

Abstract
BACKGROUND
Tight junctions form a continuous belt-like structure between cells and act to regulate paracellular signaling. Protein kinase C (PKC) has been shown to regulate tight junction assembly and disassembly and is activated by alcohol. Previous research has shown that alcohol increases the permeability of tight junctions in lung alveolar cells. However, little is known about alcohol’s effect on tight junctions in epithelium of the conducting airways. We hypothesized that long-term alcohol exposure reduces zonnula occluden-1 (ZO-1) and claudin-1 localization at the cell membrane and increases permeability through a PKC-dependent mechanism.
METHODS
To test this hypothesis, we exposed normal human bronchial epithelial cells (NHBE), cells from a human bronchial epithelial transformed cell line (Beas-2B), and Beas-2B expressing a PKCα dominant negative (DN) to alcohol (20, 50, and 100 mM) for up to 48 hours. Immunofluorescence was used to assess changes in ZO-1, claudin-1, claudin-5 and claudin-7 localization. Electrical cell substrate impedance sensing (ECIS) was used to measure permeability of tight junctions between monolayers of NHBE, Beas-2B, and DN cells.
RESULTS
Alcohol increased tight junction permeability in a concentration-dependent manner and decreased ZO-1, claudin-1, claudin-5 and claudin-7 localization at the cell membrane. To determine a possible signaling mechanism, we measured the activity of PKC isoforms (alpha, delta, epsilon, zeta). PKCα activity significantly increased in Beas-2B cells from 1–6 hours of 100 mM alcohol exposure, while PKCζ activity significantly decreased at 1 hour and increased at 3 hrs. Inhibiting PKCα with Gö-6976 prevented the alcohol-induced protein changes of both ZO-1 and claudin-1 at the cell membrane. PKCα dominant negative Beas-2B cells were resistant to alcohol-induced protein alterations.
CONCLUSIONS
These results suggest that alcohol disrupts ZO-1, claudin-1, claudin-5 and claudin-7 through the activation of PKCα, leading to an alcohol-induced “leakiness” in bronchial epithelial cells. Such alcohol-induced airway leak state likely contributes to the impaired airway host defenses associated with acute and chronic alcohol ingestion.
Keywords: Alcohol, tight junctions, ZO-1, Claudin-1, airway epithelium, lung
Background
Alcohol is the most commonly abused drug in the United States (Stinson et al., 2005). More than half the general adult population of the United States consumes alcohol and approximately 17 million individuals are alcoholics (Grant et al., 2004). Alcohol affects many tissues throughout the body including the liver, brain, heart (Lieber, 1995), lung, and conducting airways (Joshi and Guidot, 2007). Heavy alcohol consumption has long been known to impair lung defenses. Therefore, it is no surprise that alcoholics are also more likely to suffer from pulmonary diseases (Jerrells et al., 2007; Sisson, 2007) such as acute respiratory distress syndrome (ARDS; Moss and Burnham, 2003), chronic bronchitis (Suadicani et al., 2001), and pneumonia (Samokhvalov et al., 2010).
Within the airways, tight junctions play an important role in maintaining cellular polarity and homeostasis by selectively regulating the passage of water, ions, neutral molecules and inflammatory cells through the paracellular pathway (Godfrey, 1997), providing a site for cell-cell signaling (Matter et al., 2005; Schneeberger and Lynch, 2004; Shin et al., 2006) and protecting tissues from pathogenic infection (Fu et al., 2009). Tight junction disruption leads to an imbalance of fluid and ions, loss of cell polarity, impaired neural responsiveness of the airways (Godfrey, 1997), and increased exposure to pathogens (Fu et al., 2009). Importantly, impaired tight junctions have been linked to various diseases in the conducting airways such as chronic bronchitis (Wilson et al., 1996), pneumonia (Asgrimsson et al., 2006) and asthma (Holgate, 2007).
Based on the sites of common lung diseases, it is useful to divide the lung into two separate compartments: the conducting airways and the parenchyma. The conducting airways include the trachea, bronchi, and terminal bronchi. The parenchyma, or gas exchange region, is composed mainly of alveolar air spaces. Each of these compartments contain specialized cells unique to their environment. The effect of alcohol on tight junctions within these different compartments has focused predominantly on the lung parenchyma while little is known about the impact of alcohol on tight junctions in the conducting airways.
Airway “leak” characterized by increased fluid in the conducting airway epithelium, is a cardinal feature of airway diseases such as bronchitis (Thompson et al., 1993) and asthma (Aas, 1981; Fahy et al., 2000). Increased airway leak contributes to airway edema, which is a classic finding of these diseases and results in airway obstruction and impaired mucociliary clearance. Results for these studies will improve our understanding of the impact alcohol has on airway epithelial barrier functions, which are critical for the understanding and treatment of airway diseases common among alcohol abusers.
Tight junction strands are composed of both transmembrane and cytoplasmic proteins. More than 40 proteins that make up or associate with tight junction strands have been identified. The main proteins comprising the tight junction complex include the transmembrane proteins (occludins, claudins, junctional-associated adhesion molecules (JAMs) and the cytoplasmic proteins [zonnula occludens (ZO-1, ZO-2, ZO-3)], Steed et al.) For the purposes of these studies, we focus on the regulation of ZO-1, claudin-5 and claudin-7 in alcohol-induced airway epithelial leak.
Tight junction permeability is regulated through a variety of mechanisms including the modulation of protein kinase C (PKC) activity. PKC has long been known to play a role in both increasing and decreasing tight junction permeability (see for review [Gonzalez-Mariscal et al., 2008]). However, many studies demonstrate that PKC regulation of tight junctions is cell/tissue type specific (Gonzalez-Mariscal et al., 2008) rendering it difficult to universally characterize. Adding to this complexity is the fact that airway epithelium is known to express several PKC isoforms: α, ε, ζ, δ (Liedtke et al., 1997; Wyatt et al., 2009; Wyatt et al., 1998). Several studies show that chronic alcohol intake activates PKC, thus translocating it from the cytoplasm to the membrane (Newton, 2009). Previous research has suggested that alcohol, in low levels, may enhance PKCα activation through Rho GTPases (Slater et al., 2003).
Based on previous evidence demonstrating alcohol’s ability to increase the permeability of tight junctions within the lungs making the cells “leakier” and alcohol’s ability to enhance PKCα activation, we hypothesize that alcohol exposure increases the permeability of tight junctions in airway epithelial cells through a PKCα-dependent mechanism. To test this hypothesis, we exposed Beas-2B, Beas-2B cells expressing a PKCα dominant negative (DN), and normal human bronchial epithelial cells (NHBE) to alcohol and measured transepithelial resistance, PKC isoform activity, and tight junction protein localization.
Methods
Cell Culture
Beas-2B cells (American Tissue Culture Collection (ATCC), Manassas, VA) (passage 10–25), Beas-2B PKCα DN, or NHBE (passage 1–5) were grown to confluence in 75cm2 tissue culture flasks (Corning, Corning, NY) pre-coated with Vitrogen 30 (Invitrogen by Life Technologies, Carlsbad, CA) in Bronchial Epithelial Basal Media (BEBM; Lonza, Walkersville, MD) supplemented with the following additives (with the exception of gentamycin-amphotericin B) from Lonza/Clonetics Corporation as a kit: bovine pituitary extract, epinephrine, triiodothyronine, epidermal growth factor, transferrin, insulin, hydrocortisone, and retinoic acid. In addition, BEBM was supplemented with 50 U/ml penicillin and 50 U/ml streptomycin (Invitrogen, Carlsbad, CA). PKCα DN selection medium is as described above with the addition of doxycycline (1μg/ml) and G418 (100μg/ml). NHBE cells were isolated from human lung donors (International Institute for the Advancement of Medicine, Jessup, PA). Beas-2B PKCα DN cells were constructed as previously reported and characterized (Wyatt et al., 2007).
Cytotoxicity Assay
To determine cell viability of alcohol-treated and non-treated cells, LDH-Cytotoxicity Assay Kit (BioVision Incorporated, Mountain View, CA) was performed according to the manufacturer’s protocol. Briefly, 50 μl supernatant was pipetted into corresponding wells of a 90-well plate. 50 μl of reaction mixture was added to each well and incubated for 15 min at room temperature. The absorbance of all samples was taken at 490–500 nm using a microplate reader (BioRad, Hercules, CA). An additional cell viability assay was performed using trypan blue exclusion test of cell viability. Beas-2B cells were grown to confluence on 60 mm culture dishes as described above. Cells were treated with 100 mM alcohol for 24 hrs. Following 24 hrs of alcohol exposure, cells were rinsed in PBS, trypsinized, centrifuged and resuspended in 3 ml of BEBM media. 100 μl trypan blue (0.4%) and 500 μl cell solution was added to a microfuge tube and mixed thoroughly. Solution was allowed to incubate at room temperature for 5 min. Viable and non-viable cells were counted using a hemocytometer.
Alcohol Treatment
To minimize alcohol evaporation, 90% of the area on each dish was sealed with paraffin, allowing the exchange of CO2. Alcohol levels in the media were measured using HPLC to ensure minimal alcohol evaporation. For experiments carried out longer than 24 hours, fresh media with alcohol was added every 24 hours.
Alcohol concentrations used in this study were based on biologically relevant levels (100 mM). For instance, an individual is legally impaired to drive when blood alcohol concentration reaches 0.08%, which equates to a concentration of 22 mM alcohol. Higher blood alcohol concentrations, such as 50 mM (equates to 0.20% concentration) are commonly observed by individuals presenting to emergency rooms for intoxication. The highest alcohol dose we use is 100 mM, which is consistent with blood alcohol concentrations observed in patients admitted to hospitals and those who chronically abuse alcohol (Teplin et al., 1989). To control for any ethanol-induced changes in cell viability, cytotoxicity, and cell death, Beas-2B cells were exposed to alcohol (100 mM) for 24 hours. Both LDH and trypan blue cell viability assays were then performed. No significant cytotoxicity or cell death occurred in cells treated with 100 mM alcohol compared to control cells (LDH release data not shown). To further confirm cell viability, trypan blue cell viability assays were performed. Results confirmed no significant differences in cell viability of alcohol-treated cells when compared to controls cells. Both alcohol and control groups were at least 95% viable (data not shown).
Immunofluorescence
Beas-2B, Beas-2B PKCα DN, or NHBE cells were plated onto sterile glass coverslips precoated with Vitrogen 30 for indirect immunofluorescence as follows: coverslips were rinsed with PBS, fixed in 4% paraformaldehyde, permeabilized and blocked for 15 min (0.1% Triton X-100, 1% BSA, 0.02% Sodium Azide). Cells were incubated in primary antibodies (mouse anti-claudin-1, rabbit anti-claudin-7, mouse anti-claudin-5, mouse anti-ZO1, and rabbit anti-ZO-1 mid; Invitrogen by Life Technologies, Carlsbad, CA) overnight at 4°C. Following primary antibody incubation, cells were rinsed in PBS and coverslips incubated with secondary antibodies (Alexa 488 and Alexa 647 against rabbit or mouse; Invitrogen by Life Technologies, Carlsbad, CA) for 1 hr at room temperature in a humidified chamber. Following incubation, coverslips were rinsed in PBS and mounted using ProLong Gold with DAPI (Invitrogen by Life Technologies, Carlsbad, CA). Imaging was performed using an LSM 510 Meta Laser Scanning Confocal Microscope (Zeiss, Thornwood, New York). All images were collected as Z-stacks using the set parameters (laser strength, gain, etc.) for all treatments. Z sections were obtained through cells at 0.5 μm. Z-stacks were compressed for fluorescence measurement analysis.
Image J analysis
All images were obtained as Z-stacks (0.5 μm slices) using set parameters for all experimental conditions. Images were compressed using Zeiss microscope software (Zeiss, Thornwood, New York) and saved as jpeg images. Images were uploaded into ImageJ (Rasband, 1997–2009). Image analysis was completed by first subtracting background fluorescence. Then regions of interest were selected to measure fluorescent mean intensity at the membrane only. Data are presented as the mean ± SEM. Statistics were performed with GraphPad Prism software (GraphPad, La Jolla, CA) using a one-way ANOVA or t-test.
Epithelial Barrier Function
Epithelial barrier function was measured using the electric cell-substrate impedance sensing system (ECIS; Applied BioPhysics, Troy, NY). This is a non-invasive technique that allows us to measure and record transepithelial resistance (TER) changes in real time. It is also sensitive enough to determine the cell-cell interactions (tight junctions) (Hartmann et al., 2007; Wegener et al., 2000). With ECIS, cells are cultured onto gold electrodes. As cells grow and become confluent, they constrict current flow and alter impedance (Hartmann et al., 2007). ECIS is able to report impedance values or translate impedance measurements to both resistance and capacitance. This system has become a useful tool to monitor barrier function (the resistance of the paracellular pathway between cells; cell permeability) (Kilani et al., 2004; Sidhaye et al.; Tiruppathi et al., 1992).
Cell Resistance
Cell resistance was measured using ECIS continuously for as long as 7 days (alcohol and media were replenished every two days). All cells including primary cells were grown on Vitrogen 30-coated 8EW10+ arrays. 8EW10+ arrays contain 40 circular 250 μm diameter electrodes, and are capable of measuring between 2000–4000 cells (confluent monolayers). Each well has a substrate area of 0.8cm2. Cells (5×105) were seeded into each well, grown to confluence, and treated with alcohol (0 mM, 20 mM, 50 mM, 100 mM) for up to 96 hrs. Cell resistance measurements were based on changes in resistance/capacitance to current flow applied to the electrode arrays at multiple frequencies. A frequency scan was performed to test for the frequency at which the largest difference in transepithelial resistance (TER) values were obtained between the cell-covered and cell-free electrodes. The optimal frequency to study resistance in Beas-2B and NHBE cells appeared to be 250 and 400 Hz. Baseline values were established with culture media alone and compared to electrodes covered with a monolayer of cells. Experimental conditions were run in duplicate on each plate. Three separate experiments were run for each cell line.
Data were analyzed using ECIS software, resistance normalized (subsequent values divided by initial values) and compiled for each condition and presented as the mean normalized resistance ± SEM (n=3). Resistance was normalized by dividing the impedance values from electrodes confluent with cells by the corresponding quantities for the cell-free electrodes using ECIS software (Lo et al., 1995). Statistics performed were a one-way ANOVA.
PKC Activity
Beas-2B and NHBE cells were grown to confluence in 60 mm Petri dishes containing BEBM. The medium was removed, the cells gently rinsed in PBS, and 250 μl of cell lysis buffer added (35 mM Tris-HCl, 0.4 mM EGTA, 10 mM MgCl2, 0.01% Triton-X 100, 1 mM PMSF, 1 mM Sigma Protease Inhibitor). The epithelial lysates were then immediately flash-frozen in liquid nitrogen for kinase assay. Epithelial lysates were used to determine PKC activity (ε, ζ, δ, α) as previously described (Wyatt et al., 2009). PKC activity was assayed in both the supernatant and particulate fractions. Total PKC activity was measured in cytosolic fractions, which contain soluble proteins and unactivated PKC, and in particulate fractions, containing nonsoluble material (cell membranes, nuclear particles, and cytoskeletal elements) as well as all translocated and activated PKC. PKC activity assays were performed using 900 μM PKC substrate peptide (Peninsula, Belmont, CA), 24 μg/ml phorbol 12-myristate 13-acetate, 12 mM calcium-acetate, 30 mM dithiothreitol, 8 μM phosphatidyl-L-serine, 150 μM ATP, 45 mM magnesium acetate, and 10 μCi/ml [γ-32P] ATP (MP Biomedicals, Irvine, CA) in a Tris-HCl buffer (pH 7.5). Each sample (20 μl) was added to 40 μl of the reaction mixture, and incubated for 15 minutes at 30°C. Next, 50 μl of each sample was spotted onto P-81 phosphocellulose papers (Whatman, Clifton, NJ) and incubations halted. Phosphocellulose papers were then washed five times for 5 minutes in phosphoric acid (75 mM), washed in ethanol, allowed to dry, and counted in nonaqueous scintillant, as previously described (Wyatt et al., 2009). Kinase activity was standardized in relation to total cellular protein assayed, and was calculated as picomoles of phosphate incorporated per minute per milligram. For standardized comparison between isoforms, data was expressed as fold activity changes from control media.
Statistics
ECIS data are presented as the mean normalized resistance (subsequent values divided by initial values) ± SEM (n=3). Statistics performed were a one-way ANOVA for PKC activity and ECIS measurements to determine significant changes among treatment groups, using GraphPad Prism software (GraphPad, La Jolla, CA). Fluorescence mean measurements were completed using ImageJ software from NIH. Data are presented as the mean ± SEM. Statistics were performed using a one-way ANOVA or t-test.
Results
Effect of alcohol on bronchial epithelial cell permeability
To determine the effect of biologically relevant alcohol exposure on airway epithelial “leak”, cells (Beas-2B, Beas-2B PKCα DN or NHBE) were plated on specialized ECIS arrays (5×105) and treated with 0, 20, 50, or 100 mM alcohol. Barrier function and permeability measurements were obtained by measuring resistance (Kilani et al., 2004; Lo et al., 1999; Qiao et al., 2003) using ECIS. Resistance changes were maximal within the first 24–48 hours of alcohol exposure beyond which no significant increases or decreases were observed. Therefore, alcohol exposure experiments were limited to 48 hrs in subsequent experiments.
Resistance data was normalized to time zero (start of alcohol treatment) and compiled to generate ECIS graphs. Alcohol decreased TER in both Beas-2B (Figure 1A) and NHBE (Figure 1B) cells in a concentration-dependent manner, inducing significant decreases in epithelial barrier function, therefore increasing cellular “leak” in cells exposed to 50 mM and 100 mM alcohol compared to control (P<0.01).
Figure 1.
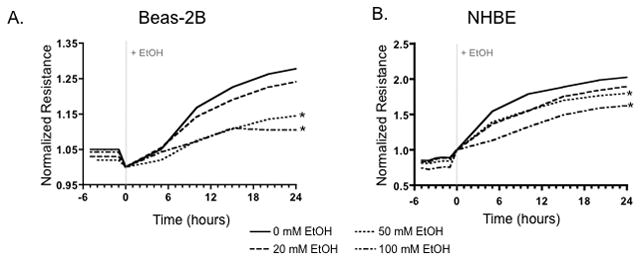
Normalized Resistance (ohms) (subsequent values divided by initial values) of Beas-2B (A) and NHBE (B) cells treated with 0, 20, 50, 100mM alcohol for 48 hours. Lower resistance measurements in Beas-2B and NHBE cells indicates an increased permeability between cells, which occurs in an alcohol dose-dependent manner. Barrier function is significantly decreased at 50 mM and 100 mM alcohol concentrations in Beas-2B and NHBE cells (n=6; * P= 0.01) compared to control.
Effect of alcohol exposure on PKC Isoform activity
Airway cells (Beas-2B, Beas-2B DN and NHBE) were treated with 100 mM alcohol for 1, 3, 6, and 24 hours to determine the activity of various PKC isoforms in response to alcohol. Following alcohol exposure, PKC α, ε, ζ, δ levels were measured as described in Materials and Methods. Activity assays revealed a significant time-dependent increase in PKCα activity at 1, 3, and 6 hrs after treatment with 100 mM alcohol (Figure 2) in Beas-2B cells. PKCα activity trended higher at 3 and 6 hrs and was significantly increased at 24 hrs in NHBE cells (Figure 3). PKCζ activity significantly decreased at 1 hr, followed by a significant increase at 3 hrs in alcohol-treated Beas-2B cells. Although we did not observe an initial decrease in PKCζ in alcohol-exposed NHBE cells, a significant increase was seen at 6 hrs. No significant activity changes were observed in PKCε or PKCδ isoforms (Figures 2 & 3) in any of the cell types tested.
Figure 2.
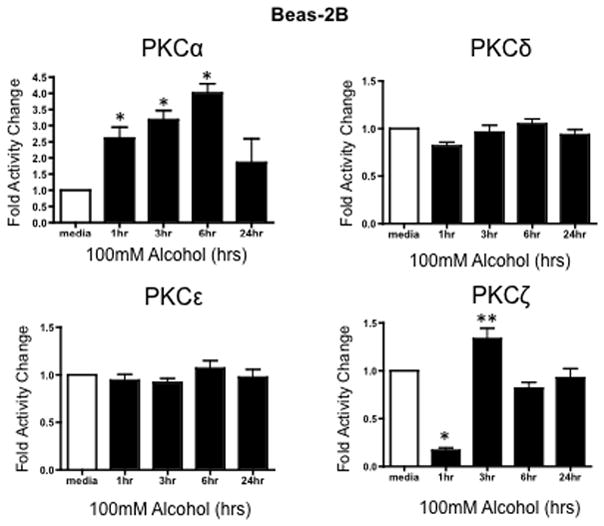
Protein kinase C isoform assay of Beas-2B cells exposed to 100mM alcohol for 1, 3, 6, and 24 hr time periods. The vertical axis represents fold activity change of PKC activity of Beas-2B cells within each exposure group (n=3). Alcohol exposure for 1 to 6 hrs caused a significant (*P ≤ 0.01) increase in the PKCα activity. A significant decrease and increase was observed in PKCζ at 1 (*P ≤ 0.01) and 3 hrs (**P ≤ 0.05). This result indicates that alcohol induces increases in PKCα and PKCζ.
Figure 3.
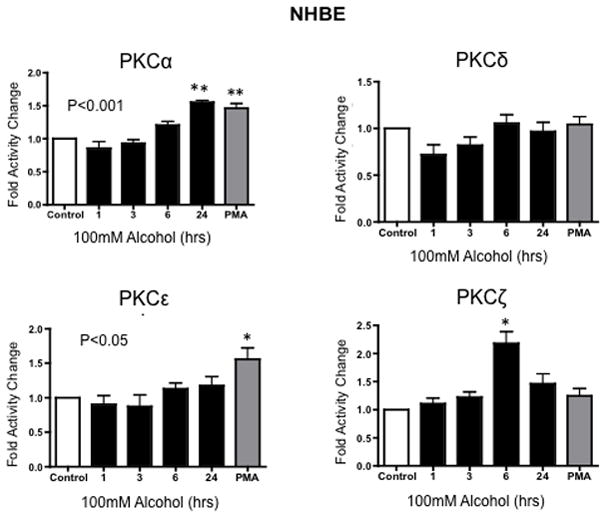
Protein kinase C isoform assay of NHBE cells exposed to 100mM alcohol for 1, 3, 6, and 24 hr time periods. The vertical axis represents the fold activity change (SEM) of PKC activity in of NHBE cells within each exposure group (n=3). Alcohol exposure for 24 hrs caused a significant (**P ≤ 0.001) increase in the PKCα activity. A significant increase was also observed in PKCζ at 6 (*P ≤ 0.01). This result indicates that alcohol induces increases in PKCα and PKCζ.
To further define PKCα as a potential mechanism for the observed alcohol-induced decrease in TER, PKC isoform activity was performed on PKCα dominant negative engineered Beas-2B cells (PKCα DN). There were no significant changes in any of the PKC isoforms in Beas-2B PKCα DN cells exposed to 100 mM alcohol at any time point measured (Figure 4), further supporting a critical role for the PKCα isoform in mediating alcohol’s effect on airway permeability. As a positive control for DN cell function, 100 ng/mL phorbol 12-myristate 13-acetate (PMA) significantly activated both PKCε and PKCδ in the PKCα DN cells (Figure 4). While PKCζ is a PMA-insensitive isoform, we observed a significant PKCζ activation in response to 10 μg/mL lipopolysaccharide (LPS) in both PKCα DN and normal Beas-2B, thus defining the suppression of only the alpha isoform in this cell line (data not shown).
Figure 4.
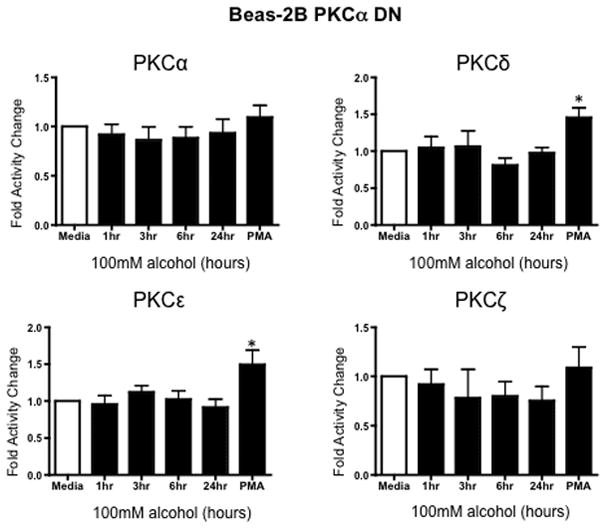
Protein kinase C isoform assay of Beas-2B PKCα DN cells exposed to 100mM alcohol for 1, 3, 6, and 24 hr time periods. The vertical axis represents fold activity change of PKC activity of Beas-2B PKCα DN cells within each exposure group (n=3). No significant increases or decreases were observed in PKC activity exposed to alcohol. A significant increase in PKCε was observed in our positive control.
Alcohol-induced barrier disruption requires PKCα activation
To determine the possible role of PKCα in regulating tight junction permeability, Beas-2B cells expressing a dominant negative PKCα (Beas-2B PKCα DN) were exposed to 0, 20, 50, and 100 mM alcohol and resistance measurements were recorded (Figure 5A). No significant changes in TER measurements were detected in Beas-2B PKCα DN. Furthermore, Beas-2B cells pretreated with a PKCα-specific inhibitor, Gö-6976, were also resistant to alcohol-induced permeability changes (Figure 5B).
Figure 5.
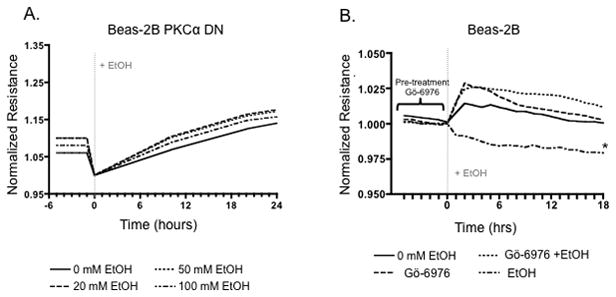
Normalized Resistance (ohms) (subsequent values divided by initial values) of Beas-2B PKCα (A) cells treated with 0, 20, 50, 100mM alcohol for 48 hrs. (B) Normalized Resistance of Beas-2B cells pretreated for 1 hr with a PKCα inhibitor, Gö-6976, and/or 100mM alcohol. Lower resistance measurements in Beas-2B cells indicate a decreased barrier function between cells. Beas-2B PKCα DN cells are resistant to alcohol-induced decreased barrier function. Similarly, wt Beas-2B cells pretreated with Gö-6976 are resistant to alcohol-induced decreased barrier function. Barrier function is significantly decreased (p<0.01) in Beas-2B cells treated with 100 mM alcohol (n=3;* P= 0.01) compared to control and Gö-6976 + EtOH treated cells.
Effect of Alcohol on ZO-1 and Claudin-1, Claudin-5 and Claudin-7 Localization
Immunofluorescence microscopy was used to determine the effect of varying concentrations of alcohol at numerous time points on the localization of ZO-1 and claudin-1 and claudin-7 in Beas-2B, Beas-2B PKCα DN, and NHBE cells. Claudin-5 localization was investigated in NHBE cells only, due to the known absence of claudin-5 in Beas-2B cells (data not shown; Merikallio et al.). No changes in protein localization occurred from 1–6 hrs in any of the alcohol concentrations (20–100 mM; data not shown). By 24 hrs, exposure to 20–100 mM alcohol reduced ZO-1, claudin-1, and claudin-7 and localization at the plasma membrane in Beas-2B (Figure 7) and NHBE cells (Figure 6). Claudin-5 was also reduced in NHBE cells. In addition, ZO-1, claudin-1, and claudin-7 appear to shift from normal membrane localization, observed in control cells, to a cytosolic localization after alcohol treatment in the NHBE cells. In contrast, no observable changes in ZO-1, claudin-1, or claudin-7 localization were detected in Beas-2B PKCα DN exposed to alcohol at any time point (Figure 9).
Figure 7.
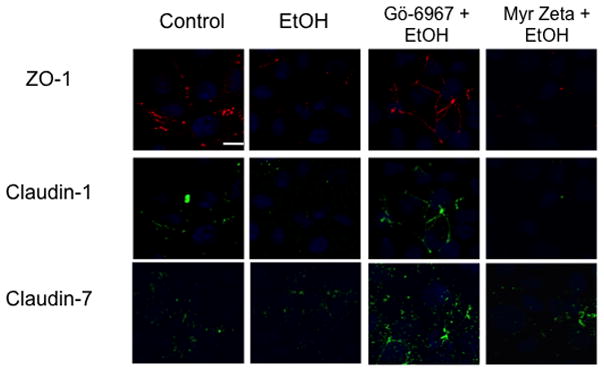
Immunofluorescence of ZO-1, claudin-1 and claudin7 in Beas-2B cells exposed to 100 mM EtOH for 24 hours ± pretreatment with PKCα inhibitor, Gö-6976 or myristolated zeta, a PKCζ inhibitor. EtOH reduced the localization of ZO-1, claudin-1 and claudin 7 at the cell membrane. Pre-treating cells with a PKCα inhibitor prevents alcohol-induced protein localization changes. Conversely, cells treated with myristolated zeta did not prevent alcohol-induced protein localization changes. Bar equals 10μm.
Figure 6.
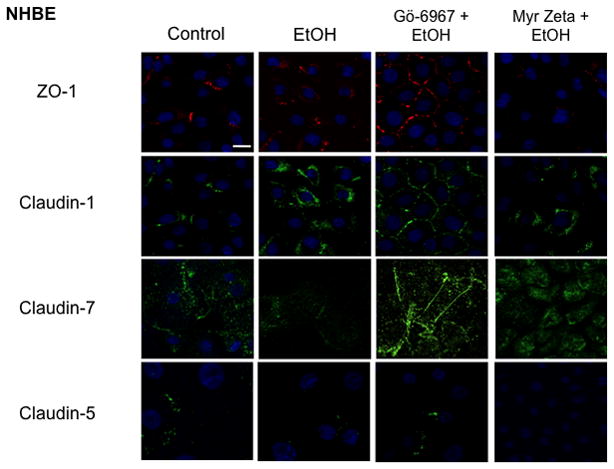
Immunofluorescence of ZO-1 (red), claudin-1, claudin-5 and claudin-7(green) in NHBE, exposed to 100mM alcohol for 24 hours ± Gö-6976, a PKCα inhibitor, or myristolated-Zeta, a PKCζ inhibitor (Note: Images are compressed Z stacks). Both ZO-1 and claudin-1 are localized to the cell membrane in control cells. Exposure to 100mM alcohol reduced ZO-1, claudin-1, claudin-5 and claudin-7 localization at the plasma membrane in alcohol-treated NHBE cells. In addition there also appears to be a shift in membrane localization of ZO-1 and claudins to a cytoplasmic localization in NHBE cells. Pretreatment with Gö-6976 prevented ZO-1, claudin-1, claudin-5, and claudin-7 alcohol-induced protein changes, while myristolated- Zeta did not. Bar equals 10μm.
Figure 9.
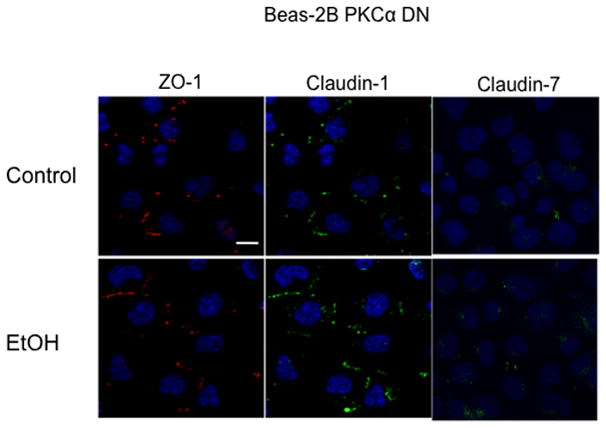
Immunofluorescence of ZO-1, claudin-1 and claudin7 in Beas-2B DN cells exposed to 100 mM EtOH for 24 hours. EtOH had no effect on the localization of ZO-1, claudin-1 and claudin 7 at the cell membrane. Bar equals 10μm.
Because PKC isoform activity assays revealed significant changes in PKCα and PKCζ activity, we sought to determine if PKCζ was also regulating alcohol-induced decreases in TER. To determine the impact of PKCα and PKCζ signaling in alcohol-impaired protein translocation, cells were pretreated with 1 μM PKCα inhibitor (Gö-6976) or 1 μM myristolated PKCζ, a cell-permeable PKCζ specific inhibitor, prior to alcohol exposure. Images revealed an alcohol-induced decrease in membrane protein localization in cells pretreated with myristolated PKCζ comparable to alcohol-only-exposed cells (Figure 8). Alternatively, ZO-1, claudin-1, and claudin-7 remained at the cell membrane in alcohol-stimulated Beas-2B and NHBE cells pretreated with Gö-6976 comparable to control cells (Figure 6 & 7). No increased membrane translocation of ZO-1, claudin-1, or claudin-7 was observed in alcohol-stimulated Beas-2B PKCα DN pretreated with Gö-6976 or myristolated PKCζ (data not shown). Quantification of membrane fluorescence determined that the significant (P<0.05) alcohol-induced translocation of ZO-1 and claudin-1 was inhibited by Gö-6976 (Figure 8).
Figure 8.
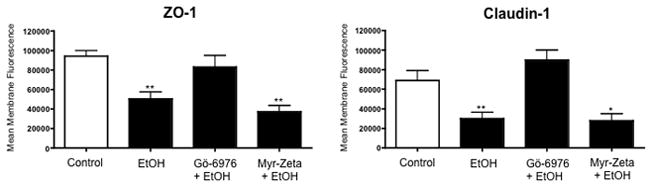
Membrane fluorescence of claudin-1 and ZO-1 in Beas-2B cells exposed to 100mM Alcohol for 24 hours and/or pretreated with Gö-6976 or myristolated zeta. ZO-1 and claudin-1 fluorescence was reduced at the plasma membrane in alcohol-treated cells compared to controls (*p < 0.05; **p < 0.01). Cells pretreated with Gö-6976, a PKCα inhibitor, are resistant to alcohol-induced protein changes. Conversely, cells pretreated with myristolated zeta, a PKCζ inhibitor, have reduced membrane fluorescence of both ZO-1 and claudin-1, comparable to alcohol alone treated cells.
Discussion
To date, studies focused on the lung alveolar compartment have shown that chronic alcohol ingestion (6 weeks) in rats increases lung epithelial permeability ~5–6 fold in vivo and promotes the flooding of alveolar spaces with proteinaceous fluid upon stress, such as sepsis (Fernandez et al., 2007). Although, the mechanisms involved in alcohol-induced alveolar leak are still not fully understood, Fernandez et al. (2007) observed decreased claudin-1 and -7 in whole lung and alveolar epithelial monolayers in alcohol-fed rats and increased claudin-5. Our observed reduction in claudin-1 and claudin-7 in our airway epithelial cells resembles those of Fernandez et al. (2007). Contrary to Fernandez’s findings of increased claudin-5, we observed a decrease in claudin-5 in the NHBE cells. Fernandez et al. were utilizing alveolar epithelial monolayers from alcohol-fed rats in their studies, while our studies involved primary human conducting airway epithelial cells treated with alcohol. This observed difference in claudin-5’s response to alcohol is likely due to differences that are often observed between methods of alcohol administration (i.e. drinking rats vs. alcohol-exposed cells), species and cell types.
While both ZO-1 and claudin proteins are important in the formation and maintenance of functional tight junctions (Fanning et al., 2007; Lal-Nag and Morin, 2009; Tsukita et al., 2009), it is normal for cells and tissues to exhibit various permeability properties through the addition or reduction of tight junctions (Mitic et al., 2000; Tsukita et al., 2001). When tissues or cells begin to lose the ability to manipulate these tight junction properties appropriately, they become more susceptible to various injuries, such as inflammation, viral and bacterial infections, and injury due to inhaled particles (Holgate et al., 2009).
Our goal was to determine the effect of alcohol on tight junction permeability in airway epithelial cells and the primary signaling mechanism involved. We initially found that alcohol increases permeability in a concentration-dependent manner in Beas-2B and NHBE cells; whereas, alcohol had no effect on permeability in Beas-2B cells expressing a PKCα DN phenotype. These results indicated that alcohol is likely acting through a PKCα-dependent mechanism. The inability of alcohol to induce changes in cells expressing a DN PKCα or wild type Beas-2B pretreated with an inhibitor of PKCα suggests that alcohol requires the presence of activatable PKCα to increase tight junction permeability. Indeed, PKC isoform assays in Beas-2B and NHBE cells revealed a significant increase in PKCα activity occurring at 1–6 hrs and 24 hrs in alcohol-treated cells. Interestingly, we did observe differences in the time points of maximal PKC activity in Beas-2B vs. NHBE cells. In addition, NHBE cells demonstrated more alcohol-induced cytoplasmic localization of ZO-1 and claudin-1 than did Beas-2B cells. These observations are likely due to inherent differences between immortalized and primary cells. However, the trend of alcohol-induced activation of PKC and redistribution of ZO-1 and claudin-1 were consistent for both these different cells. We suspect that PKCζ is an important component in tight junction assembly in airway epithelial cells. The initial decrease in PKCζ activity at 1 hour suggests that the cells are responding to an insult, thus reducing tight junction assembly promotion. Increased PKCζ activity at 3 hours is likely a compensatory mechanism in response to the insult. While our understanding of tight junction regulation in general is poorly understood, we can only speculate as to the regulation of tight junctions under altered conditions. Although we detected modest changes in PKCζ activity, we hypothesize that PKCα is the major mechanism through which alcohol is acting due to its ability to indirectly activate PKCα (Slater et al., 2003) and other studies implicating PKCα as a major component in increasing tight junction permeability (Clarke et al., 2000; Mullin et al., 1997). Our protein localization studies using PKCα and PKCζ-specific inhibitors prior to alcohol exposure support this hypothesis.
To determine if alcohol disrupts the translocation of ZO-1 and claudin-1 to the plasma membrane, cells were stained for ZO-1 and claudin-1. Fluorescence microscopy revealed a significant decrease in ZO-1 and claudin-1 at the plasma membrane in alcohol-treated cells. This alcohol-impaired membrane protein localization did not occur in cells pretreated with a PKCα inhibitor. No changes were observed in ZO-1 or claudin-1 in cells pretreated with myristolated-PKCζ prior to alcohol exposure compared to alcohol-only-treated cells (without pretreatment). Knowing that ZO-1 and claudin-1 proteins are important for the proper formation of tight junctions and permeability control, we think that the observed decrease in junction protein localization at the membrane corresponds to alcohol-induced airway “leak.”
PKCα and PKCζ have been shown in other cell types to both increase and decrease tight junction permeability (Gonzalez-Mariscal et al., 2008). While our PKC isoform-specific kinase activity data suggest that both PKCα and PKCζ can be activated by alcohol, our inhibitor experiments indicate that PKCα is the more likely mechanism through which alcohol is inducing epithelial leak. Increased epithelial permeability and PKCα activity, as well as altered ZO-1 and claudin-1 protein localization, occurred at the plasma membrane in alcohol-treated cells. Most importantly, all of these alcohol-induced changes were prevented in Beas-2B cells pre-treated with Gö-6976. Because our pharmacologic inhibition of PKCα by Gö-6976 may also result in the activation of other calcium-dependent PKC isoforms, such as PKCβ, we chose to confirm our findings using Beas-2B cells transfected to express only a dominant negative (and thus catalytically inactive) PKCα. Likewise, we observed no alcohol-induced permeability in the Beas-2B DN cells. The increase in PKCα activity, its ability to increase permeability, and the inhibition of alcohol-induced changes by PKCα inhibition all implicate this kinase as an important regulator of alcohol-induced airway leak.
In summary, our results demonstrate that alcohol increases the permeability of tight junctions in airway epithelial cells in a concentration-dependent manner, interferes with membrane localization of ZO-1, claudin-1, claudin-7 and claudin-5 proteins and increases the activities of PKCα and PKCζ. Beas-2B cells expressing a dominant negative PKCα phenotype were unaffected by alcohol, implicating this PKC isoform as the critical regulator. Furthermore, Beas-2B and NHBE cells pretreated with a PKCα inhibitor prior to alcohol exposure were also unaffected by alcohol-induced changes, while PKCζ inhibitor did not prevent alcohol-induced changes. These findings suggest that alcohol increases tight junction permeability in airway epithelial cells through a PKCα-dependent mechanism. We speculate that alcohol-induced airway permeability impairs bronchial airflow and interferes with airway host defenses, thus potentially increasing alcohol-exposed individuals susceptibility to airway infection.
Acknowledgments
Grant support: Funding: NIH 5R37AA008769-20 (JHS), 5R01AA017993-02 (TAW), VA Merit (TAW)
We thank Janice A. Taylor and James R. Talaska of the Confocal Laser Scanning Microscope Core Facility at the University of Nebraska Medical Center for providing assistance with confocal microscopy and the Nebraska Research Initiative and the Eppley Cancer Center for their support of the Core Facility. We thank Ms. Lisa Chudomelka for expert editorial assistance in preparing the manuscript.
References
- Aas K. Heterogeneity of bronchial asthma. Sub-populations--or different stages of the disease. Allergy. 1981;36(1):3–14. doi: 10.1111/j.1398-9995.1981.tb01818.x. [DOI] [PubMed] [Google Scholar]
- Asgrimsson V, Gudjonsson T, Gudmundsson GH, Baldursson O. Novel effects of azithromycin on tight junction proteins in human airway epithelia. Antimicrob Agents Chemother. 2006;50(5):1805–12. doi: 10.1128/AAC.50.5.1805-1812.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke H, Ginanni N, Laughlin KV, Smith JB, Pettit GR, Mullin JM. The transient increase of tight junction permeability induced by bryostatin 1 correlates with rapid downregulation of protein kinase C-alpha. Exp Cell Res. 2000;261(1):239–49. doi: 10.1006/excr.2000.5035. [DOI] [PubMed] [Google Scholar]
- Fahy JV, Corry DB, Boushey HA. Airway inflammation and remodeling in asthma. Curr Opin Pulm Med. 2000;6(1):15–20. doi: 10.1097/00063198-200001000-00004. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Little BP, Rahner C, Utepbergenov D, Walther Z, Anderson JM. The unique-5 and -6 motifs of ZO-1 regulate tight junction strand localization and scaffolding properties. Mol Biol Cell. 2007;18(3):721–31. doi: 10.1091/mbc.E06-08-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AL, Koval M, Fan X, Guidot DM. Chronic alcohol ingestion alters claudin expression in the alveolar epithelium of rats. Alcohol. 2007;41(5):371–9. doi: 10.1016/j.alcohol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LS, Ko YH, Lin KW, Hsu JY, Chu JJ, Chi CS. Dioscorin protects tight junction protein expression in A549 human airway epithelium cells from dust mite damage. J Microbiol Immunol Infect. 2009;42(6):457–63. [PubMed] [Google Scholar]
- Godfrey RW. Human airway epithelial tight junctions. Microsc Res Tech. 1997;38(5):488–99. doi: 10.1002/(SICI)1097-0029(19970901)38:5<488::AID-JEMT5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778(3):729–56. doi: 10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, Pickering RP. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004;74(3):223–34. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Hartmann C, Zozulya A, Wegener J, Galla HJ. The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: an in vitro study. Exp Cell Res. 2007;313(7):1318–25. doi: 10.1016/j.yexcr.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Holgate ST. Epithelium dysfunction in asthma. J Allergy Clin Immunol. 2007;120(6):1233–44. doi: 10.1016/j.jaci.2007.10.025. quiz 1245–6. [DOI] [PubMed] [Google Scholar]
- Holgate ST, Roberts G, Arshad HS, Howarth PH, Davies DE. The role of the airway epithelium and its interaction with environmental factors in asthma pathogenesis. Proc Am Thorac Soc. 2009;6(8):655–9. doi: 10.1513/pats.200907-072DP. [DOI] [PubMed] [Google Scholar]
- Jerrells TR, Pavlik JA, DeVasure J, Vidlak D, Costello A, Strachota JM, Wyatt TA. Association of chronic alcohol consumption and increased susceptibility to and pathogenic effects of pulmonary infection with respiratory syncytial virus in mice. Alcohol. 2007;41(5):357–69. doi: 10.1016/j.alcohol.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PC, Guidot DM. The alcoholic lung: epidemiology, pathophysiology, and potential therapies. Am J Physiol Lung Cell Mol Physiol. 2007;292(4):L813–23. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- Kilani MM, Mohammed KA, Nasreen N, Hardwick JA, Kaplan MH, Tepper RS, Antony VB. Respiratory syncytial virus causes increased bronchial epithelial permeability. Chest. 2004;126(1):186–91. doi: 10.1378/chest.126.1.186. [DOI] [PubMed] [Google Scholar]
- Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009;10(8):235. doi: 10.1186/gb-2009-10-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS. Medical disorders of alcoholism. N Engl J Med. 1995;333(16):1058–65. doi: 10.1056/NEJM199510193331607. [DOI] [PubMed] [Google Scholar]
- Liedtke CM, Cole T, Ikebe M. Differential activation of PKC-delta and -zeta by alpha 1-adrenergic stimulation in human airway epithelial cells. Am J Physiol. 1997;273(3 Pt 1):C937–43. doi: 10.1152/ajpcell.1997.273.3.C937. [DOI] [PubMed] [Google Scholar]
- Lo CM, Keese CR, Giaever I. Impedance analysis of MDCK cells measured by electric cell-substrate impedance sensing. Biophys J. 1995;69(6):2800–7. doi: 10.1016/S0006-3495(95)80153-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CM, Keese CR, Giaever I. Cell-substrate contact: another factor may influence transepithelial electrical resistance of cell layers cultured on permeable filters. Exp Cell Res. 1999;250(2):576–80. doi: 10.1006/excr.1999.4538. [DOI] [PubMed] [Google Scholar]
- Matter K, Aijaz S, Tsapara A, Balda MS. Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr Opin Cell Biol. 2005;17(5):453–8. doi: 10.1016/j.ceb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Merikallio H, Kaarteenaho R, Paakko P, Lehtonen S, Hirvikoski P, Makitaro R, Harju T, Soini Y. Impact of smoking on the expression of claudins in lung carcinoma. Eur J Cancer. 47(4):620–30. doi: 10.1016/j.ejca.2010.10.017. [DOI] [PubMed] [Google Scholar]
- Moss M, Burnham EL. Chronic alcohol abuse, acute respiratory distress syndrome, and multiple organ dysfunction. Crit Care Med. 2006;31(4 Suppl):S207–12. doi: 10.1097/01.CCM.0000057845.77458.25. [DOI] [PubMed] [Google Scholar]
- Mitic LL, Van Itallie CM, Anderson JM. Molecular physiology and pathophysiology of tight junctions I. Tight junction structure and function: lessons from mutant animals and proteins. Am J Physiol Gastrointest Liver Physiol. 2000;279(2):G250–4. doi: 10.1152/ajpgi.2000.279.2.G250. [DOI] [PubMed] [Google Scholar]
- Mullin JM, Kampherstein JA, Laughlin KV, Saladik DT, Soler AP. Transepithelial paracellular leakiness induced by chronic phorbol ester exposure correlates with polyp-like foci and redistribution of protein kinase C-alpha. Carcinogenesis. 1997;18(12):2339–45. doi: 10.1093/carcin/18.12.2339. [DOI] [PubMed] [Google Scholar]
- Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2009;298(3):E395–402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J, Huang F, Lum H. PKA inhibits RhoA activation: a protection mechanism against endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L972–80. doi: 10.1152/ajplung.00429.2002. [DOI] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. U.S. National Institutes of Health; Bethesda, Maryland, USA: 1997–2009. [Google Scholar]
- Samokhvalov AV, Irving HM, Rehm J. Alcohol consumption as a risk factor for pneumonia: a systematic review and meta-analysis. Epidemiol Infect. 2010:1–7. doi: 10.1017/S0950268810000774. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286(6):C1213–28. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol. 2006;22:207–35. doi: 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- Sidhaye VK, Chau E, Breysse P, King LS. Septin-2 Mediates Airway Epithelial Barrier Function in Physiologic and Pathologic Conditions. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2010-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisson JH. Alcohol and airways function in health and disease. Alcohol. 2007;41(5):293–307. doi: 10.1016/j.alcohol.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater SJ, Cook AC, Seiz JL, Malinowski SA, Stagliano BA, Stubbs CD. Effects of ethanol on protein kinase C alpha activity induced by association with Rho GTPases. Biochemistry. 2003;42(41):12105–14. doi: 10.1021/bi034860e. [DOI] [PubMed] [Google Scholar]
- Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol. 20(3):142–9. doi: 10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Stinson FS, Grant BF, Dawson DA, Ruan WJ, Huang B, Saha T. Comorbidity between DSM-IV alcohol and specific drug use disorders in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Drug Alcohol Depend. 2005;80(1):105–16. doi: 10.1016/j.drugalcdep.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Suadicani P, Hein HO, Meyer HW, Gyntelberg F. Exposure to cold and draught, alcohol consumption, and the NS-phenotype are associated with chronic bronchitis: an epidemiological investigation of 3387 men aged 53–75 years: the Copenhagen Male Study. Occup Environ Med. 2001;58(3):160–4. doi: 10.1136/oem.58.3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplin LA, Abram KM, Michaels SK. Blood alcohol level among emergency room patients: a multivariate analysis. J Stud Alcohol. 1989;50(5):441–7. doi: 10.15288/jsa.1989.50.441. [DOI] [PubMed] [Google Scholar]
- Thompson AB, Huerta G, Robbins RA, Sisson JH, Spurzem JR, von Essen S, Rickard KA, Romberger DJ, Rubinstein I, Ghafouri M, et al. The bronchitis index. A semiquantitative visual scale for the assessment of airways inflammation. Chest. 1993;103(5):1482–8. doi: 10.1378/chest.103.5.1482. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci U S A. 1992;89(17):7919–23. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Furuse M, Itoh M. Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol. 2001;2(4):285–93. doi: 10.1038/35067088. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Katsuno T, Yamazaki Y, Umeda K, Tamura A. Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann N Y Acad Sci. 2009;1165:44–52. doi: 10.1111/j.1749-6632.2009.04056.x. [DOI] [PubMed] [Google Scholar]
- Wegener J, Hakvoort A, Galla HJ. Barrier function of porcine choroid plexus epithelial cells is modulated by cAMP-dependent pathways in vitro. Brain Res. 2000;853(1):115–24. doi: 10.1016/s0006-8993(99)02317-3. [DOI] [PubMed] [Google Scholar]
- Wilson R, Dowling RB, Jackson AD. The effects of bacterial products on airway cells and their function. Am J Respir Crit Care Med. 1996;154(4 Pt 2):S197–201. doi: 10.1164/ajrccm/154.4_Pt_2.S197. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Slager RE, Devasure J, Auvermann BW, Mulhern ML, Von Essen S, Mathisen T, Floreani AA, Romberger DJ. Feedlot dust stimulation of interleukin-6 and -8 requires protein kinase Cepsilon in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293(5):L1163–70. doi: 10.1152/ajplung.00103.2007. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Slager RE, Heires AJ, Devasure JM, Vonessen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am J Respir Cell Mol Biol. 2009;42(6):706–15. doi: 10.1165/rcmb.2009-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol. 1998;275(4 Pt 1):L827–35. doi: 10.1152/ajplung.1998.275.4.L827. [DOI] [PubMed] [Google Scholar]