

Abstract
BACKGROUND AND PURPOSE
We hypothesized that proteinase-activated receptor-2 (PAR2)-mediated vasorelaxation in murine aorta tissue can be due in part to the release of adipocyte-derived relaxing factors (ADRFs).
EXPERIMENTAL APPROACH
Aortic rings from obese TallyHo and C57Bl6 intact or PAR2-null mice either without or with perivascular adipose tissue (PVAT) were contracted with phenylephrine and relaxation responses to PAR2-selective activating peptides (PAR2-APs: SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2), trypsin and to PAR2-inactive peptides (LRGILS-NH2, 2-furoyl-OLRGIL-NH2 and LSIGRL-NH2) were measured. Relaxation was monitored in the absence or presence of inhibitors that either alone or in combination were previously shown to inhibit ADRF-mediated responses: L-NAME (NOS), indomethacin (COX), ODQ (guanylate cyclase), catalase (H2O2) and the K+ channel-targeted reagents, apamin, charybdotoxin, 4-aminopyridine and glibenclamide.
KEY RESULTS
Endothelium-intact PVAT-free preparations did not respond to PAR2-inactive peptides (LRGILS-NH2, LSIGRL-NH2, 2-furoyl-OLRGIL-NH2), whereas active PAR2-APs (SLIGRL-NH2; 2-furoyl-LIGRLO-NH2) caused an L-NAME-inhibited relaxation. However, in PVAT-containing preparations treated with L-NAME/ODQ/indomethacin together, both PAR2-APs and trypsin caused relaxant responses in PAR2-intact, but not PAR2-null-derived tissues. The PAR2-induced PVAT-dependent relaxation (SLIGRL-NH2) persisted in the presence of apamin plus charybdotoxin, 4-aminopyridine and glibenclamide, but was blocked by catalase, implicating a role for H2O2. Surprisingly, the PAR2-inactive peptides, LRGILS-NH2 and 2-furoyl-OLRGIL-NH2 (but not LSIGRL-NH2), caused relaxation in PVAT-containing preparations from both PAR2-null and PAR2-intact (C57Bl, TallyHo) mice. The LRGILS-NH2-induced relaxation was distinct from the PAR2 response, being blocked by 4-aminopyridine, but not catalase.
CONCLUSIONS
Distinct ADRFs that may modulate vascular tone in pathophysiological settings can be released from murine PVAT by both PAR2-dependent and PAR2-independent mechanisms.
Keywords: adipocyte-derived relaxing factor, catalase, hydrogen peroxide, ion channels, mouse, PAR2, perivascular adipose tissue, potassium channels, protease-activated receptors, proteinase-activated receptors, relaxation
Introduction
Perivascular adipose tissue (PVAT) is now recognized as a regulator of vascular function because of its release of adipocyte-derived relaxing factors (ADRFs) that diminish the contractile actions of vasoconstrictors such as phenylephrine, 5-HT, angiotensin II and U46619 (Soltis and Cassis, 1991; Lohn et al., 2002; Verlohren et al., 2004; Gao et al., 2005; Gao et al., 2007; Lee et al., 2009). Although the release of ADRFs from adipose tissue has been detected in arterial preparations from rats (aorta; mesenteric) mice (mesenteric) and humans (internal thoracic), the majority of work so far has been done with rat aortic tissue. Routinely, the release of ADRF activity has been monitored in terms of a reduction in the contractile action of an agonist like phenylephrine observed relative to the increased contraction of a comparable fat-free preparation from the same vessel (Soltis and Cassis, 1991; Lohn et al., 2002; Verlohren et al., 2004). As summarized in Table 1, and reviewed in more detail elsewhere (Gollasch, 2011) the differences between the various ADRFs have been characterized primarily in terms of the effect of a variety of inhibitors on their anticontractile actions. For instance, the endothelium-dependent NO-mediated relaxant effect of angiotensin (1-7), identified as an ADRF found in Wistar rat aortic adipose tissue, is blocked by either the NOS inhibitor, Nω-nitro-L-arginine (L-NNA) or the KCa-channel blockers, apamin and charybdotoxin, but not by 4-aminopyridine and glibenclamide (Gao et al., 2007; Lee et al., 2009). The endothelium-independent ADRF described by these investigators was blocked by ODQ (guanylyl cyclase inhibitor) and catalase (blocks H2O2 action) (Gao et al., 2007). In contrast, the anticontractile ADRF described by Lohn et al. (2002) in Sprague-Dawley aortic tissue was inhibited by glibenclamide (KATP-channel blocker) and genistein (tyrosine kinase inhibitor), but not by L-NNA or indomethacin (COX inhibitor) (Lohn et al., 2002); its release from adipose tissue was, however, blocked by the kinase A inhibitor, H89 and the tyrosine kinase inhibitor, tyrphostin A25/AG82 (Dubrovska et al., 2004). In contrast, the rat anticontractile ADRF described by the same laboratory in Sprague-Dawley mesenteric arteries was blocked by 4-aminopyridine, but not by glibenclamide (Verlohren et al., 2004). Thus, according to the inhibitor data, the ‘ADRFs’ that are released by perivascular fat to reduce the contractile action of contractile agonists appear to be distinct (i) in aortic tissue derived from different rat strains and (ii) in different vascular preparations from the same animal.
Table 1.
Effects of inhibitors on ADRF action/release in rat aorta and mesenteric preparations
| Inhibitor | Tissue & species | Effect on ADRF action release | Agonist | Comment | Reference |
|---|---|---|---|---|---|
| L-NNA | Aorta, Sprague | None | 5-HT | Lohn et al., 2002 | |
| L-NNA | Aorta, Wistar | Blocks | PE | Endothelium dependent | Gao et al., 2007 |
| ODQ | Aorta, Wistar | Blocks | PE | Endothelium independent | Gao et al., 2007 |
| Indomethacin | Aorta, Sprague | None | 5-HT | Endothelium independent | Lohn et al., 2002 |
| TEA | Aorta, Sprague | None | 5-HT | Endothelium independent | Lohn et al., 2002 |
| TEA | Aorta, Wistar | Blocks | PE | Endothelium dependent | Gao et al., 2007 |
| Charybdotoxin-apamin | Aorta, Wistar | Blocks | PE | Endothelium dependent | Gao et al., 2007 |
| Glibenclamide | Aorta, Sprague | Blocks | 5-HT | Endothelium independent | Lohn et al., 2002 |
| Glibenclamide | Aorta, Wistar | None | 5-HT | Endothelium independent | Lee et al., 2009 |
| Glibenclamide | Mesenteric, Sprague | None | PE | Endothelium dependent | Verlohren et al., 2004 |
| 4-Aminopyridine | Mesenteric, Sprague | Blocks | 5-HT | Verlohren et al., 2004 | |
| 4-Aminopyridine | Mesenteric NZO and adiponectin-1 null mice | Blocks | 5-HT | ADRF not adiponectin; endothelium-dependence not determined | Fésüs et al., 2007 |
| Genistein | Aorta, Sprague | Blocks | 5-HT, PE | Affects ADRF release, not action | Lohn et al., 2002; Dubrovska et al., 2004 |
| Tyrphostin A25-AG82 | Aorta, Sprague | Blocks | 5-HT | Dubrovska et al., 2004 | |
| H89 | Aorta, Sprague | Blocks | 5-HT | Affects ADRF release, not action | Dubrovska et al., 2004 |
| Catalase | Aorta, Wistar | Blocks | PE | Endothelium independent | Gao et al., 2007 |
| Minus Endothelium | Aorta, Sprague | None | 5-HT | Endothelium independent | Lohn et al., 2002 |
| Minus Endothelium | Aorta, Wistar | Partial block | PE | Endothelium independent | Gao et al., 2007 |
PE, phenylephrine; mesenteric, mesenteric artery.
In our own work with rats and mice, we have focused on the vasorelaxation caused by proteinase-activated receptor-2 (PAR2) when triggered by trypsin or by receptor-selective peptide agonists (Al-Ani et al., 1995; Hollenberg et al., 1996; 1997; Ramachandran and Hollenberg, 2008). In the aorta, PAR2 agonists cause an endothelium-dependent NO-mediated (blocked by L-NAME) vasorelaxation. In carefully cleaned endothelium-free aorta preparations, PAR2 agonists cause neither a contractile nor a relaxant response (Al-Ani et al., 1995). As we have worked routinely with trimmed, adipose tissue-free vascular preparations, we wondered if PAR2, like the other G-protein coupled agonists (e.g. phenylephrine), might also trigger a similar PVAT-dependent relaxation. We used a mouse model to evaluate this possibility, because we currently employ PAR2-null mice on a C57Bl background for our work dealing with physiological roles for PAR2. These PAR2-null mice are, nonetheless, have a fully functional vascular thrombin receptor, PAR1. Further, because of differences in ADRFs observed for different rat strains summarized above and in Table 1, we wished to evaluate ADRF release in at least two different mouse strains. For a comparison with the C57Bl, we chose the TallyHo mouse (Kim et al., 2001), because this strain exhibits an obese insulin-resistant phenotype along with hyperglycaemia and endothelial-vascular dysfunction (Cheng et al., 2007). It was our initial prediction that the obese TallyHo mouse-derived tissue would release and/or respond to ADRFs differently from vascular tissue from the C57Bl strain. Using the two mouse strains, we tested the hypothesis that in addition to the established endothelium-dependent NO-mediated relaxation caused by PAR2 activation, this receptor could also cause vasorelaxation by releasing ADRF from PVAT. We therefore assessed the PAR2-mediated release of ADRF in the absence and presence of inhibitors (e.g. L-NAME, ODQ, indomethacin, 4-aminopyridine, combined apamin + charybdotoxin, glibenclamide, genistein, H89 and catalase) that had been previously reported to block ADRF action as summarized above and in Table 1 (Lohn et al., 2002; Dubrovska et al., 2004; Verlohren et al., 2004; Gao et al., 2007; Lee et al., 2009).
PAR2 is one of four novel receptor family members (PARs 1, 2, 3 and 4) that belong to the GPCR superfamily. PARs are cleaved and activated proteolytically by a unique mechanism involving the unmasking of a cryptic N-terminal receptor sequence that acts as a ‘tethered ligand’. The revealed tethered ligand triggers the receptor by binding to its extracellular domains (Vu et al., 1991; Nystedt et al., 1994; Hollenberg and Compton, 2002; Coughlin, 2005; Ramachandran and Hollenberg, 2008). Like the receptors for thrombin (PARs 1 and 4), PAR2 can be activated without proteolysis by synthetic peptides corresponding to the enzymatically exposed N-terminal tethered ligand sequence. The receptor-selective PAR2-activating peptides (PAR2-APs) have proved to be valuable receptor probes, because they are able to activate the receptor without causing the other complex effects that proteinases can trigger in vivo. The PAR2-APs, SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2, along with the reverse-sequence PAR2-inactive peptides (e.g. LRGILS-NH2, LSIGRL-NH2 and 2-furoyl-OLRGIL-NH2) that cannot activate PAR2 have been used to determine the physiological effects of PAR2 activation both in vitro and in vivo (Steinhoff et al., 2005; Ramachandran and Hollenberg, 2008).
To date, the G-protein coupled agonists that have been documented to release ADRF from PVAT (e.g. phenylephrine, 5-HT, angiotensin II) are contractile vascular agonists that act directly on the smooth muscle. An advantage of the PAR2 agonist peptides that we used for our work (SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2) is that they are non-contractile in the aorta preparation. Thus, we were able to test our hypothesis that PAR2 activation can release ADRF by evaluating the relaxant activity of the PAR2-APs in phenylephrine-contracted arterial preparations that did or did not contain PVAT. Our data obtained from the two strains of mice using these reagents show that distinct ADRFs that appear to differ in terms of their mechanism of action both from each other and from those characterized previously in rat aorta tissues can be released from murine PVAT by PAR2-derived peptides via both a PAR2-dependendent and a PAR2-independent mechanism. Further, our characterization of the ADRFs showed no differences between the TallyHo and C57Bl mice, suggesting that the obesity and endothelial dysfunction present in the TallyHo is not directly linked to an ADRF-related mechanism.
Methods
Animals
Two strains of mice were used: C57Bl and the so-called ‘TallyHo’. The TallyHo mice represent an animal obesity model of non-insulin-dependent diabetes that exhibits endothelial dysfunction (Kim et al., 2001; Cheng et al., 2007). The overt hyperglycaemia in TallyHo mice has been attributed to a mutation in a major diabetes susceptibility locus on chromosome 19, which interacts with additional genes to lead to an obese diabetic phenotype. The rapid development of obesity in these animals enabled us to test conveniently the impact of perivascular fat on vascular responsiveness and to obtain significant amounts of perivascular fat for analysis. The C57Bl mice were used because of the availability of PAR2-null mice on the C57Bl background. Breeding pairs for both the TallyHo and PAR2-null animals were obtained from Jackson Laboratories (Bar harbor, ME, USA) and in-house bred animals were used. Wild-type and PAR2-null C57Bl mice, for which breeding pairs were also obtained as a gift through the courtesy of Dr Patricia Andrade-Gordon (Damiano et al., 1999: Johnson & Johnson Pharmaceutical Research & Development L. L. C., Spring House, PA, USA), were also in-house bred and used interchangeably for comparison with the Jackson Laboratory-purchased mice. Data obtained with tissues from the PAR2-null mice were compared with data obtained from wild-type littermates.
All of the mice used were 14–18 weeks of age. Mice were housed at room temperature with a 12 h light/12 h dark cycle room and had free access to food and water. The development of hyperglycaemia in the male TallyHo mice was followed with glucometer measurements (Life Scan Inc., Milpitas CA, USA). All animal care and experimental procedures were approved by the Animal Resources Committee at the University of Calgary and were in accordance with the guidelines of the Canadian Council for Animal Care in Research.
Peptides and other reagents
Drug/molecular target nomenclature used for all compounds and receptors corresponds to this journal's Guide to Receptors and Channels (Alexander et al., 2009). Amino acids are abbreviated by their one-letter codes (e.g. A, alanine, R, arginine, etc.). PAR2-selective activating peptides: 2-furoyl-LIGRLO-NH2, SLIGRL-NH2; PAR2-inactive control reverse-sequence peptides: LSIGRL-NH2, LRGILS-NH2, 2-furoyl-OLRGIL-NH2. All peptides were synthesized as carboxy amides (>95% purity, assessed by HPLC and mass spectrometry) by the Peptide Core Facility at the University of Calgary, Calgary, AB, Canada (peplab@ucalgary.ca). The PAR2-selective receptor-activating peptides were: SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2; the PAR2-inactive reverse-sequence peptides were: LRGILS-NH2, LSIGRL-NH2 and 2-furoyl-OLRGIL-NH2. All other chemicals were purchased either from Sigma-Aldrich (St. Louis, MO, USA) or from Calbiochem (La Jolla, CA, USA). Peptides were dissolved in buffer, pH 7.4, containing 25 mM HEPES. Vasoconstrictor phenylephrine, vasodilator ACh, NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME), apamin and charbdotoxin were dissolved in distilled water. The COX inhibitor, indomethacin, was prepared with absolute ethanol and the stock solution of the inhibitor of guanylate cyclase, 1H-[1,2,4] oxadiazolo[4,3,a]quinoxalin-1-one (ODQ: Garthwaite et al., 1995), was prepared using dimethyl sulphoxide. Catalase 8882 U mg−1, 4-aminopyridine and glibenclamide (5-chloro-N-(4-[N-(cyclohexylcarbamoyl)sulphamoyl]phenethyl)-2-methoxybenzamide) were also from Sigma.
Preparation of aortic tissue for bioassay
Mice were killed by cervical dislocation. The thoracic aortae were removed and dissected out into ice-cold Krebs solution of the following composition (in mM): NaCl 120, NaHCO3 25, KCl 4.8, NaH2PO4 1.2, dextrose 11.0, and CaCl2 1.8 aerated with 95% O2 and 5% CO2. Aortae were cut into four sections (4 mm each). For each set of tissues, one group had the PVAT removed to serve as a PVAT-free control, and the other set were isolated with the adherent PVAT for assays of ADRF release. In total the project used approximately 100 TallyHo mice and 40 C57Bl strain mice, comprising 20 wild-type and 20 PAR2-nulls.
Wire myograph studies
Isometric tension studies using wire myography were performed as described previously (Waldron et al., 1999). In brief, PVAT-intact and PVAT-free aortae were mounted in a Mulvany–Halpern myograph organ bath (5 mL volume, 610 multi-myograph system; J.P. Trading, Copenhagen, Denmark). Isometric tension was recorded online via serial connection to a computer hard drive. Resting tension (4.5 mN) was fixed for an initial equilibration period of 1 h. All experiments were performed at 37°C in Krebs buffer of the above composition, gassed with 95% O2 and 5% CO2. Software for data acquisition and analysis (Myodaq 2.01/Myodata 2.02) were designed by J.P. Trading for the 610 multi-myograph system.
Bioassay protocols
Tissues were routinely contracted with 100 mM potassium chloride (KCl) to test their viability. Then, after re-equilibration for 20 min in fresh buffer, tissues were contracted with 1 µM of phenylephrine and a test concentration of 1 µM ACh was added and the presence or absence of a relaxant response was monitored to verify the presence or absence of an intact functional endothelium. The contractile response to phenylephrine was expressed as a percentage of the contractile response caused by 100 mM KCl (% KCl). Upon standardizing the preparation with the use of KCl and ACh, the effects of added SLIGRL-NH2, 2-furoyl-LIGRLO-NH2, LRGILS-NH2, LSIGRL-NH2 and 2-furoyl-OLRGIL-NH2 on the tension of the phenylephrine-contracted preparations (1 µM phenylephrine) was monitored for tissues with/without an intact endothelium and with/without adherent PVAT. Relaxation (%) was expressed as a percentage reduction of the plateau tension developed in the presence of phenylephrine. The effects of the inhibitors (L-NAME, ODQ, indomethacin, 4-aminopyridine, combined apamin + charybdotoxin, glibenclamide, genistein, H89 and catalase) were measured by treating the tissues with the inhibitors for 15 min before their contraction with 1 µM phenylephrine, then followed by the addition of test concentrations of SLIGRL-NH2, 2-furoyl-LIGRLO-NH2, LRGILS-NH2, LSIGRL-NH2 and 2-furoyl-OLRGIL-NH2. In most experiments evaluating a role for PAR2, SLIGRL-NH2 was used at a concentration of 20 µM to ensure selectivity for PAR2. This concentration proved to be twofold higher than the EC50 for causing ADRF release (see below, Figure 2A). LRGILS-NH2 was regularly used as a PAR2-inactive ‘control’ peptide at a concentration of 50 µM. This concentration of the LRGILS-NH2‘control’ peptide was used because it was a supra-maximal concentration in terms of its ability to release ADRF via a non-PAR2 mechanism (see below, Figure 2B). Routinely, the inhibitor effects were monitored in the combined presence of L-NAME (100 µM), ODQ (10 µM) and indomethacin (10 µM). Contractile/relaxant assays were also conducted in the presence of catalase (1200 U·mL−1) and 4-aminopyridine (1 mM). Values in the Figures represent the means ± SEM (bars) for measurements made with five to ten individual aorta preparations obtained independently from a minimum of five different animals.
Figure 2.
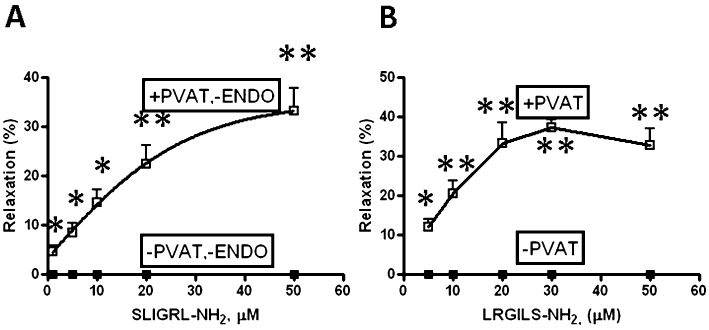
Comparison of the concentration-effect curves for SLIGRL-NH2- versus LRGILS-NH2-mediated relaxation in the absence (−PVAT) or presence (+PVAT) of PVAT. TallyHo-derived (A) and C57Bl-derived (B) aortic rings were contracted with phenylephrine (1 µM) in the absence of inhibitors and the relaxation responses to increasing concentrations of (A) SLIGRL-NH2 and (B) LRGILS-NH2 were measured. The action of SLIGRL-NH2 was assessed in endothelium-free preparations, because this peptide stimulates NO release in endothelium-intact TallyHo aortic rings; (B) LGRILS-NH2 in C57BL6 aortic rings. Comparable data were obtained for SLIGRL-NH2 in C57BL6 and for LRGILS-NH2 in TallyHo aortic rings. Relaxation (%) was expressed as a percentage reduction of the plateau tension developed in the presence of phenylephrine. Data represent the average relaxation (±SEM: bars, n = 5) at each peptide concentration. The asterisks denote statistically significant differences (*P < 0.05; **P < 0.01) for values observed in the presence, compared with the absence of PVAT.
Isolation of PVAT and adipocytes for RNA isolation and RT-PCR detection of mRNA for PARs 1 and 2
Perivascular adipose tissue for RNA isolation was obtained from both aorta and mesenteric artery preparations. Isolated adipocytes were freed from mesenteric adipose tissue by collagenase digestion in keeping with previously described methods (Van and Roncari, 1977), with minor modifications. In brief, the mesenteric adipose tissue was washed in Hank's balanced salt solution pH 7.4 (Invitrogen) and then minced and treated with collagenase. Collagenase digestion for adipocyte isolation was performed in 15 mL tubes with Hanks buffered saline containing 1 mg·mL−1 of collagenase type II from Clostridium histolyticum (Catalogue number C6885 from Sigma-Aldrich, St. Louis, MO, USA) for 45 to 60 min at 37°C with shaking at 120 r.p.m. until tissue was broken down. The digested cells were freed from tissue fragments by passage through a 40 µm nylon mesh and adipocytes were harvested by flotation after centrifugation at 500×g for 10 min. To isolate RNA from adipose tissue or adipocytes, samples were either extracted immediately from freshly prepared cells or were snap-frozen in liquid nitrogen and stored at −80°C until processed.
Total RNA was extracted using the RNeasy Lipid Tissue Mini Kit according to manufacturer's instructions (Qiagen, Hilden, Germany). Complementary DNA from RNA was synthesized by a reverse transcriptase reaction using Superscript II (Invitrogen, Carlsbad, CA, USA). RT-PCR detection of mRNA for PARs 1 and 2 was achieved using the following primer pairs: for PAR1, forward: GCGGGCAGCCTTGGGACAAT; reverse: ATGAAGGGAGGAGGCGGCGT; expected PCR product size: 296 bp. For PAR2, forward: CCACGTCCGGGGATGCGAAG, reverse: GCACAGGGCCTCCCCGTAGA; expected size of PCR product: 462 bp. The intensity of PCR signals for the PARs were compared with the PCR bands for β-actin obtained from the same samples for the semi-quantitative analysis of the expression level. Primer pairs for actin, designed to span an actin intron, were: forward, CACCCGCGAGCACAGCTTCT; reverse, CCTCAGGGCATCGGAACCGC. The expected size of the intron-free actin PCR product was 842 bp. The temperature and number of cycles of the PCR reaction were: 94°C, 58°C and 72°C for 30 to 35 cycles using Platinum Taq DNA polymerase from Invitrogen. Kodak Image Station 4000MM Pro was used for PCR detection of the PAR and actin mRNA PCR products. The sequences of the PAR PCR products were verified by the University of Calgary Core DNA service.
Statistical analysis
Data are presented as means ± SEM (bars in figures) for the assays done with vascular tissues obtained from the numbers of mice recorded in the figure legends. Each data point represents the average of measurements obtained from five to 10 individual tissues derived from a minimum of five different animals. The comparisons of mean values for each parameter were made using a one-way anova calculation followed by the Student–Newman–Keuls post hoc tests. Differences in mean values were considered significant when a P value was equal to or less than 0.05. In the figures, the asterisks denote statistical significance: *P < 0.05; **P < 0.01.
Results
Responses of TallyHo aorta with or without PVAT to phenylephrine and ACh
As illustrated in the representative tracing in Figure 1A and the averaged data in Figure 1B, the plateau contractile response to phenylephrine of the PVAT-containing TallyHo aorta tissue was diminished by almost 50% (lower tracing, Figure 1A), compared with PVAT-free preparations (upper tracing, Figure 1A). In contrast, in comparable endothelium-intact preparations with or without PVAT, the relaxant effect of ACh was the same (representative relaxation shown in Figure 1A; averaged data in Figure 1C). Further, the relaxant action of ACh in tissues with PVAT was blocked by L-NAME (Figure 1C).
Figure 1.
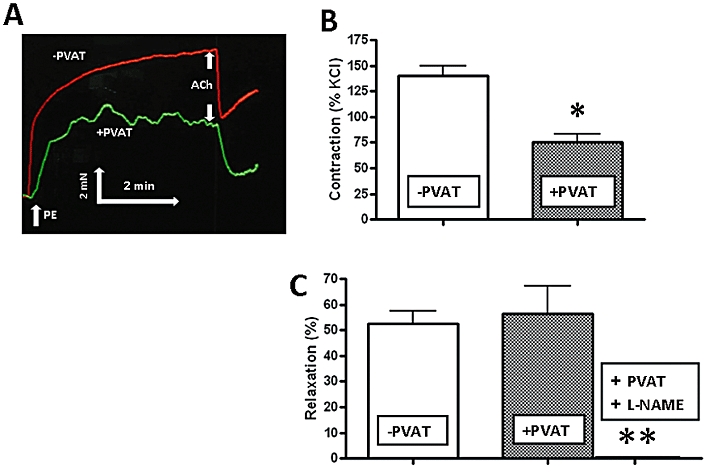
Perivascular adipose tissue (PVAT) reduces the contractile action of phenylephrine but has no effect on the relaxant action of ACh in aorta tissue from TallyHo mice. (A) Representative tracing of tension developed in the presence of phenylephrine (PE: 1 µM) and the relaxation observed upon adding ACh (1 µM) monitored in the absence of L-NAME, ODQ and indomethacin and either in the absence (−PVAT, upper tracing) or presence (+PVAT, lower tracing) of PVAT. (B) PE-induced contractile responses observed either in the absence (−PVAT) or presence (+PVAT) of PVAT. Histograms represent the average maximal contractile tension (as in the representative tracing in A), measured relative to the contraction caused in the same preparation by 100 mM KCl (% KCl). (C) ACh (1 µM)-mediated relaxation as illustrated by the representative tracings in (A) was measured in the same preparations without (−PVAT); with (+PVAT) PVAT after contraction by phenylephrine (1 µM); or +PVAT and in the presence of L-NAME (100 µM). Relaxation (%) was expressed as a percentage reduction of the plateau tension developed in the presence of phenylephrine. Values for (B) and (C) represent the means ± SEM (bars) for five replicate measurements on different vascular preparations. *P < 0.05 for phenylephrine contraction in the absence versus presence of PVAT.
Relaxant effects of PAR2-APs: endothelium-dependence and effects of L-NAME, ODQ and indomethacin in preparations without and with PVAT
We next evaluated the ability of PAR2-APs to cause vasorelaxation in the presence and absence of an intact endothelium and in the presence or absence of PVAT. We found that in endothelium-intact, fat-free aorta ring preparations from TallyHo and C57Bl mice, the PAR2-selective receptor-activating peptides SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2 caused an endothelium-dependent relaxation that, like the response to ACh, was inhibited by L-NAME and therefore mediated by NO (not shown). As neither SLIGRL-NH2 nor 2-furoyl-OLRGIL-NH2 caused a relaxation in endothelium-free aorta preparations that were free of fat, we used the endothelium-free rings first in the absence of L-NAME or other inhibitors (see below) to assess ADRF release caused by PAR2 activation. In the endothelium-free aorta preparations, SLIGRL-NH2 caused a concentration-dependent relaxation in preparations only in the presence of PVAT [+PVAT, −ENDO (endothelium): Figure 2A]. The PAR2 agonist, 2-furoyl-LIGRLO-NH2 mimicked the action of SLIGRL-NH2, but with a twofold higher potency (not shown and see below). Comparable data were obtained from tissues derived from C57Bl mice (see below). Further, in comparable fat-free preparations from all strains of mice that were either endothelium-intact or endothelium-denuded, the reverse-sequence PAR2-inactive peptides that cannot activate PAR2 (LRGILS-NH2, LSIGRL-NH2 and 2-furoyl-OLRGIL-NH2) caused neither a relaxant nor a contractile response (not shown). Based on SLIGRL-NH2 causing a concentration-dependent relaxation with an EC50 of 10 µM in endothelium-free preparations in the presence of PVAT (Figure 2A), we used 20 µM as a suitable ‘test’ concentration of peptide in subsequent experiments designed to evaluate the effects of potential inhibitors of ADRF release/action.
To facilitate the further evaluation of PAR2-mediated ADRF release, we used endothelium-intact preparations in which the effect of both eNOS and COX were inhibited in the combined presence of L-NAME, ODQ and indomethacin. Under these conditions, the PAR2 agonists cannot cause relaxation in PVAT-free aorta tissue and a role for NO and prostanoids can be ruled out. When L-NAME, ODQ and indomethacin were present in the fat-containing preparations, phenylephrine caused a more rapid initial rise in tension than in untreated tissues. However, the plateau tension developed in the presence of PVAT was diminished (compared with PVAT-free tissue) to an extent comparable to the reduced response of tissues that were not treated with these inhibitors (compare tracings in Figure 1A with the representative tracing in Figure 3A). In contrast to the relaxant effect of ACh, which was completely blocked by L-NAME alone in PVAT-containing preparations (Figure 1C), ADRF relaxant activity triggered by the PAR2 agonist, SLIGRL-NH2 (present only in PVAT-containing tissues) was observed in the presence of L-NAME, either without (not shown) or along with the combined presence of ODQ and indomethacin (Figure 3B; representative tracings shown in Figure 3A). We found no statistical difference between the ADRF-mediated relaxation measured in the presence of these inhibitors in endothelium-intact preparations (Figure 3B, plus PVAT: ‘+’), compared with the ADRF relaxant response observed in the endothelium-denuded preparations in the absence of the inhibitors (Figure 2A, upper curve). As expected, in the presence of L-NAME, ODQ and indomethacin, SLIGRL-NH2 did not cause a relaxation in the adipose tissue-free endothelium-intact preparations; (Figure 3B, minus PVAT: ‘−’). Our data thus indicate that PAR2 agonists trigger the release of ADRF by a mechanism that is endothelium-independent and is not mediated by either NO or prostanoids. Moreover, the data validated our ability to assess the release of ADRF using endothelium-intact preparations that were treated with L-NAME, ODQ and indomethacin.
Figure 3.
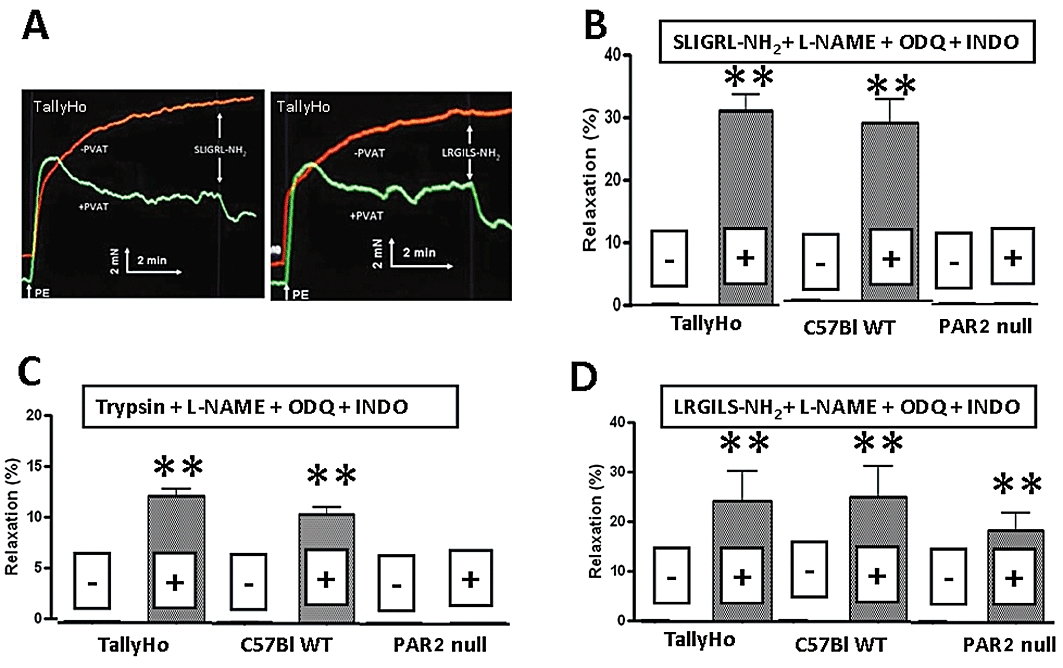
Comparison of the effects of SLIGRL-NH2 (20 µM), LRGILS-NH2 (50 µM) and trypsin (10 nM; 5 U·mL−1) on PVAT-dependent relaxation in aortic rings from TallyHo, C57Bl and C57Bl PAR2-null mice. All studies were performed in phenylephrine (PE) contracted (1 µM) tissues in the combined presence of L-NAME (100 µM), ODQ (10 µM) and indomethacin (INDO, 10 µM). (A) Representative tracing for the relaxation caused by SLIGRL-NH2 (left panel: 20 µM) and LRGILS-NH2 (right panel: 50 µM) in PVAT-containing preparations (+PVAT, lower tracings) compared with PVAT-free preparations (−PVAT, upper tracings). The inset shows the scale for time (min) and tension (mN). (B) Effects of SLIGRL-NH2 (20 µM) on PVAT-intact (+) and PVAT-minus (−) aortic rings from TallyHo, C57Bl6 and C57Bl6 PAR2-null mice. (C) Effects of trypsin (10 nM; 5 U·mL−1) on PVAT-intact (+) and PVAT-minus (−) aortic rings from TallyHo, C57BL6 and C57Bl6 PAR2-null mice. (D) Effects of LRGILS-NH2 (50 µM) on PVAT-intact (+) and PVAT-minus (−) aortic rings from TallyHo, C57BL6 and C57Bl6 PAR2-null mice.
Is ADRF release PAR2-dependent?
To verify that the ADRF release caused by the PAR2-APs was due to PAR2 activation and not to a non-specific effect of the peptides acting on a non-PAR2 target, we turned to strategies that (A) made use of our C57Bl PAR2-null mice instead of the TallyHo animals and (B) used a PAR-activating peptide PAR2 structure-activity approach. Thus, we employed: (i) a comparative evaluation of PVAT-dependent ADRF release caused by the PAR2-APs and trypsin in PAR2 wild-type versus PAR2-null mice as well as in TallyHo animals that express wild-type PAR2; (ii) a comparison of the relative potencies of the PAR2-selective receptor-activating peptides, 2-furoyl-LIGRLO-NH2 and SLIGRL-NH2 for PAR2-triggered ADRF release [2-furoyl-LIGRLO-NH2 is known to be more potent than SLIGRL-NH2 for PAR2 activation (McGuire et al., 2004)]; and (iii) the use of reverse sequence PAR2-inactive peptides that cannot activate PAR2 (LRGILS-NH2, 2-furoyl-ORLGIL-NH2, and LSIGRL-NH2).
To this point, we had established (i) that in endothelium-intact PVAT-free vascular rings from wild-type C57Bl mice and from TallyHo mice that were contracted with phenylephrine, the receptor-selective PAR2-APs, SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2, caused an endothelium-dependent relaxation that was completely blocked by L-NAME either alone or in the combined presence of ODQ and indomethacin (not shown); and (ii) that the reverse-sequence PAR2-inactive peptide, LRGILS-NH2 had no effect in comparable fat-free preparations either with or without an intact endothelium (not shown). Thus, we next went on to evaluate the impact of PVAT on PAR2 relaxation in the C57Bl tissues (both PAR2 wild-type and PAR2-null) triggered either by a receptor-activating peptide or by the PAR2-activating proteinase, trypsin. The experiments were done in the presence of L-NAME, ODQ and indomethacin to minimize any possible contribution of the endothelium-derived NO and COX metabolites to the relaxant response.
In the PVAT-containing aorta tissue from the wild-type C57Bl mice, in the combined presence of L-NAME, ODQ and indomethacin, SLIGRL-NH2did cause a relaxation response (Figure 3B, middle histograms) comparable to that observed with the TallyHo-derived aorta tissue (left-hand shaded histogram, Figure 3B). Similarly, in PVAT-containing tissue from TallyHo and wild-type C57Bl mice (but not in PVAT-free preparations), the PAR2-activating proteinase, trypsin, caused a relaxant effect (Figure 3C). The relaxation caused by trypsin and due to PAR2 activation (see below) was comparable in tissues derived from the TallyHo and C57Bl mice (Figure 3C). Moreover, we found that for ADRF-induced relaxation in PVAT-containing preparations, 2-furoyl-LIGRLO-NH2 was more than twofold more potent (relative EC50s) than SLIGRL-NH2 (not shown), in keeping with the order of potency for these two peptides for activating recombinantly expressed PAR2.
In contrast with the data obtained with the PAR2-expressing tissues (TallyHo and wild-type C57Bl), SLIGRL-NH2 and trypsin did not cause a relaxant effect either in PVAT-free or PVAT-containing aorta preparations from the PAR2-null mice (experiments done in the combined presence of L-NAME, ODQ and indomethacin: Figure 3B and C, right-hand histograms). We were thus able to conclude that the release of ADRF by SLIGRL-NH2 and trypsin was due to the activation of PAR2 in the tissues from the PAR2-expressing mice and that the effect of ADRF released via PAR2 on vascular tone was comparable in the TallyHo and C57Bl mice.
The complete- and partial-reverse-sequence PAR2-inactive peptides, LRGILS-NH2, LSIGRL-NH2 and 2f-OLRGIL-NH2 that cannot regulate PAR2 were next tested for their ability to generate ADRF in tissues from both TallyHo and C57Bl animals. The partial reverse sequence PAR2-inactive peptide, LSIGRL-NH2 (50 µM) failed to cause a relaxant response in preparations either with or without PVAT (not shown). However, to our surprise, either without or in the combined presence of L-NAME, ODQ and indomethacin, both PAR2-inactive peptides, LRGILS-NH2 and 2-furoyl-OLRGIL-NH2 caused a relaxant response in the PVAT-containing aorta tissue from both TallyHo and C57Bl PAR2 wild-type mice, but not in PVAT-free preparations (Figure 3D, shaded histograms; representative tracing, Figure 3A, right panel; and data not shown for 2-furoyl-OLRGIL-NH2). In the PVAT-containing TallyHo and wild-type C57Bl aorta tissue, the relaxant responses to LRGILS-NH2 were not sigificantly different from the relaxation caused by SLIGRL-NH2 (Figure 3, B vs. D).
The concentration-response curve for the action of the reverse sequence peptide LRGILS-NH2 showed that in the presence of PVAT, its potency for causing relaxation (EC50 of 9.5 µM) was comparable to that of SLIGRL-NH2 (Figure 2B). In contrast, in PVAT-free aorta tissue from the wild-type C57Bl mice either in the absence (Figure 2B) or presence of L-NAME, ODQ and indomethacin, concentrations of this PAR2-inactive peptide up to 50 µM were not able to cause a relaxation (Figures 2B and 3D). Comparable data were obtained with the TallyHo-derived tissues (not shown and Figure 3D).
We then studied the actions of the reverse-sequence peptides in aorta tissue from the PAR2-null C57Bl mice. As shown in Figure 3D, the PAR2-inactive peptide, LRGILS-NH2, caused a relaxant response in PVAT-containing tissues from the PAR2-null mice, but had no effect in PVAT-free tissues. Therefore, in contrast to the receptor-selective PAR2-AP SLIGRL-NH2 and the PAR2-activating proteinase, trypsin, which did not cause a relaxant effect in PVAT-containing tissues from PAR2-null mice, the reverse-sequence peptides, LRGILS-NH2 and -furoyl-OLRGIL-NH2 (but not LSIGRL-NH2) were able to release ADRF from PVAT-containing tissues from both the PAR2-wild-type and the PAR2-null animals. Thus, LRGILS-NH2 caused the release of ADRF via a PAR2-independent mechanism.
Is PAR2 present in PVAT?
Given that the release of ADRF caused by the PAR2-APs and trypsin was shown both genetically and pharmacologically to be due to PAR2 activation only in PVAT-containing preparations, we wished to verify that PAR2 was indeed present in the adipose tissue. To this end, we assessed, using RT-PCR, if mRNA for PAR2 was present in both intact adipose tissue and in isolated adipocytes. As shown in Figure 4, the PCR signals that were of the expected size based on the primer pairs indicated that mRNA for both PAR2 and PAR1 could be detected both in intact murine PVAT from aorta and from collagenase-dissociated isolated adipocytes obtained from PVAT. We also detected PAR1 mRNA in samples from the PAR2-null mice (not shown) in which the PAR1 receptor is known to be activated by both thrombin and PAR1-activating peptides. However, from our PCR data we were not able to know if PAR1 was up-regulated in the tissues from the PAR2-null mice. Sequencing of the PCR products confirmed that they represented PARs 1 and 2.
Figure 4.
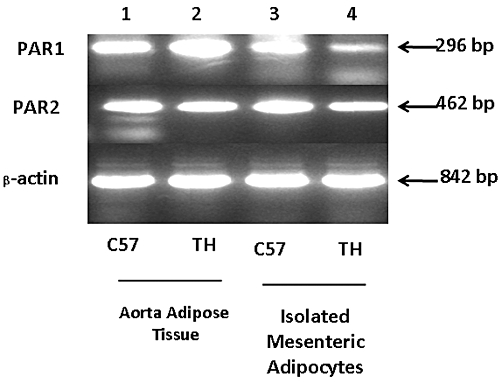
Detecting PARs 1 and 2 in perivascular adipose tissue and isolated adipocytes by RT-PCR. The composite image shows the PCR signals for PARs 1 and 2 and actin detected either in aorta-derived adipose tissue from TallyHo (TH) or wild-type PAR2 C57Bl mice (C57) or in isolated adipocytes obtained from TallyHo and wild-type PAR2 C57Bl mesenteric artery preparations. The size of the PCR product (in base-pairs: bp) is shown on the right.
Are the ADRFs released by SLIGRL-NH2 and LRGILS-NH2 the same; and do they differ from previously characterized ADRFs?
We hypothesized that the murine ADRFs released by PAR2-AP, SLIGRL-NH2, and by the PAR2-inactive peptide, LRGILS-NH2 might differ from each other and might be different from previously described ADRFs (Table 1). As outlined by the data in the following sections, we used a number of K+-channel blockers and catalase, along with the kinase inhibitors, H89 and genistein: (i) to distinguish the actions of the ADRF(s) we had detected from other previously described ADRFs; and (ii) to distinguish the ADRF(s) released by SLIGRL-NH2 from the one(s) released by LRGILS-NH2. As the ADRF responses of the tissues derived from the TallyHo and C57Bl mice were essentially interchangeable, the continuing work described in the following sections was done primarily with the TallyHo tissues due to the more efficient accessibility to adipose-containing aorta tissue in these mice, compared with the C57Bl strain.
Roles of K+ channels
To assess a role for the small and intermediate conductance calcium-activated K+ channels known to be involved in the actions of endothelium-derived relaxing factors, we evaluated the impact of both apamin and charybdotoxin on the relaxant effects of SLIGRL-NH2 and LRGILS-NH2. In the combined presence of these two inhibitors, along with L-NAME, ODQ and indomethacin, we found that the PVAT-dependent relaxant action of both SLIGRL-NH2 and LRGILS-NH2 persisted (not shown and Figure S1). Similarly, glibenclamide, that inhibits ATP-sensitive K+ channels, did not block ADRF-triggered relaxation mediated by either SLIGRL-NH2 or LRGILS-NH2 (not shown and Figure S1). We thus concluded that the ADRF released by both SLIGRL-NH2 and LRGILS-NH2 did not involve either the small and intermediate conductance calcium-regulated K+ channels or the sulphonylurea-sensitive ATP-sensitive K+ channels.
To evaluate a role for voltage-dependent K+ channels, we tested the ability of SLIGRL-NH2 and LRGILS-NH2 to release ADRF in the presence of 4-aminopyridine. As shown in Figure 5, 4-aminopyridine did not affect ADRF release caused by SLIGRL-NH2, but inhibited the action of LRGILS-NH2 in both the TallyHo and C57Bl-derived tissues. Thus, the ADRFs released by the two peptides differed in terms of the involvement of 4-aminopyridine-blocked voltage-dependent K+ channels.
Figure 5.
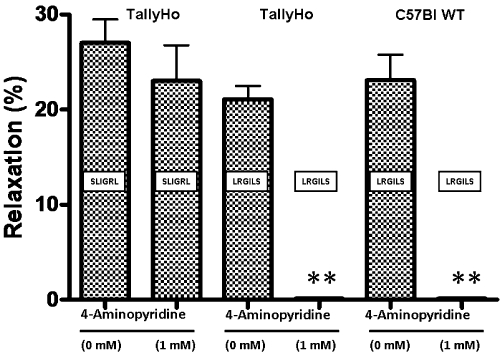
4-Aminopyridine blocks relaxation caused by LRGILS-NH2 but not SLIGRL-NH2-mediated ADRF release from PVAT-containing TallyHo and C57Bl aorta rings. TallyHo and C57Bl wild-type (WT) mouse aorta rings with adherent perivascular adipose tissue were contracted with phenylephrine (1 µM) and the relaxant responses to either SLIGRL-NH2 (SLIGRL, 20 µM) or LRGILS-NH2 (LRGILS, 50 µM) were measured in the combined presence of L-NAME (100 µM), ODQ (10 µM) and indomethacin (10 µM), either without (0 mM) or with added 4-aminopyridine (1 mM). Values for each histogram represent the average relaxation (%, mean ± SEM, n = 5) relative to the plateau tension developed in the presence of phenylephrine. **P < 0.01 for the comparison of 4-aminopyridine-treated tissues versus controls.
Role of peroxide
We evaluated a potential role for peroxide as an ‘ADRF’ by testing the effect of catalase on the PVAT-associated relaxant activity triggered by both SLIGRL-NH2 and LRGILS-NH2. As shown in Figure 6, the presence of catalase fully blocked the relaxant action of SLIGRL-NH2 and trypsin in both TallyHo and C57Bl mice, whereas the relaxant action of the PAR2-inactive peptide, LRGILS-NH2 persisted in the presence of catalase. Thus, the ADRF released by SLIGRL-NH2 and trypsin in both TallyHo and C57Bl-derived tissues via PAR2 involved a peroxide-mediated process, whereas the PAR2-independent LRGILS-NH2-released ADRF was peroxide-independent.
Figure 6.
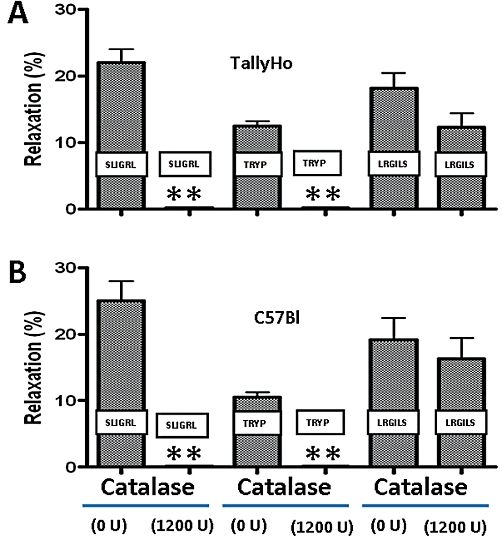
Catalase blocks SLIGRL-NH2 and trypsin-mediated ADRF release but does not affect LRGILS-NH2-mediated relaxation in PVAT-containing TallyHo and PAR2 wild-type C57Bl aorta tissue. TallyHo and C57Bl wild-type aortic rings with adherent perivascular adipose tissue were contracted with phenylephrine (1 µM) and the relaxant responses to either SLIGRL-NH2 (SLIGRL, 20 µM), trypsin (TRYP: 10 nM; 5 U·mL−1) or LRGILS-NH2 (LRGILS, 50 µM) were measured in the combined presence of L-NAME (100 µM), ODQ (10 µM) and indomethacin (10 µM), either without (0 U·mL−1) or with added catalase (1200 U·mL−1). Values for each histogram represent the average relaxation (%, mean ± SEM, n = 5) relative to the plateau tension developed in the presence of phenylephrine. **P < 0.01 for the comparison of catalase-treated tissues versus controls.
Protein kinase A and tyrosine kinase pathways
Potential roles for tyrosine kinases and protein kinase A in mediating ADRF responses were tested with the non-selective tyrosine kinase inhibitor, genistein, and the kinase A inhibitor, H89. Neither H89 (5 µM) nor genistein (10 µM) diminished the relaxant responses caused by either SLIGRL-NH2 or LRGILS-NH2 in the adipose tissue-containing TallyHo preparations that were concurrently exposed to L-NAME, ODQ and indomethacin (not shown and Figure S1). Figure 7 provides an overview of the inhibition of ADRFs released by the PAR-derived peptides, SLIGRL-NH2 (ADRFSL) and LRGILS-NH2 (ADRFLR).
Figure 7.
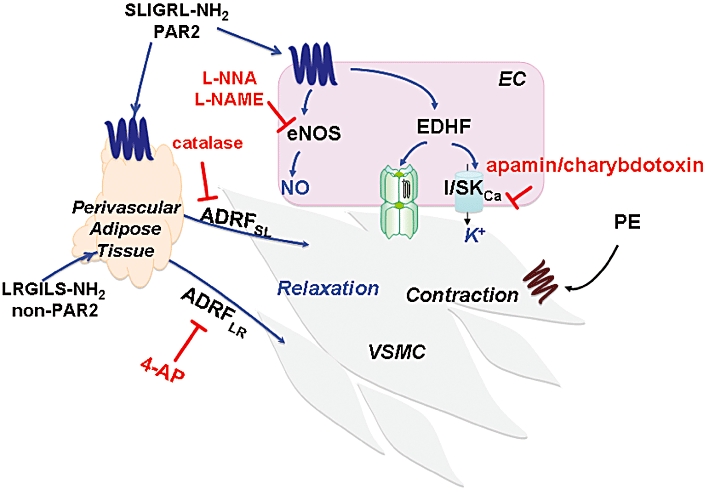
Distinct ADRFs released by SLIGRL-NH2 (ADRFSL) and LRGILS-NH2 (ADRFLR). A scheme is shown summarizing the data for ADRFs released by PAR2-derived peptides from perivascular adipose tissue (blue arrows) that are either catalase-sensitive (ADRFSL) or 4-aminopyridine (4-AP)-sensitive (ADRFLR). PVAT-independent effects of SLIGRL-NH2 that also result in relaxation of vascular smooth muscle cells (VSMC) via the PAR2-mediated release of endothelium-dependent relaxing factors (NO/EDHF) are also shown (blue script). Contraction is attributed to the activation of α1-adrenoceptors by phenylephrine (PE). The ability of L-NNA, L-NAME and apamin/charybdotoxin to block relaxation caused by the non-ADRF endothelial cell (EC)-derived mediators (NO, EDHF) in the preparation is also illustrated.
Discussion and conclusions
PAR2 agonist- and LRGILS-NH2-stimulated ADRFs are distinct from each other
Our main finding, summarized schematically in Figure 7 was that distinct ADRFs can be released from murine aorta perivascular fat by either a PAR2-dependent or a PAR2-independent process. The distinct ADRFs are characterized by their differential blockade by either catalase or 4-aminopyiridine (Figure 7). The data obtained with the C57Bl-derived preparations paralleled the data obtained with tissues from the TallyHo mice. This result indicates that in contrast to differences in ADRFs released from different rat strains (Sprague vs. Wistar: Table 1), there was no difference in ADRFs between the two mouse strains that we studied. Further, the results show that the obese hyperglycaemic phenotype of the TallyHo mouse does not influence the contribution and/or effects of ADRFs on vascular function. The distinct PAR peptide-released ADRFs (the one released by SLIGRL-NH2 designated as ADRFSL and the one released by LRGILS-NH2 designated as ADRFLR in Figure 7) differ in terms of their mechanisms of action. For instance, the PAR2-triggered PVAT-dependent relaxation stimulated by SLIGRL-NH2 in both the TallyHo and C57Bl-derived tissues (ADRFSL, Figure 7) was inhibited by catalase, but not by 4-aminopyridine. Although peroxide may be a ‘final common mediator’ for several ‘ADRFs’, each peroxide-mediated factor that results in relaxation may be distinct. The mechanism whereby the PAR2-released ADRF acts via peroxide (catalase inhibited) remains to be determined, but differs from the ODQ-sensitive endothelium-independent ADRF described by Gao et al. (2007) that was also blocked by catalase (Table 1). In contrast, the PAR2-independent relaxation caused by LRGILS-NH2 (ADRFLR, Figure 7) was blocked by 4-aminopyridine, but not by catalase. This result points to a role for voltage-activated K+ channels, in keeping with data obtained previously for a rat mesenteric ADRF and for a murine ‘non-adiponectin’ ADRF (Verlohren et al., 2004; Fésüs et al., 2007: Table 1). Thus, the murine aorta ADRFs released by SLIGRL-NH2 and LRGILS-NH2 are mechanistically different from each other. We suggest that our data distinguishing the PAR2 versus non-PAR2-released ADRFs relate to the different effects of the ADRFs on vascular K+ channels. Given that our data showed no inhibition of the PAR2 peptide-released ADRF action by glibenclamide or a combination of apamin and charybdotoxin, an effect on either the small and medium calcium-activated K+ channels known to be affected by endothelium-derived relaxing factors, endothelium-derived hyperpolarizing factor, or the sulphonylurea-inhibited ATP-sensitive K+ channels can be ruled out (Waldron and Garland, 1994). We have not been able to detect ADRF-like activity from PAR peptide-treated adipose tissue supernatants, as has been reported for other ADRFs (Lohn et al., 2002; Dubrovska et al., 2004; Gao et al., 2007). Thus, we cannot yet determine if the effect of the inhibitors we used is on either: (i) the release of the ADRFs from the PVAT, or (ii) the action of the ADRFs directly on the vascular smooth muscle to prevent ADRF-mediated relaxation.
The inhibitor profiles distinguish the PAR peptide-released ADRFs from those released by other agonists
Taken together, the distinct ‘inhibitor profiles’ for both of the PAR peptide-released ADRFs (Figure 7) distinguish them from the phenylephrine-released ADRFs described previously (Table 1). Thus, unlike other ADRFs, described so far (Lohn et al., 2002; Dubrovska et al., 2004; Gao et al., 2007; Lee et al., 2009: Table 1), the factors we detected (both PAR2-dependent and PAR2-independent) act in an endothelium-independent manner, that persists in the combined presence of L-NAME, indomethacin, ODQ, the combined KCa inhibitors, apamin and charybdotoxin and the KATP-channel inhibitor, glibenclamide. Further, ADRF release caused by SLIGRL-NH2 and LRGILS-NH2 was not affected by the kinase A and tyrosine kinase inhibitors H89 and genistein, as is the case for the ADRF released from rat aorta PVAT by phenylephrine and 5-HT (Lohn et al., 2002; Dubrovska et al., 2004: Table 1). Additionally, our inhibitor data distinguish the actions of the PAR peptide-released ADRFs from endothelium-derived hyperpolarizing factor-mediated vascular relaxation due to non-NO/non-PGI2 endothelium-dependent mechanisms (Adeagbo and Triggle, 1993; Waldron and Garland, 1994; Garland and Plane, 1996; Edwards et al., 1998; McGuire et al., 2001). Of interest is that, in contrast to our aorta-derived data, a glibenclamide-sensitive ADRF has been shown to regulate microvascular tone in the obese ob/ob insulin-resistant mouse (Xiang and Hester, 2009) as was found for 5-HT-induced ADRF released in rat aorta (Lohn et al., 2002). Thus, the ADRFs released by common agonists from different adipose depots in the same species (e.g. aorta vs. mesenteric in the mouse) or by different agonists from the same location in different species (e.g. rat vs. mouse aorta ADRFs triggered by either phenylephrine or PAR peptides) would appear to differ. That said, it is feasible that the same ADRF released in different vascular beds might act via different mechanisms on the smooth muscle. Thus, although it is most likely that molecularly distinct ADRFs are involved in these situations, conclusive evidence for the chemical differences between the ADRFs obtained from different species or from different vascular beds in the same animal will require their purification and characterization.
Detecting ADRFs: might the paradigm for monitoring ADRF release identify different compounds?
Our study uses a somewhat different experimental paradigm to detect ADRF, compared with all other works published to date, except for angiotensin (1-7), an endothelium-dependent, NO-mediated ADRF substance released by phenylephrine from Wistar rat PVAT (Gao et al., 2007; Lee et al., 2009). In all other previous studies (e.g. Lohn et al., 2002; Dubrovska et al., 2004; Verlohren et al., 2004), the presence of ADRF has been monitored in terms of a reduced response to a contractile agonist that is observed in the presence of PVAT compared with an increased contraction observed in PVAT-free tissue (e.g. for phenylephrine: Figure 1A). In contrast with agonists tested for ADRF release so far, the PAR2-derived peptides that also release ADRF do not contract aorta preparations either with or without an intact endothelium (Al-Ani et al., 1995 and data not shown). Thus, we were able to use a contracted vessel as a ‘reporter’ of ADRF release/action. Under the conditions we used (either endothelium-free preparations, or endothelium-intact preparations treated with L-NAME combined or not with ODQ, indomethacin and KCa-channel blockers), a relaxant response was observed only in the presence of PVAT. As our inhibitor data argue against the participation of previously described ADRFs, we suggest that it is very likely there are significant differences in the ADRF substance(s) released from murine versus rat versus human PVAT. We suggest that our approach for monitoring ADRF release by measuring the relaxation of a contracted vessel may single out factors that differ from those detected by a diminished contractile response. Given that more than one type of ADRF has been demonstrated in other work (Gao et al., 2005; Lee et al., 2009: Table 1), it is likely that adipose tissue-related vasorelaxation involves a number of different compounds, depending on the tissue, agonist and species. This potential heterogeneity of ADRFs that are detected by different bioassay approaches will need to be accommodated in future studies.
Involvement of PAR2
Our original hypothesis was that PAR2 activation would release ADRF in vascular tissue. This process could thus play a role in tissue inflammation known to involve PAR2. Indeed, the ability of two different PAR2-selective peptide agonists (SLIGRL-NH2 and 2-furoyl-LIGRLO-NH2) as well as trypsin to cause a PVAT-dependent relaxation supports this hypothesis as do (i) the lack of effect of the PAR2-inactive peptide, LSIGRL- NH2 and (ii) the inability of trypsin or SLIGRL-NH2 to elicit ADRF release in tissues from PAR2-null mice (Figure 3B and C). These results are supported by the finding of PAR2 mRNA in both intact adipose tissue and in isolated adipocytes. Although PAR2 is clearly involved, the precise target for the PAR2-APs or trypsin in the adipose tissue (e.g. adipocytes per se, vs. non-adipocyte vascular adventitial elements or other stromal cells) remains to be ascertained.
The non-PAR2 ‘LRGILS-NH2’ receptor
The ability of the PAR2-inactive reverse-sequence peptides, LRGILS-NH2 and 2-furoyl-OLRGIL-NH2 (but not the LSIGRL-NH2 peptide) to cause a relaxation response only in the presence of PVAT in both wild-type and PAR2-null mice (Figure 3D) indicates that a peptide receptor other than PAR2 can also release ADRF. This conclusion that LRGILS-NH2 and 2-furoyl-OLRGIL-NH2 act via a PAR2-independent mechanism is supported not only by the ability of LRGILS-NH2 and 2-furoyl-OLRGIL-NH2 to release ADRF from the PAR2-null tissues (Figure 3D and data not shown) but also by the lack of a relaxant action of either LRGILS-NH2 or 2-furoyl-OLRGIL-NH2 in endothelium-intact adipose tissue-free preparations from PAR2 wild-type animals (not shown). The novel non-PAR2 receptor activated by LRGILS-NH2 and 2-furoyl-OLRGIL-NH2, but not by LSIGRL-NH2 remains to be identified (McGuire et al., 2002).
Proteinases, ADRF release and adipose tissue function
Trypsin's ability to mimic the actions of the PAR2-APs in releasing ADRF only from PAR2 wild-type and not from PAR2-null mice indicates that its action is via PAR2. However, further work must be done to establish if proteinases of adipose-tissue origin might be able to regulate ADRF release via PAR2 in vivo. Nonetheless: (i) the presence in fat of the trypsin-related enzyme adipsin; (ii) the ability of trypsin to cause ‘insulin-like’ responses in both adipocyte and diaphragm target tissues (Rieser and Rieser, 1964; Cuatrecasas, 1971; Kono and Barham, 1971); and (iii) the ability of trypsin to cause the release of ADRF from PVAT adds fat-derived serine proteinases to the list of potential regulators of adipose tissue function.
In summary, our work demonstrates the release of multiple murine PVAT-derived factors that cause relaxation of contracted vessels via mechanisms that differ from each other and are, with respect to PAR2 activation, also different from those ADRFs described so far. That the release of murine ADRFs can occur via a PAR2-dependent as well as a PAR2-independent mechanism suggests that a number of receptors yet to be described in PVAT may be able to release comparable ADRFs. Further, our work suggests a possible role for adipose tissue-generated serine proteinases (e.g. adipsin) as potential regulators of vascular function via an action on PVAT. It will be of importance to determine if the ADRFs play a pathophysiological role in the setting of vascular inflammation, atherosclerosis, obesity or diabetes, where their release may modulate vessel function. The chemical nature of the factors we have detected and the receptors other than PAR2 that may be involved are important topics for future work in this area.
Acknowledgments
This work was supported principally by grants from the Canadian Institutes for Health Research (to M. D. H. and C. R. T.), the Qatar Foundation National Priorities Research Program (NPRP 08 165-3-054: to M. D. H., C. R. T. and H. D.) and the Heart and Stroke Foundation of Alberta and Nunavut (to M. D. H.). Other funds were also provided by grants from the Canadian Diabetes Association and Alberta Cancer Board (to D. C. W. L.). We are grateful to Dr Pierre-Yves von der Weid for his suggestions and for his critical reading of our manuscript.
Glossary
Abbreviations
- ADRF
adipocyte-derived relaxing factor
- genistein
5,7-dihydroxy-3-(4-hydroxyphenyl)chromen-4-one
- glibenclamide
5-chloro-N-(4-[N-(cyclohexylcarbamoyl)sulphamoyl]phenethyl)-2-methoxybenzamide
- H89 (protein kinase A inhibitor)
N-[2-((3-(4-bromophenyl)-2-propenyl)-amino)-ethyl]-5- isoquinolinesulphonamide, di-hydrochloride
- L-NAME
NOS inhibitor, Nω-nitro-L-arginine methyl ester
- L-NNA
NOS inhibitor, Nω-nitro-L-arginine
- ODQ
guanylyl cyclase inhibitor, 1H-[1,2,4] oxadiazolo[4,3,a]quinoxalin-1-one
- PAR
proteinase-activated receptor
- PAR2-AP
proteinase-activated receptor-2-activating peptide
- PVAT
perivascular adipose tissue
Conflicts of interests
The authors declare that they have no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Lack of effect of inhibitors on ADRF-relaxant responses caused by SLIGRL-NH2 and LRGILS-NH2. PVAT-containing aorta rings from TallyHo mice were constricted with phenylephrine (1 μM) and the relaxant responses to either SLIGRL-NH2 or LRGILS-NH2 were monitored in the absence or presence of the inhibitors (A) glibenclamide (5 μM), (B) genistein (10 μM) and (D) H89 (5 μM). Also shown is (C) the persistence of the ADRF relaxant response to the two peptides in the presence of added apamin (1 μM) and charybdotoxin (0.1 μM) supplementing the combined presence of L-NAME (100 μM), ODQ (10 μM) and indomethacin (10 μM). Values for each histogram represent the average relaxation (%, mean ± SEM, n = 5) relative to the plateau tension developed in the presence of phenylephrine (1 μM).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adeagbo AS, Triggle CR. Varying extracellular [K+]: a functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J Cardiovasc Pharmacol. 1993;21:423–429. [PubMed] [Google Scholar]
- Al-Ani B, Saifeddine M, Hollenberg MD. Detection of functional receptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol. 1995;73:1203–1207. doi: 10.1139/y95-172. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng ZJ, Jiang YF, Ding H, Severson D, Triggle CR. Vascular dysfunction in type 2 diabetic TallyHo mice: role for an increase in the contribution of PGH2/TxA2 receptor activation and cytochrome p450 products. Can J Physiol Pharmacol. 2007;85:404–412. doi: 10.1139/y07-010. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- Cuatrecasas P. Perturbation of the insulin receptor of isolated fat cells with proteolytic enzymes. J Biol Chem. 1971;246:6522–6531. [PubMed] [Google Scholar]
- Damiano BP, Cheung WM, Santulli RJ, Fung-Leung WP, Ngo K, Ye RD, et al. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J Pharmacol Exp Ther. 1999;288:671–678. [PubMed] [Google Scholar]
- Dubrovska G, Verlohren S, Luft FC, Gollasch M. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1107–H1113. doi: 10.1152/ajpheart.00656.2003. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Fésüs G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, et al. Adiponectin is a novel humoral vasodilator. Cardiovasc Res. 2007;75:719–727. doi: 10.1016/j.cardiores.2007.05.025. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I, et al. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thorac Cardiovasc Surg. 2005;130:1130–1136. doi: 10.1016/j.jtcvs.2005.05.028. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Lu C, Su LY, Sharma AM, Lee RMKW. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–331. doi: 10.1038/sj.bjp.0707228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Plane F. Relative importance of endothelium-derived hyperpolarizing factor for the relaxation of vascular smooth muscle in different arterial beds. In: Vanhoutte PM, editor. Endothelium-Derived Hyperpolarizing Factor. Amsterdam: Harwood Academic Publishers; 1996. pp. 173–179. [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- Gollasch M. Vasodilator signals from perivascular adipose tissue. Br J Pharmacol. 2011 doi: 10.1111/j.1476-5381.2011.01430.x. DOI: 10.1111/j.1476-5381.2011.01430.x. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev. 2002;54:203–217. doi: 10.1124/pr.54.2.203. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B. Proteinase-activated receptor-2 in rat aorta: structural requirements for agonist activity of receptor-activating peptides. Mol Pharmacol. 1996;49:229–233. [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- Kim JH, Sen S, Avery CS, Simpson E, Chandler P, Nishina PM, et al. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics. 2001;74:273–286. doi: 10.1006/geno.2001.6569. [DOI] [PubMed] [Google Scholar]
- Kono T, Barham FW. Insulin-like effects of trypsin on fat cells. Localization of the metabolic steps and the cellular site affected by the enzyme. J Biol Chem. 1971;246:6204–6209. [PubMed] [Google Scholar]
- Lee RM, Lu C, Su LY, Gao YJ. Endothelium-dependent relaxation factor released by perivascular adipose tissue. J Hypertens. 2009;27:782–790. doi: 10.1097/HJH.0b013e328324ed86. [DOI] [PubMed] [Google Scholar]
- Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–1063. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Ding H, Triggle CR. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s) Can J Physiol Pharmacol. 2001;79:443–470. [PubMed] [Google Scholar]
- McGuire JJ, Dai J, Andrade-Gordon P, Triggle CR, Hollenberg MD. Proteinase-activated receptor-2 (PAR2): vascular effects of a PAR2-derived activating peptide via a receptor different than PAR2. J Pharmacol Exp Ther. 2002;303:985–292. doi: 10.1124/jpet.102.040352. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Saifeddine M, Triggle CR, Sun K, Hollenberg MD. 2-furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J Pharmacol Exp Ther. 2004;309:1124–1131. doi: 10.1124/jpet.103.064584. [DOI] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Hollenberg MD. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153(Suppl 1):S263–S282. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieser P, Rieser CH. Anabolic responses of diaphragm muscle to insulin and to other pancreatic proteins. Proc Soc Exp Biol Med. 1964;116:669–671. doi: 10.3181/00379727-116-29339. [DOI] [PubMed] [Google Scholar]
- Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13:277–296. doi: 10.3109/10641969109042063. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Buddenkotte J, Shpacovitch V, Rattenholl A, Moormann C, Vergnolle N, et al. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr Rev. 2005;26:1–43. doi: 10.1210/er.2003-0025. [DOI] [PubMed] [Google Scholar]
- Van RL, Roncari DA. Isolation of fat cell precursors from adult rat adipose tissue. Cell Tissue Res. 1977;181:197–203. doi: 10.1007/BF00219980. [DOI] [PubMed] [Google Scholar]
- Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, et al. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004;44:271–276. doi: 10.1161/01.HYP.0000140058.28994.ec. [DOI] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Waldron GJ, Garland CJ. Effect of potassium channel blockers on L-NAME insensitive relaxations in rat small mesenteric artery. Can J Physiol Pharmacol. 1994;72(Suppl 1):P1.7.26. [Google Scholar]
- Waldron GJ, Ding H, Lovren F, Kubes P, Triggle CR. Acetylcholine-induced relaxation of peripheral arteries isolated from mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 1999;128:653–658. doi: 10.1038/sj.bjp.0702858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L, Hester RL. Adipocyte-derived factor reduces vasodilatory capability in ob-/ob- mice. Am J Physiol Heart Circ Physiol. 2009;297:H689–H695. doi: 10.1152/ajpheart.01327.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.