequency of N1 among sensitive parasites, as a random variable and the heterozygosity as a function of R0. Let us write
| (70) |
Given the initial frequency of the neutral allele N1 is R0, the beneficial mutation occurs initially in association with allele N1 with probability R0, and in association with allele N2 with probability (1 − R0). Hence, we have Q0 = 1 with probability R0 and Q0 = 0 with probability (1 − R0). Therefore, the average heterozygosity given R0 is calculated to be
| (71) |
Hence, according to the theorem of total probability, the average heterozygosity is calculated to be
| (72) |
The initial heterozygosity is given by H0 = 2R0(1 − R0). Hence, the fraction of the expected equilibrium heterozygosity over the initial heterozygosity is given by
| (73) |
which is independent of the distribution of R0. Since A(0) = 0 we have H(0) = 0.
If we further approximate Q̂ by B̃ given by (52), we obtain
| (74) |
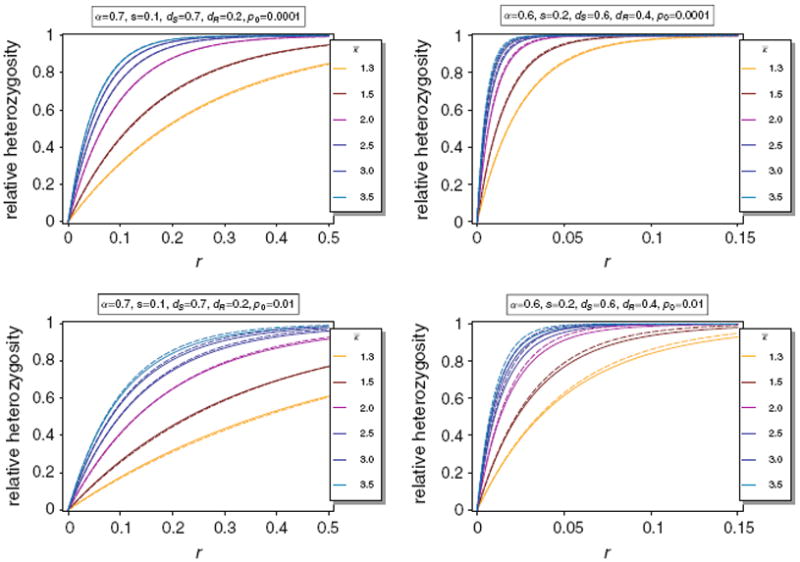
From (74) it is seen that H(κ) shows a strong genome-wide reduction if κ1 is large and selection is sufficiently strong. The approximation is illustrated in Fig. 8.
Fig. 8.
Relative expected heterozygosity H(κ) (thin lines) of the neutral allele N1 versus the approximation H̃(κ) (thick dashed lines) for different distributions κ. In all cases we assume a truncated exponential distribution with range 1–10 and mean κ̅. By truncated we mean that the probability of m = 10 is the probability that m ≥ 10 for a poisson distribution with mean κ̅. The parameters are as in Fig. 3
For the special case that m is constant the above reduces to which is increasing as a function of m. The reason is that ϑ̃(m) is monotone increasing in m, either if it is given by (58) or in the case of host heterogeneity by (64).
It was mentioned in Schneider and Kim (2010) that accounting for host heterogeneity results in a more pronounced hitchhiking compared to the basic model if the values of λ and μ coincide, or, more precisely, if and . The reason is that, for every m, under these assumptions ϑ̃(m) given by (64) is smaller than if it is given by (58). Hence, this holds also for the corresponding values of ϑ̃(κ), and consequently (74) is smaller if host heterogeneity is incorporated. We shall summarize this as a remark and prove it in the appendix.
Remark 4
Host heterogeneity leads to an increased hitchhiking effect, i.e., to a stronger reduction in relative heterozygosity H̃(κ)(r), compared to the basic model with only one class of treated and one class of untreated hosts with corresponding fitness parameters, i.e., , and .
For fixed large m the differences in H̃(m) become very small and vanish in the limit m → ∞ because H̃(m) approaches the classical approximation for standard hitchhiking.
Note, that the last statement of the remark is delicate, because the approximation H̃(m) will be inaccurate for very large m.
We can use (73), or (74) to calculate the maximum recombination distance for which a given reduction in relative heterozygosity can be observed. This is relevant for predicting the width of the valley of relative heterozygosity for given selection parameters. By comparison of (73), or (74) with empirical data on the relative heterozygosity, it is possible to re-evaluate or validate parameter estimates (e.g., for α, λ, μ, etc.).
From (73) we obtain that the maximum recombination rate, r̂, for which the relative heterozygosity is smaller than β by solving the equation H(r̂) = β. If we solve this equation first with respect to A(r̂), we obtain
| (75) |
By using the approximation according to (74), we obtain
| (76) |
Figure 9 illustrates the valley of reduced heterozygosity as a function of α for different distributions κ of m. Such illustrations can be used to determine the range of parameters that lead to a given reduction in relative heterozygosity.
Fig. 9.
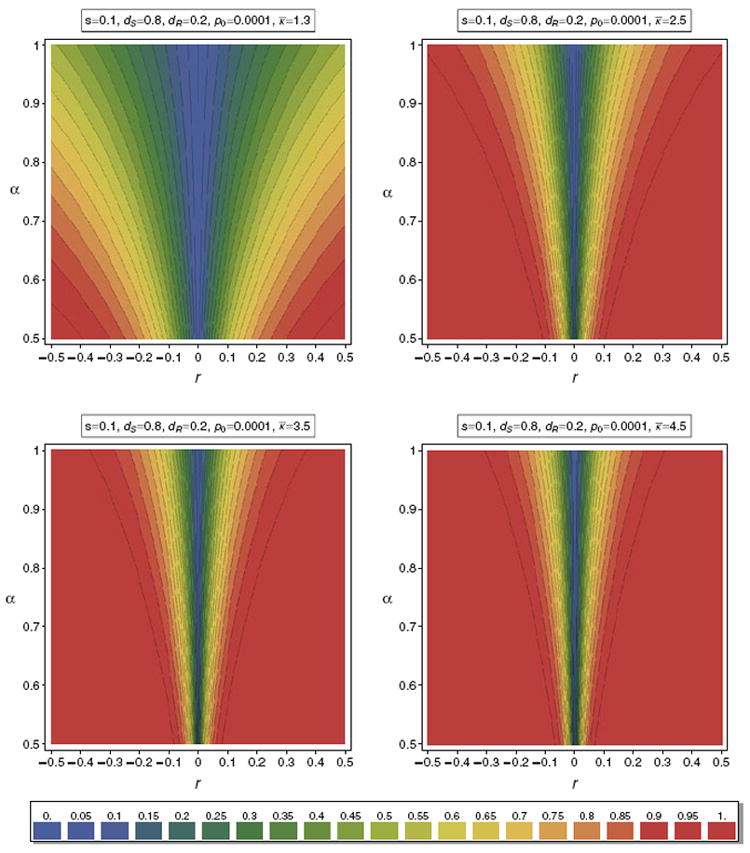
Contour plot of the valley of reduced heterozygosity for different distributions κ. In all cases we assume a truncated exponential distribution with range 1–10 and mean κ̅. By truncated we mean that the probability of m = 10 is the probability that m ≥ 10 for a poisson distribution with mean κ̅. In all panels the same selection parameters are assumed. The parameters are summarized in the boxes above the panels
6 Discussion
We obtained a closed-form approximation for the expected heterozygosity shaped by genetic hitchhiking in the model of antimalarial drug-resistance evolution proposed in Schneider and Kim (2010). This model aims to capture the effect of multiple infections per host (m) and drug-treatment rate (α), which are considered the most important epidemiological parameters that characterize geographic differences in the dynamics of drug resistance (Escalante et al. 2009), as well as the complex malaria transmission cycle on the pattern of selective sweeps caused by drug-resistant mutations (Daily 2006; Prugnolle et al. 2009). Due to the model complexity the exact solution is expressed by the summation of infinite (or a very large number of) terms. Here, we provided numerous approximate solutions with varying degree of accuracy. Notably, using the assumption that the starting frequency of the resistant allele under positive directional selection is low, we could obtain a solution that is simple enough to allow clear biological interpretations regarding the effects of epidemiological parameters. Furthermore, our approximations are flexible enough to incorporate arbitrary distributions of hosts with different infection rates and/or host heterogeneity, e.g., arbitrary distributions of hosts with different drug concentrations. The latter condition, arising due to a slow decay of antimalarial drugs in the bodies of treated patients, was demonstrated to be crucial for the initiation of drug resistance evolution (Hastings et al. 2002). For more discussion on how this model can be used to predict the spread of resistance and its speed, and how it can be used to design ‘optimal’ treatment strategies we refer to Schneider and Kim (2010).
The mean fitness of the resistant parasites, λ, and of the sensitive parasites, μ, are crucial in the considered model. If λ and μ are not too different, log λ − log μ corresponds to the selection coefficient of the beneficial (resistant) mutation. Then, our approximation (74) is basically identical to that under the standard model of hitchhiking obtained by Maynard Smith and Haigh (1974) except the modifying factors of the recombination rate, , and of the initial frequency, . Therefore, the dynamics of hitchhiking unique to this malaria model is summarized by these factors. We have because for all host class j and all m. The latter fraction represents the probability that a given resistant gametocyte pairs with a sensitive gametocyte in the body of a mosquito which took its blood meal from a j host, when the frequency of resistant allele is low. (As with the standard hitchhiking model, the final heterozygosity is predominantly determined by the dynamics of the resistant allele at its early stage.) If the drug in the host is very effective ( ), it will greatly reduce . Namely, the strength of selection determines the effective rate of recombination (decay of association between beneficial and neutral allele), unlike the case of standard hitchhiking model in which the two factors are decoupled. Moreover, the approximation (74) will be more accurate even for larger r and p0 compared to the standard hitchhiking model because of the two adjustment factors and .
Assume m is constant. It is also obvious from the above that the number, m, of independent parasite strains infecting a host determines the effective recombination rate. A small value of m thus increases the hitchhiking effect. In the extreme case of m = 1, genetic variation in the population is completely wiped out as parasites reproduce effectively asexually. On the other hand, with m → ∞ our approximation approaches that of standard hitchhiking. Note, however, that in the exact solution is still less than one, even with m → ∞, because the approximation leading to ϑ̃(m) (Eq. 58) assumes that the frequency of the resistant allele is low and hence that the probability that a given host is infected by two or more resistant strains is negligible. However, if m increases to a large number, such chance is no longer negligible. Therefore, the combined effect of strong drug pressure and the limited number of clones in hosts can greatly amplify the effect of genetic hitchhiking beyond the level predicted by the standard model. If m is not constant, the hitchhiking effect is more pronounced for more left-skewed distributions κ. It will be in particular pronounced if a large fraction of single infections occurs.
Our approximation also reveals another important departure of our model from the previous models that assume random mating and homogeneous selective pressures. In the standard model, the allele-frequency trajectory of a beneficial mutation is necessary and sufficient information for predicting its hitchhiking effect on the linked neutral variation (Betancourt et al. 2004; Chevin and Hospital 2008). For example, the speed with which the beneficial mutation increases in the population determines the size of the genomic regions affected by hitchhiking. However, in our model with host heterogeneity, different combinations of parameter values (j and ) that specify the same allele frequency trajectory of the resistance allele may generate different hitchhiking effects. Schneider and Kim (2010) showed that the changes of sensitive and resistant allele frequencies are uniquely determined by their absolute fitness, μ and λ, respectively. With host heterogeneity, the fitness is simply the mean of or weighted by the frequencies of host classes (αj). The modifying factor of effective recombination rate, (ore more generally ), however is not a linear function of or . As a result, as shown in Remark 4, (or )) decreases as one introduces host heterogeneity while keeping λ and μ constant. This makes our simplest approximation assuming no host heterogeneity a conservative predictor of the hitchhiking effect.
Comparison of approximate solutions with empirical observation of local reductions of genetic variation around the loci of drug resistance mutations, combined with other genetic (e.g. recombination rate, the frequency change of drug resistance) and epidemiological (e.g. the mean number of independent parasite clones per host) information, will greatly advance our understanding of antimalarial drug-resistance evolution. Especially, if empirical data for the reduction in heterozygosity is available, our results can be used to determine possible ranges for parameters that are unknown and/or infeasible to measure. It should be noted that the fraction λ/μ can be easily estimated from retrospective genetic data. Since , log λ/μ is just the slope of the linear regression of the logarithm of the ratio of resistant over sensitive parasites measured at different time points. However, it is difficult to scale time, because the number of transmission cycles per year is difficult to quantify. Moreover, also the parameter α should be easy to measure. On the opposite, the distribution of κ of m will be difficult to estimate. However, the distribution of m, and especially single infections, lead to a genome-wide reduction in relative heterozygosity if selection is sufficiently strong. This genome-wide reduction might be used to estimate the distribution κ. However, applying our results to real data lies beyond the scope of this article and will be accomplished in a follow-up paper.
Acknowledgments
This work was funded by the National Institute of Health grant R01GM084320. We want to thank Prof. Ananias Escalante for helpful comments on an earlier draft of this work. We gratefully acknowledge the fruitful discussions with him on this and similar topics. We also want to thank two anonymous reviewers.
Appendix A
A.1 Bounds and approximations
Proof of Remark 2
If for l ∈ ℕ we just need to adapt the proof of Theorem 1. The derivation of G1 assumed . If this assumption is violated we have to adjust the derivation. In this case we have and we can replace (24) by
Hence, we have to replace (25) in the proof of Theorem 1 by
Thus, we have
| (77) |
| (78) |
and
| (79) |
Combining the above with (26), the definitions of ξ, η, c, (31), and (32) yields
| (80) |
and
| (81) |
respectively. Note that we have
| (82) |
| (83) |
| (84) |
| (85) |
| (86) |
Hence, (80) and (81) simplify to
| (87) |
and
| (88) |
which equal (36) and (35), respectively.
Now, let us regard g(x) given by (17) as a function in x and a and write ga(x) for it. Clearly for x ≥ 0, ga(x) is monotone increasing in a, and for x < 0 it is monotone decreasing. Since by Theorem 1 the integral exists for x̃ ∈ {−1, 0} and , it follows by the Theorems of monotone convergence and, in case x̃ = −1, also by the theorem of dominated convergence that . By setting it follows that (36) and (35) are the continuations of (16) and (15) in the limit .
If x0 ≤ 0, we do not need the function G1, and hence the derivations from Theorem 1 and Remark 1 hold for ψ. The same holds for Ψ if x0 ≤ −1. This finishes the proof.
Proof of Theorem 2
For κ ≠ −l(l ∈ ℕ) we have
| (89) |
First assume for l ∈ ℕ, i.e., .
Setting and z = −c gives
| (90) |
whereas setting and gives
| (91) |
Moreover, setting and z = −1 yields
| (92) |
whereas by setting and z = −1 we obtain
| (93) |
First, combining (93), (90), and (92) with (31), and using this approximation in (20) yields (39) by using the definitions of ξ and η. Similarly, we obtain (40) by combining (93), (91), (92), with (32) and (21).
Clearly, (39) and (40) are continuous especially also at . Since ψ(a) and Ψ(a) have continuations at for (l ∈ ℕ) according to Remark 2, we have ϕ(a) ≈ ψ(a) and Φ(a) ≈ Ψ(a) for all a. This finishes the proof.
Proof of Remark 3
We obtain (41) by combining (33) with (89) for and , whereas we obtain (42) by combining (34) with (89) for and .
Clearly (39), (40), (41), and (42) are continuous functions in p0. For it is easily seen that (39) equals (41), whereas for (40) equals (42). Hence, (41) and (42) are the continuations in p0 of (39) and (40), respectively.
Proof of Theorem 3
Note that we have
| (94) |
where
| (95) |
Furthermore, is monotone increasing in x and . Since λ > μ and , we have . Hence, we obtain
and we have
| (96) |
which follows from the fact that can be written as a geometric series.
Therefore,
| (97) |
For C > 0 we obtain is monotone increasing in x, because . Furthermore, we have limx→0 f (x) = log C. By choosing , we see that
is negligible compared with
| (98) |
Furthermore, also
| (99) |
is negligible compared with (98). Therefore,
| (100) |
A.2 Relative heterozygosity
Proof of Remark 4
Let for x, y ∈ ℝ+. Its Hessian matrix is calculated to be
Clearly, we have and det H = 0, i.e., the leading minors of H are non-positive. Hence, f is concave but not strictly concave (note that f (x, x) = x/2). Hence, for positive random variables X and Y defined on a probability space (Ω, A, P) and a sub-σ algebra B the Jensen’s inequality for higher dimensions yields
Now, choose (Ω, A, P) = (ℕ, P(ℕ), P), where P(j) = αj. Moreover, choose and B = {0̷, U, T, ℕ}. Then the Jensen’s, inequality gives
Similarly, we obtain
Combination of the above yields
| (101) |
i.e., that ϑ̃(m) given by (64) is smaller than if it is given by (58).
Clearly in the limit m → ∞ equality holds in (101). Moreover, ϑ̃(m) → λ. Hence, in the limit case H̃(m) reduced to the approximation for standard hitchhiking.
The proof is finished by applying the argument formulated above the remark.
Contributor Information
Kristan A. Schneider, Email: kristan.schneider@asu.edu, School of Life Sciences, Arizona State University, 1711 South Rural Road, Tempe, AZ 85287, USA; Department of Mathematics, University of Vienna, Nordbergstrasse 15, UZA 4, 1090 Vienna, Austria; CEMI/Biodesign Institute, Arizona State University, P. O. Box 875301, Tempe, AZ 85287-5301, USA.
Yuseob Kim, School of Life Sciences, Arizona State University, 1711 South Rural Road, Tempe, AZ 85287, USA; Center for Evolutionary Medicine and Informatics, Biodesign Institute, 1001 S. McAllister Ave., Tempe, AZ 85281, USA.
References
- Barton NH. Genetic hitchhiking. Philos Trans R Soc Lond B Biol Sci. 2000;355(1403):1553–1562. doi: 10.1098/rstb.2000.0716. http://rstb.royalsocietypublishing.org/content/355/1403/1553.abstract. [DOI] [PMC free article] [PubMed]
- Betancourt AJ, Kim Y, Orr HA. A pseudohitchhiking model of X vs. autosomal diversity. Genetics. 2004;168(4):2261–2269. doi: 10.1534/genetics.104.030999. http://www.genetics.org/cgi/content/abstract/168/4/2261. [DOI] [PMC free article] [PubMed]
- Brooks DR, Wang P, Read M, Watkins WM, Sims PFG, Hyde JE. Sequence variation of the hydroxymethyldihydropterin pyrophosphokinase: dihydropteroate synthase gene in lines of the human malaria parasite Plasmodium falciparum with differing resistance to sulfadoxine. Eur J Biochem. 1994;224(2):397–405. doi: 10.1111/j.1432-1033.1994.00397.x. http://dx.doi.org/10.1111/j.1432-1033.1994.00397.x. [DOI] [PubMed]
- Chevin L-M, Hospital F. Selective sweep at a quantitative trait locus in the presence of background genetic variation. Genetics. 2008;180(3):1645–1660. doi: 10.1534/genetics.108.093351. http://www.genetics.org/cgi/content/abstract/180/3/1645. [DOI] [PMC free article] [PubMed]
- Cowman AF, Morry MJ, Biggs BA, Cross GA, Foote SJ. Amino acid changes linked to pyrimethamine resistance in the dihydrofolate reductase-thymidylate synthase gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1988;85(23):9109–9113. doi: 10.1073/pnas.85.23.9109. http://www.pnas.org/content/85/23/9109.abstract. [DOI] [PMC free article] [PubMed]
- Daily JP. Antimalarial drug therapy: the role of parasite biology and drug resistance. J Clin Pharmacol. 2006;46(12):1487–1497. doi: 10.1177/0091270006294276. http://jcp.sagepub.com/cgi/content/abstract/46/12/1487. [DOI] [PubMed]
- Escalante AA, Smith DL, Kim Y. The dynamics of mutations associated with anti-malarial drug resistance in plasmodium falciparum. Trends Parasitol. 2009;25(12):557–563. doi: 10.1016/j.pt.2009.09.008. http://www.sciencedirect.com/science/article/B6W7G-4XJ9BS6-1/2/04d13d69d2006be02c12ef0d051cc5c6. [DOI] [PMC free article] [PubMed]
- Hastings IM. Response to: the puzzling links between malaria transmission level and drug resistance. Trends Parasitol. 2003;19(4):160–161. doi: 10.1016/s1471-4922(03)00054-0. http://www.sciencedirect.com/science/article/B6W7G-483SMKD-1/2/680a5da4f74f112c6e1b87c270a9921d. [DOI] [PubMed]
- Hastings IM. Complex dynamics and stability of resistance to antimalarial drugs. Parasitology. 2006;132(05):615–624. doi: 10.1017/S0031182005009790. http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=428221&fulltextType=RA&fileId=S0031182005009790. [DOI] [PubMed]
- Hastings IM, Mackinnon MJ. The emergence of drug-resistant malaria. Parasitology. 1998;117(05):411–417. doi: 10.1017/s0031182098003291. http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=24263&fulltextType=RA&fileId=S0031182098003291. [DOI] [PubMed]
- Hastings IM, Watkins WM, White NJ. The evolution of drug-resistant malaria: the role of drug elimination half-life. Philos Trans R Soc Lond B Biol Sci. 2002;357(1420):505–519. doi: 10.1098/rstb.2001.1036. http://rstb.royalsocietypublishing.org/content/357/1420/505.abstract. [DOI] [PMC free article] [PubMed]
- Hedrick PW. Hitchhiking: a Comparison of Linkage and Partial Selfing. Genetics. 1980;94(3):791–808. doi: 10.1093/genetics/94.3.791. http://www.genetics.org/cgi/content/abstract/94/3/791. [DOI] [PMC free article] [PubMed]
- Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169(4):2335–2352. doi: 10.1534/genetics.104.036947. http://www.genetics.org/cgi/content/abstract/169/4/2335. [DOI] [PMC free article] [PubMed]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160(2):765–777. doi: 10.1093/genetics/160.2.765. http://www.genetics.org/cgi/content/abstract/160/2/765. [DOI] [PMC free article] [PubMed]
- Korenromp E, Miller J, Nahlen B, Wardlaw T, Young M. World malaria report 2005. World Health Organization (WHO); Geneva: 2005. [Google Scholar]
- Mackinnon MJ, Hastings IM. The evolution of multiple drug resistance in malaria parasites. Trans R Soc Tropical Med Hygiene. 1998;92(2):188–195. doi: 10.1016/s0035-9203(98)90745-3. http://www.sciencedirect.com/science/article/B75GP-4BY31N7-MS/2/bfd1e32b1cf5fd0f23e10978a356bd36. [DOI] [PubMed]
- Marsh K. Malaria disaster in africa. Lancet. 1998;352(9132):924–924. doi: 10.1016/S0140-6736(05)61510-3. http://www.sciencedirect.com/science/article/B6T1B-4FWV357-JX/2/c9facab67b868b8fddd289f050bfae19. [DOI] [PubMed]
- Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23(01):23–35. http://journals.cambridge.org/action/displayAbstract?fromPage=online&aid=1754360&fulltextType=RA&fileId=S0016672300014634. [PubMed]
- McCollum AM, Basco LK, Tahar R, Udhayakumar V, Escalante AA. Hitchhiking and selective sweeps of Plasmodium falciparum sulfadoxine and pyrimethamine resistance alleles in a population from Central Africa. Antimicrob Agents Chemother. 2008;52(11):4089–4097. doi: 10.1128/AAC.00623-08. http://aac.asm.org/cgi/content/abstract/52/11/4089. [DOI] [PMC free article] [PubMed]
- Mita T, Tanabe K, Takahashi N, Tsukahara T, Eto H, Dysoley L, Ohmae H, Kita K, Krudsood S, Looareesuwan S, Kaneko A, Bjorkman A, Kobayakawa T. Independent evolution of pyrimethamine resistance in Plasmodium falciparum isolates in Melanesia. Antimicrob Agents Chemother. 2007;51(3):1071–1077. doi: 10.1128/AAC.01186-06. http://aac.asm.org/cgi/content/abstract/51/3/1071. [DOI] [PMC free article] [PubMed]
- Nair S, Nash D, Sudimack D, Jaidee A, Barends M, Uhlemann A-C, Krishna S, Nosten F, Anderson TJC. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol Biol Evol. 2007;24(2):562–573. doi: 10.1093/molbev/msl185. http://mbe.oxfordjournals.org/cgi/content/abstract/24/2/562. [DOI] [PubMed]
- Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann J-P, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJC. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol. 2003;20(9):1526–1536. doi: 10.1093/molbev/msg162. http://mbe.oxfordjournals.org/cgi/content/abstract/20/9/1526. [DOI] [PubMed]
- Nash D, Nair S, Mayxay M, Newton PN, Guthmann J-P, Nosten F, Anderson TJ. Selection strength and hitchhiking around two anti-malarial resistance genes. Proc R Soc B Biol Sci. 2005;272(1568):1153–1161. doi: 10.1098/rspb.2004.3026. http://rspb.royalsocietypublishing.org/content/272/1568/1153.abstract. [DOI] [PMC free article] [PubMed]
- Prugnolle F, Durand P, Renaud F, Rousset F. Effective size of the hierarchically structured populations of the agent of malaria: a coalescent-based model. Heredity. 2009:1–7. doi: 10.1038/hdy.2009.127. http://dx.doi.org/10.1038/hdy.2009.127. [DOI] [PubMed]
- Schneider KA, Kim Y. An analytical model for genetic hitchhiking in malaria parasites caused by drug resistance. Theor Popul Biol. 2010 doi: 10.1016/j.tpb.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwobel B, Alifrangis M, Salanti A, Jelinek T. Different mutation patterns of atovaquone resistance to Plasmodium falciparum in vitro and in vivo: rapid detection of codon 268 polymorphisms in the cytochrome b as potential in vivo resistance marker. Malaria J. 2003;2(1):5. doi: 10.1186/1475-2875-2-5. http://www.malariajournal.com/content/2/1/5. [DOI] [PMC free article] [PubMed]
- Stephan W, Wiehe THE, Lenz MW. The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor Popul Biol. 1992;41(2):237–254. http://www.sciencedirect.com/science/article/B6WXD-4F1Y9N0-3M/2/1245281bba0c6b542457fdd75c343edf.
- Triglia T, Cowman AF. Primary structure and expression of the dihydropteroate synthetase gene of Plasmodium falciparum. Proc Natl Acad Sci USA. 1994;91(15):7149–7153. doi: 10.1073/pnas.91.15.7149. http://www.pnas.org/content/91/15/7149.abstract. [DOI] [PMC free article] [PubMed]
- WHO. WHO Expert Committee on malaria. World Health Organ Tech Rep Ser. 2000;892:1–74. http://www.genetics.org/cgi/content/abstract/160/2/765. [PubMed]
- Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ. Su X-z 07 2002 Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 418(6895):320–323. doi: 10.1038/nature00813. http://dx.doi.org/10.1038/nature00813. [DOI] [PubMed]