

Abstract
Actinobacteria such as streptomycetes are renowned for their ability to produce bioactive natural products including nonribosomal peptides (NRPs) and polyketides (PKs). The advent of genome sequencing has revealed an even larger genetic repertoire for secondary metabolism with most of the small molecule products of these gene clusters still unknown. Here, we employed a “protein-first” method called PrISM (Proteomic Investigation of Secondary Metabolism) to screen 26 unsequenced actinomycetes using mass spectrometry-based proteomics for the targeted detection of expressed nonribosomal peptide synthetases or polyketide synthases. Improvements to the original PrISM screening approach (Nature Biotechnology, 2009, 27, 951 – 956), e.g. improved de novo peptide sequencing, have enabled the discovery of ten NRPS/PKS gene clusters from six strains. Taking advantage of the concurrence of biosynthetic enzymes and the secondary metabolites they generate, two natural products were associated with their previously ‘orphan’ gene clusters. This work has demonstrated the feasibility of a proteomics-based strategy for use in screening for NRP/PK production in actinomycetes (often >8 Mbp, high GC genomes) versus the bacilli (2–4 Mbp genomes) used previously.
Keywords: Nonribosomal peptide, polyketide, natural product biosynthesis, Actinobacteria, de novo sequencing, Fourier-Transform Mass Spectrometry
Introduction
Natural products have been recognized as a major source for medicinal agents and therapeutics by both academe and industry for decades1. There is a continuing interest in discovering new natural products with novel scaffolds or clinically relevant properties, as well as in characterizing their biosynthesis for the purpose of designing new natural product derivatives2. The traditional bioassay-guided strategy for natural product discovery, where bioactive components are iteratively fractionated and concentrated from culture extracts, tends to rediscover known compounds. On the other hand, genomics based approaches that use DNA sequences as a guidance for natural product discovery, such as ‘genome mining’, have evolved to offer new natural product discovery strategies3,4. Nonetheless, much of the genetic capacity of would-be natural product producers is ‘cryptic’, since the natural products encoded by them are not produced under lab conditions. This gap between the genetic potential in the microbial world and the actual expression of the natural products remains a major barrier in drug discovery today. Recently, a proteomic-based approach, called PrISM (Proteomic Investigation of Secondary Metabolism) was developed in our laboratory5 to complement genetics, genomics, and metabolomics. PrISM initiates the process of natural product discovery from the detection of biosynthetic enzymes, and uses proteomic information to direct the discovery of expressed biosynthetic genes and their corresponding natural products.
PrISM permits targeted screening for nonribosomal peptides (NRPs) and polyketides (PKs), two of the most important classes of natural products produced by microorganisms. NRPs and PKs are synthesized by nonribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), which are often multimodular enzymes larger than 200 kDa6. In PrISM, microorganisms are screened under varying culture conditions for the expression of high molecular weight proteins (>200 kDa). This approach has proven capable of identifying a novel natural product and its biosynthetic pathway in Bacilli7.
Previous proteomic studies of secondary metabolism in microorganisms were largely performed in a whole proteomic format8, 9, until the development of two proteomic approaches for targeted profiling of NRPS/PKS: PrISM and OASIS (Orthogonal Active Site Identification System)10. PrISM and OASIS used different methods for the targeting or enrichment of PKS and NRPS: PrISM selects NRPS/PKS by their large sizes, and OASIS chemically reacts with the active sites of NRPS/PKS for affinity enrichment.
Members of the phylum Actinobacteria are high G + C content containing, Gram-positive bacteria, well-known for the wide range of natural products they produce. About 66% of all the known antibiotics and 40% of other bioactive metabolites are produced by Actinobacteria11. Recent genome sequencing projects have revealed that most of the natural product-rich Actinobacteria have large genomes (>8 Mbp) that encode for an average of ~20–30 gene clusters for secondary metabolism, about half of which are NRPS/PKS12. In the present study, a proteomics-based screening for actively expressed NRPS/PKS proteins was performed using an improved PrISM platform, including de novo peptide sequencing to increase the detection sensitivity of expressed NRPSs/PKSs within unsequenced actinomycetes. Out of 26 strains screened, we detected the expression of ten NRPS/PKS gene clusters from six strains. Starting from the NRPS/PKS peptides detected by LC-MS/MS, the corresponding biosynthetic gene clusters were identified and natural products were either detected or dereplicated. We were able to correlate two gene clusters that had no previous functional studies with their natural products. This work represents the first proteomics-based screen of Actinobacteria for NRP/PK biosynthesis.
Material and Methods
Chemicals, media and reagents
All chemicals were from Sigma unless otherwise noted. Growth media were from BD Biosciences. Oligonucleotides were purchased from Integrated DNA Technologies and listed in Supplementary Table 3. Stable-isotope labeled amino acids were from Cambridge Isotope Laboratories. Sequencing grade modified trypsin was from Promega.
Actinobacteria growth and proteome preparation
The basic workflow of the PrISM approach has been described elsewhere using Bacilli as the proof-of-concept5. In this study, Actinobacteria strains from Agricultural Research Service culture collection, United States Department of Agriculture were allowed to grow in 5 mL culture contained in a ~40 mL glass culture tube, under 250 rpm shaking. Single colonies were used to inoculate 5 mL ATCC 172 medium (10 g/L glucose, 20 g/L soluble starch, 5 g/L yeast extract, 5 g/L N-Z amine type A, 1 g/L CaCO3) and grow at 30°C for 3 days before transferring at 1:10 ratio to 4×R2A medium (2 g/L peptone, 2 g/L starch, 2 g/L glucose, 2 g/L yeast extract, 2 g/L casein hydrolysate, 1.2 g/L K2HPO4, 1.2 g/L sodium pyruvate, 0.1 g/L MgSO4) or mineral base medium13 supplemented with 10 mM sodium succinate and 0.05% Casamino acids. Cultures were harvested after 24 or 48 hours for SDS-PAGE analysis. Cells were lysed by heating in 4×SDS-PAGE loading buffer at 60°C for 60 min., followed by 0.1 mm glass bead (MO BIO Laboratories, CA) beating for 30 min. Alternatively, cells were resuspended in lysis buffer (50 mM Tris-HCl, 200 mM NaCl, pH 7.4, with protease inhibitor), sonicated on ice for 5×1 min.. The proteome was separated by a 10% T SDS-PAGE followed by staining with Coomassie Brilliant Blue G250.
Trypsin digestion and LC-MS/MS
In-gel trypsin digestion was done following a published protocol14. The entire region above 150 kDa was excised, cut into three gel slices, and separately trypsinized. The treated peptides were subsequently separated on a self-packed nano-capillary column (5µm Jupiter C18, 100 mm × 75 µm) using a nanoLC-Ultra system (Eksigent, Dublin, CA). LC solvents included 5% acetonitrile in water with 0.1% formic acid (mobile phase A) and 95% acetonitrile with 0.1% formic acid (mobile phase B). The LC gradient was set as follows: 0 min., 0% B; 55 min., 45% B; 63 min., 80% B; 67 min., 0% B with re-equilibration of 0% B until 90 min. Peptides were eluted into a nanoelectrospray ionization (nESI) source on a 7 Tesla LTQ-FT ICR mass spectrometer (Thermo Fisher Scientific). Peptides were first detected in a FT ICR cell with resolving power setting of 100,000 (at 400 m/z). Intact peptide data were collected in the 400–2000 m/z range, and MS/MS spectra were acquired using data dependent mode where the top five most abundant peaks from the FTMS full scan were selected for collision induced dissociation (CID) fragmentation followed by mass analysis of the fragment ions in the linear ion trap.
LC-MS/MS data analysis
LC-MS/MS raw data files were converted to dta files by Compass 1.0.4.415, assuming precursor charge states of 2+, 3+ and 4+. The dta files generated from the same strain were merged into a single file and searched against the non-redundant protein database NCBInr using Open Mass Spectrometry Search Algorithm (OMSSA) version 2.1.116, which uses a Poisson-based statistical model for probability-based identification as outlined by Meng et al.17 The following parameters were applied during database searching: 0.01 Da precursor mass error tolerance, peptide charge states allowed were 2+ to 4+, minimum charge to start using multiple charged products was 3+; 0.5 Da fragment mass error tolerance, maximum charge state allowed for product ions was 3+, and out of the top 6 most intense peaks, at least one must match. Variable modifications included carbamidomethylation for cysteines (+57 Da) and oxidation of methionines (+16 Da); two missed cleavage sites and two variable modification combinations were allowed. An Expectation (E-value) cutoff of 0.01 was used for initial filtering.
De novo peptide sequencing and homology–based searching
In addition to database searching using OMSSA, the same raw data files were also subjected to de novo peptide sequencing and homology searching using PEAKS Studio 5.218 (Bioinformatics Solutions Inc., ON, Canada). MS scans (with 10 ppm MS1 tolerance) were summed over 1 minute to reduce the amount of data for downstream processing. For de novo sequencing, the precursor mass tolerance was 10 ppm and the fragment mass tolerance was 0.5 Da. Variable modifications of carbamidomethylation of cysteine and oxidation of methionine were used and two combined modifications were allowed for each peptide. De novo peptide sequences were searched against a custom database by homology match. This database contains 326,109 entries built by combining all sequences of bacterial proteins, proteins containing PFAM domains associated with NRPSs and PKSs (regardless of origin), human keratins and porcine trypsin from NCBI followed by removing redundant GI identifiers. The homology-based search used the same mass error tolerance and modification settings as de novo sequencing, and assumed equivalency between leucine and isoleucine and lysine and glutamine. Peptide hits identified as homologous to NRPS/PKS were first sorted by PEAKS Spider scores and then manually examined.
PCR amplification of target region
Peptides identified as deriving from NRPS or PKS genes, from either OMSSA or PEAKS homology searches, were manually examined for sequence tags with high quality and confidence as well as low codon degeneracy. The peptide sequence tags were then reverse translated to nucleotide sequences using the Expasy Reverse Translate tool (http://www.bioinformatics.org/sms2/rev_trans.html), using the codon usage table for Streptomyces coelicolor A3(2). Primers were designed based on both the most likely codons and degenerate codons. Primers designed from reverse translation were paired either with each other or with degenerate primers for the A3 and A7 conserved regions of NRPSs or KS1 and M6 conserved regions of PKSs for PCR reactions19.
Genomic DNA of strains to be analyzed was isolated from 5 mL cultures using a DNeasy Blood and Tissue DNA kit (Qiagen). PCR reactions were performed using GoTaq® Green Master Mix (Promega) in 25 µL volume. PCR reaction annealing temperature was set to 66°C, and the elongation time depended on the expected PCR product length. Successful PCR products were sequenced using the same primer pairs.
Domain organization analysis of NRPS/PKS enzymes was performed using ‘NRPS PKS analysis’ software20. ‘NRPS predictor’21 was used for predicting the substrate specificity of NRPS adenylation domains.
Re-searching LC-MS/MS data against translated sequences of PCR products
In order to confirm the specificity of the PCR reactions, sequences of the amplicons were first analyzed using BLASTX to find the correct translation frame, and these translated sequence fragments were added into the protein database. The LC-MS/MS data were re-searched against the new database by OMSSA using the same parameters as above to confirm that the PCR amplicon corresponds to the protein detected by LC-MS/MS.
Natural product detection using LC-MS
Strains were inoculated into 5 mL of ATCC 172 medium for 3 days at 30°C before transferring to 5 mL of the desired media at 1:10 ratio and culture aliquots were taken every day for 5 days. For metabolic labeling of secondary metabolites, amino acid precursors with stable isotopes specified in text below were added to the media every day to a final concentration of 0.5 mM. One hundred µL of the 0.2 µm filtered culture supernatants were injected on to a reversed phase Gemini®-NX 5µm C18 110Å LC column 150 × 2mm (Phenomenex, Torrance, CA) and separated using a linear gradient from 0 to 100% of acetonitrile with 0.1% formic acid over 50 min. An Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) was used to monitor the effluent, detecting in positive mode and displaying m/z 400 to 1500. When necessary, the same culture supernatant was fractionated by a Gemini® 3µm C18 110Å LC column 150 × 4.6 mm using the same gradient. Fractions containing the desired products were subjected to a 7 Tesla LTQ-FT ICR for fragmentation by CID.
Shotgun genome sequencing of strain F-6133
The draft genome sequences of NRRL F-6133 was produced using paired-end sequencing with a 100 bp run on an Illumina HiSeq22 at the Roy J. Carver Biotechnology Center at the University of Illinois. The library was prepared using the Nextera DNA Sample Prep Kit and pooled with eleven other samples into one lane. The library for NRRL F-6133 was covered by 5,211,308 paired reads prior to quality trimming. Assembly was performed using a custom pipeline that combined Velvet23, SOAPdenovo24, and EULER-SR25 assemblies in gsAssembler26 using 400,000 paired-end reads reformatted for gsAssembler as additional input, followed by final scaffolding with SSPACE27. This resulted in a total of 8,207,899 bp assembled into 358 scaffolds built from 2371 contigs, with an N50 scaffold value 102,086 bp. Prodigal28 was used for ORF prediction, resulting in 8,993 ORFs.
Results and discussion
Screening for high molecular weight NRPS/PKS proteins
The PrISM workflow is initiated by screening for high molecular weight NRPS/PKS proteins as shown in Figure 1a. First, bacterial proteomes were separated by size using SDS-PAGE. The large sizes of modular NRPSs and PKSs compared to other bacterial proteins makes SDS-PAGE a convenient approach for targeted screening of expressed NRPS/PKS enzymes from a complex proteome. Since mass spectrometry is >100 times superior to Coomassie staining in terms of sensitivity of detection, the entire region above 150 kDa instead of visible protein bands was excised for in-gel trypsin digestion and LC-MS/MS analysis, in order to utilize the enhanced sensitivity of mass spectrometry.
Figure 1.

(a) The work and logic flow for PrISM used in this study. High molecular weight proteins (>150 kDa) from a bacterial proteome are subjected to in-gel trypsin digestion and LC-MS/MS analysis. The data were analyzed by both database searching (top) and de novo peptide sequencing (bottom). Peptides identified as coming from NRPSs/PKSs were reverse translated into nucleotide sequences for PCR amplification of the target region. The PCR products guided the identification of biosynthetic gene clusters as well as discovery of the natural products. (b) Decision tree for LC-MS/MS data analysis. The LC-MS/MS data were first searched against NCBInr using OMSSA, and strains with NRPS/PKS identifications were divided into two categories based on their sequence similarity to known NRPS/PKS proteins, which was represented by the number of peptides identified and sequence coverage for each protein identification. Strains with low similarity to known NRPSs/PKSs were subjected to de novo peptide sequencing and homology-based searching by PEAKS Studio. Peptides identified by either search engines were used for PCR amplification. After confirming the specificity of the PCR products, they were used to guide the identification of biosynthetic gene clusters.
Using the PrISM platform above, we screened 26 Actinobacteria strains, and peptides that were identical or homologous to known NRPSs or PKSs were confidently identified from six of these (Supplementary Table 1), while the other 20 strains did not show NRPS/PKS expression under the screening conditions. Considering that the genomic sequences of these strains were unavailable (except the one described later), we followed the decision tree shown in Figure 1b, which is dependent on the similarity of the detected proteins with known NRPS/PKS proteins in the database. The raw LC-MS/MS data was first searched against NCBInr, a highly redundant database composed of many homologs, orthologs and paralogs, which maximized the number of identifications of perfectly matching peptides from unsequenced organisms. The OMSSA search identified multiple peptides from the same NRPS/PKS gene clusters, e.g. F-6133 (PKS), F-6143, F-6134 and F-6652. Each protein identification had more than four peptides, translating to peptide maps with >2% coverage of primary sequences (Supplementary Table 2c). In other cases, peptides were identified as coming from NRPS/PKS proteins from disparate gene clusters, e.g. F-6133 (NRPS), F-6562 and F-6556, and the sequence coverage for each protein identification was less than 2% (Supplementary Table 2a). The low sequence coverage was due to the lack of genome sequences for these strains in the database, and the low sequence homology with known NRPSs/PKSs.
In order to obtain more protein sequence information from NRPSs/PKSs not well represented in the database, we performed de novo sequencing and homology-based searching using PEAKS Studio for those strains lacking confident protein identifications. Using sequence tags derived from MS/MS spectra de novo, homology-based searches against a tailored database (see Materials and Methods) identified nine and six peptides homologous to known NRPS/PKS proteins from strains F-6133 and F-6556, respectively (Supplementary Table 2b and Supplementary Figure 2a–2b).
PCR amplification based on peptide identifications
For strains expressing new gene clusters, i.e. with low homology to known NRPSs/PKSs, it is difficult to accurately predict the domain organization and function of the target proteins by analyzing the peptides alone. To get more information about the NRPS and PKS biosynthetic machinery from the peptides identified, high quality peptides were reverse translated to DNA sequences as PCR primers to amplify the coding genes for the NRPSs and PKSs (Supplementary Table 3). NRPSs and PKSs are modularly organized proteins composed of conserved domains most often including a condensation, adenylation, and thiolation domain for NRPS, and a ketosynthase, acyltransferase and acyl carrier protein for PKS (Supplementary Figure 1)6. These domains possess conserved regions (e.g. A3 and A7 for adenylation, KS1 for ketosynthase and M6 for methyl-malonyl-CoA transferase)19. Thus, each detected peptide can be mapped to the domain context and its relative position to A3/A7/KS1/M6 conserved regions can be predicted. Overall, the primers designed from peptides were either paired with each other or more often paired with primers designed based on A3/A7/KS1/M6 sequences for PCR (Supplementary Figure 1, Supplementary Table 4).
In order to confirm the PCR probe specificity, i.e., that the DNA sequence amplified by PCR corresponds to the detected protein, we re-searched the LC-MS/MS raw files using an expanded database with the translated nucleotide sequences of the PCR products added. Supplementary Table 5 shows several successful PCR reactions, the primers used for each reaction, the number of peptides detected and the closest homolog based on BLASTX results. All the PCR products exhibited <80% sequence identity to their highest-scoring homologs at the protein level. This was consistent with the low sequence coverage of proteins identified by OMSSA-based searching, and suggested the potential novelty of each biosynthetic gene cluster.
Functional analysis of expressed gene clusters
Genes encoding for biosynthesis of nonribosomal peptides and polyketides are located within a cluster on a chromosome. Figure 2 lists all NRPS and PKS gene clusters identified either by protein identifications or by their homologs based on PCR product sequences. NRPS and/or PKS gene clusters were correlated to a potential natural product structure by analyzing the NRPS/PKS domain organization and predicted substrate specificity. For example, although only three NRPS peptides were identified from strain F-6556 as coming from Thermobifida fusca YX, the PCR products showed the detected protein is most similar (50–70% sequence identity at protein level) to the gene cluster at locus Tfu_1855–1873 from Thermobifida fusca YX (Supplementary Table 5, Figure 2X). This gene cluster has been reported recently to produce the new peptide-based siderophore, fuscachelin29.
Figure 2.

NRPS/PKS gene clusters detected by PrISM and their corresponding natural products. For each gene cluster, the NRRL number of strain, the type of biosynthetic enzymes and a diagram of the genes are shown. Gene sequences were based on the most homologous gene cluster as shown. The sequence of the NRPS gene cluster identified from strain F-6133 was based on shotgun genome sequencing and partially assembled into contigs. The natural product produced by each gene cluster is shown at right.
In another case, several PCR products from strain F-6562 matched with a region in Streptomyces pristinaespiralis ATCC 25486 with ~80% identity at the protein level. It is predicted to produce a peptidic siderophore since there are several siderophore transporter genes in the gene cluster (Figure 2 VIII). Another PCR product from F-6562 matched with an NRPS from Streptomyces sp. SA3_actE with 60% sequence identity, and is highly likely to encode for enterobactin biosynthesis based on the NRPS domain organization (Figure 2IX).
Investigation of strain F-6133 led to two successful PCR products that showed moderately low sequence homology to known proteins (45% and 71% sequence identity to two different proteins), indicating a potentially uncharacterized gene cluster. To get the full sequence of the gene cluster, shotgun genome sequencing was performed on strain F-6133. By mapping the two PCR sequences to the genome sequencing result, an NRPS gene cluster was discovered containing enzymes for dihydroxybenzoate (DHB) biosynthesis and siderophore transporters, turning small molecule dereplication towards peptidic siderophores containing dihydroxybenzoate (Figure 2 II). Thorough small molecule dereplication based on these substructures is discussed below.
The PKS gene cluster detected in F-6133 has been linked to bafilomycin biosynthesis30 (Figure 2I). The NRPS detected in F-6143 is predicted for siderophore production due to the siderophore transporters in the gene cluster, though the genome sequencing gaps in that region blocked analysis of NRPS domain organization (Figure 2 III). In strain F-6134, the PKS gene cluster has been associated with filipin, a polyene natural product biosynthesis31 (Figure 2 IV), and the NRPS gene cluster is responsible for actinomycin biosynthesis32 (Figure 2 V). Lastly, two NRPS/PKS gene clusters were detected from strain F-6652, both highly similar to gene clusters from Streptomyces flavogriseous ATCC33331. However, neither of these clusters have been annotated with corresponding natural products. Based on genetic information, we predict the NRPS cluster from F-6652 encodes a yet-unknown lipohexapeptide, and the hybrid PKS-NRPS is highly similar to a group of gene clusters that produces tetramate macrolactams, such as heat-stable antifungal factor (HSAF)33 (Figure 2 VI, VII).
Natural product detection and structure determination
The DNA sequence information about NRPS and PKS gene clusters was used to deduce structural features that guided natural product detection by LC-MS. Since the active production of the natural products is believed to correlate reasonably well with expression of their biosynthetic enzymes, strains were grown in the same conditions that NRPS/PKS proteins were identified. Culture supernatants were collected at multiple time points for LC-MS analysis and several known compounds were identified, including bafilomycin B1 and C1 (1), several actinomycin derivatives (4), and two siderophores - fuscachelin (8) and enterobactin (7) (Table 1).
Table 1.
LC-MS detection of the natural products produced by expressed NRPS/PKS gene clusters. The compound structures are shown in Figure 2.
| Strain NRRL# |
Gene Cluster Type |
Proposed Product | Predicted Structure |
Mass Detected (Da) |
Theoretical Mass (Da) |
Compound Name |
|---|---|---|---|---|---|---|
| F-6133 | PKS | bafilomycin | 1 | 815.4462 720.4095 |
815.4456 720.4085 |
bafilomycin B1 bafilomycin C1 |
| NRPS | siderophore | 2 | 798.387 | 798.3872 | antibiotic S213L | |
| F-6143 | NRPS | siderophore | Unknown | NDa | NAb | NA |
| F-6134 | PKS | filipin | 3 | ND | NA | NA |
| NRPS | actinomycin | 4 | 1270.6207 1240.6116, 1282.6216 1268.6085 |
1270.6234 1240.6128 1282.6234 1268.6077 |
actinomycin XOβ/ XOδ actinomycin D0 actinomycin Pip 1δ actinomycin X2 |
|
| F-6652 | NRPS | lipopeptide | unknown | ND | NA | NA |
| PKS-NRPS | tetramate macrolactam | 5 | ND | NA | NA | |
| F-6562 | NRPS | siderophore | 6 | 575.2558 | 575.2551 | foroxymithine |
| NRPS | enterobactin | 7 | 669.1475 | 669.1442 | enterobactin | |
| F-6556 | NRPS | fuscachelin | 8 | 1047.4368 | 1047.437 | fuscachelin B |
ND: not detected;
NA: not available
After dereplicating known natural products and their biosynthetic enzymes, we focused on the gene clusters that have no known natural products reported (so-called ‘orphan’ clusters). The gene cluster NRPS1 from strain F-6562 is predicted to produce a peptidic siderophore. NRPS domain architecture suggested the final product is likely to incorporate at least two ornithines and a serine (Figure 3a). From the LC-MS chromatogram of the culture supernatant, we detected a peak with m/z 576.2631 ([M+H]+, 575.2558 Da) coeluting with its iron bound form (m/z 629.1726, [M-2H++Fe]+, Figure 3b). The mass is within 2 ppm of a known peptidic siderophore foroxymithine (6, cal. 575.2551 Da). CID MS/MS on m/z 576.3 gave the correct fragmentation pattern predicted for foroxymithine, and feeding the strain with 13C6-labeled arginine confirmed the incorporation of three ornithine residues (Figure 3c–d). Although the stereochemistry of the product still needs to be investigated by NMR and Marfey’s method, the MS1 and MS2 as well as isotopic labeling results gave enough information to support the structure of foroxymithine. Foroxymithine was found in 1985 as an angiotensin-converting enzyme inhibitor34, and its biosynthetic pathway has yet to be elucidated. The NRPS gene cluster identified from strain F-6562 is entirely consistent with the biosynthesis of foroxymithine based on the domain organization of the NRPS protein (Figure 3e). A compound with a similar structure to foroxymithine, erythrochelin, has recently been reported with biosynthetic analysis using a ‘genome mining’ strategy35. The difference between the two compounds is that two δ-N-formyl groups in 6 are replaced by δ-N-acetyl groups in erythrochelin. We predict the structural difference is coming from the difference in these biosynthetic gene clusters: while the δ-N-formyl group in foroxymithine is likely formed by a formyltransferase (orf2) in the gene cluster, the δ-N-acetyl group in erythrochelin has been shown to be condensed by a remote acetyltransferase acting in trans35.
Figure 3. Association of foroxymithine with its biosynthetic pathway.
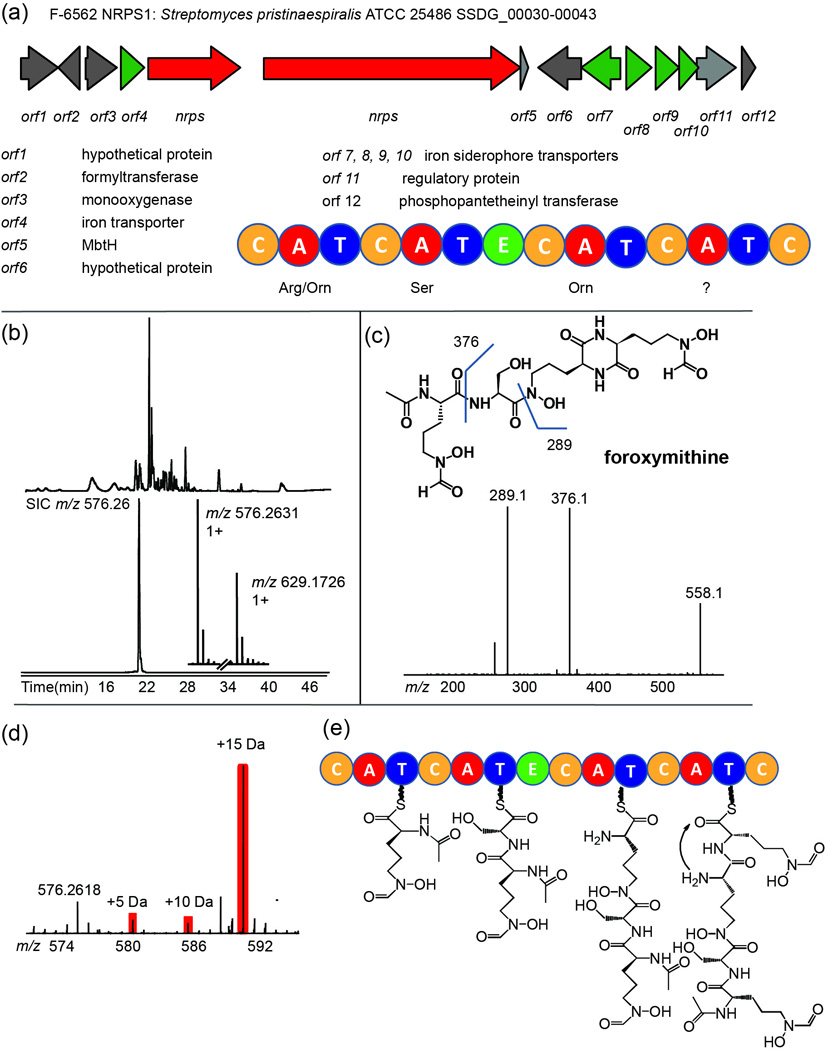
(a) The foroxymithine gene cluster identified from strain F-6562, using the DNA sequence from Streptomyces pristinaespiralis ATCC 25486 as a template. Domain organization and substrate specificity of adenylation domains of the NRPS were predicted by bioinformatics. C, condensation domain; A, adenylation domain; T, thiolation domain; E, epimerization domain. (b) LC-MS Base peak chromatogram (top) of F-6562 culture supernatant produced by cells grown in 4×R2A medium for 4 days and selected ion chromatogram (SIC, bottom) of m/z 576.2631; inset, mass spectrum of species at m/z 576.2631 (iron free form) and 629.1726 (iron bound form). (c) Structure of foroxymithine with predicted fragment masses (top). The CID fragmentation spectrum on m/z 576.26 is shown at bottom. (d) 13C6-arginine feeding of strain F-6562 shows three ornithines are incorporated into the species at m/z 576.2618 as evidenced by a mass shift of 15 Da. Arginine can be converted to ornithine in vivo by losing the terminal -C(NH)NH2 group. (e) Predicted biosynthetic mechanism for foroxymithine based on the NRPS domain organization.
As discussed above, the NRPS gene cluster from strain F-6133 does not have a closely related homolog in the database. An NRPS gene cluster discovered by genomic sequencing suggests it is likely producing a peptidic siderophore that contains dihydroxybenzoate (DHB). Bioinformatic analysis of the NRPS indicated that it is likely to incorporate an ornithine residue (Figure 4a). From its culture supernatant, we were able to detect a peak with m/z 799.3942 ([M+H]+, 798.3869 Da, Figure 4b), together with its iron bound form (m/z 852.3055, [M-2H++Fe]+). The mass is within 1 ppm of a patented compound called antibiotic S 213L (2, cal. 798.3872 Da). Fragment ion patterns from MS/MS of m/z 799.4 also agreed with the expected pattern for 2 (Figure 4c). Antibiotic S 213L is reported to have antifungal activity36, and using PrISM platform, we have discovered its biosynthetic gene cluster. Further genomic and biochemical work is ongoing to elucidate the full biosynthesis of the natural product.
Figure 4. Discovery of the biosynthetic gene cluster for antibiotic S 213L in strain F-6133.

(a) The partial gene cluster determined by shotgun genome sequencing of strain F-6133. orf 12 NRPS contains peptides identified by LC-MS/MS and two PCR products from F-6133. Gene annotations were based on BLAST analysis. (b) SIC for m/z 799.39 detected in a culture supernatant from strain F-6133. The mass spectrum of the SIC peak is shown as an inset. Two masses separated by 52.9 Da (mass difference of Fe and 3 × H+) were shown, suggesting iron free and iron bound forms of the same species. (c) Structure of antibiotic S 213L. The labeled fragment ions were observed by tandem MS of the species at m/z 799.39.
Conclusion
Actinomycetes are well known for their biosynthesis of diverse natural products, including a great number of nonribosomal peptides and polyketides. In this study, we extended the recently developed PrISM platform to actinomycetes, screening for endogenous NRPS/PKS enzymes by proteomics using mass spectrometry. By surveying only 26 actinomycetes, we identified six strains harboring NRPS/PKS proteins, several with multiple gene clusters expressed at high levels. Using protein identifications as well as PCR amplification of the target region, a total of ten NRPS/PKS gene clusters were found, and the natural products they produced were accordingly predicted. Analysis of the culture supernatant confirmed four NRPs and PKs that have been reported before, and discovered natural products for two gene clusters where no previous reports were available.
As one of the recently developed proteomic approaches for targeted profiling of NRPS/PKS, PrISM has proven capable of identifying both known and novel natural products5, 7. In this study, we implemented technical improvements, such as improved database search protocols and bypassing the need for protein staining on gels. The limit of detection for Coomassie staining is ~100 ng (equivalent to 0.5 pmol for a 200 kDa protein), whereas mass spectrometry is able to detect <50 fmol of in-gel digested proteins. This translates to an ability to detect a protein copy number of ~30/cell if the equivalent of ~109 cells are loaded on gel for analysis. We anticipate detection of lower abundant biosynthetic proteins would lead eventually to the discovery of novel and more potent bioactive compounds.
The goal of PrISM as a proteomics based approach is multi-faceted: to find new natural products, new biosynthetic pathways, new biosynthetic transformations, and to associate gene clusters with the metabolites they produce. With the current state of art, we anticipate PrISM can be extended to larger scale screening of environmental strains (up to hundreds to thousands of strains) for the discovery of potentially new biosynthetic pathways and natural products. PrISM is also valuable for screening different culture conditions for teasing sequenced organisms into expressing their cryptic gene clusters. Most importantly, PrISM dereplicates the well-characterized systems early at the protein sequence level, allowing more effort to be focused uncharacterized systems.
Future efforts should continue to enhance the sensitivity of detection and to improve the overall efficiency of detecting uncharacterized gene clusters. We anticipate that with improved instrumentation (e.g., using electron-based methods for tandem mass spectrometry), longer peptide sequence stretches with better accuracy will facilitate PrISM-based discovery. With such technical advancements, this protein-first strategy will gain a more visible role in microbial proteomics, allowing larger scale screening and extension to other types of metabolism. For secondary metabolites, it should also be applicable to fungi and mixed microorganism communities, perhaps even without the need for culturing to enable a new field of ‘metaproteomics’ to complement metagenomics.
Supplementary Material
Acknowledgment
We thank the Agricultural Research Service, United States Department of Agriculture for providing us the actinobacteria strains. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer. This work was supported in part by the National Institute of Health (NIH) grants (GM 067725 and GM 077596). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS or NIH. L.Z. was also supported by NIH-NIDA grant (F30 DA026672) and NSF grant (DMS 080063).
Footnotes
Associated content
Supplementary Table 1 lists the taxonomy of the six actinobacteria strains that yielded NRPS/PKS identifications, along with their growing conditions. Supplementary Table 2 lists all the NRPS/PKS peptide or protein identifications from the six actinobacteria strains. Supplementary Table 3 lists the primers for PCR amplification of target region for strains F-6133, F-6562 and F-6556. Supplementary Table 4 lists the PCR reactions completed for analysis of the genomes of strains F-6133, F-6562 and F-6556. Supplementary Table 5 lists the PCR amplicons from strains F-6133, F-6562 and F-6556 whose translated nucleotides were identified by OMSSA search. Supplementary Figure 1 shows a scheme for PCR amplification of target NRPS/PKS genes using reverse translated peptide sequences. Supplementary Figure 2 shows all MS/MS spectra of peptides identified by PEAKS Studio.
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 2.Wilkinson B, Micklefield J. Mining and engineering natural-product biosynthetic pathways. Nat Chem Biol. 2007;3:379–386. doi: 10.1038/nchembio.2007.7. [DOI] [PubMed] [Google Scholar]
- 3.Lautru S, Deeth RJ, Bailey LM, Challis GL. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol. 2005;1:265–269. doi: 10.1038/nchembio731. [DOI] [PubMed] [Google Scholar]
- 4.Van Lanen SG, Shen B. Microbial genomics for the improvement of natural product discovery. Curr Opin Microbiol. 2006;9:252–260. doi: 10.1016/j.mib.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Bumpus SB, Evans BS, Thomas PM, Ntai I, Kelleher NL. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27:951–956. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 7.Evans BS, Ntai I, Chen Y, Robinson SJ, Kelleher NL. Proteomics-based discovery of koranimine, a cyclic imine natural product. J Am Chem Soc. 2011;133:7316–7319. doi: 10.1021/ja2015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schley C, Altmeyer MO, Swart R, Muller R, Huber CG. Proteome analysis of Myxococcus xanthus by off-line two-dimensional chromatographic separation using monolithic poly-(styrene-divinylbenzene) columns combined with ion-trap tandem mass spectrometry. J Proteome Res. 2006;5:2760–2768. doi: 10.1021/pr0602489. [DOI] [PubMed] [Google Scholar]
- 9.Udwary DW, Gontang EA, Jones AC, Jones CS, Schultz AW, Winter JM, Yang JY, Beauchemin N, Capson TL, Clark BR, Esquenazi E, Eustaquio AS, Freel K, Gerwick L, Gerwick WH, Gonzalez D, Liu WT, Malloy KL, Maloney KN, Nett M, Nunnery JK, Penn K, Prieto-Davo A, Simmons TL, Weitz S, Wilson MC, Tisa LS, Dorrestein PC, Moore BS. Significant natural product biosynthetic potential of actinorhizal symbionts of the genus frankia, as revealed by comparative genomic and proteomic analyses. Appl Environ Microbiol. 2011;77:3617–3625. doi: 10.1128/AEM.00038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem Biol. 2009;4:948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical streptomyces genetics. Norwich: John Innes Foundation; 2000. [Google Scholar]
- 12.Nett M, Ikeda H, Moore BS. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stanier RY, Palleroni NJ, Doudoroff M. The aerobic pseudomonads: a taxonomic study. J Gen Microbiol. 1966;43:159–271. doi: 10.1099/00221287-43-2-159. [DOI] [PubMed] [Google Scholar]
- 14.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 15.Wenger CD, Phanstiel DH, Lee MV, Bailey DJ, Coon JJ. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 17.Meng F, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 18.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 19.Ayuso-Sacido A, Genilloud O. New PCR primers for the screening of NRPS and PKS-I systems in actinomycetes: detection and distribution of these biosynthetic gene sequences in major taxonomic groups. Microb Ecol. 2005;49:10–24. doi: 10.1007/s00248-004-0249-6. [DOI] [PubMed] [Google Scholar]
- 20.Bachmann BO, Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 21.Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentley DR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Yang H, Wang J. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–272. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaisson MJ, Brinza D, Pevzner PA. De novo fragment assembly with short mate-paired reads: Does the read length matter? Genome Res. 2009;19:336–346. doi: 10.1101/gr.079053.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 28.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dimise EJ, Widboom PF, Bruner SD. Structure elucidation and biosynthesis of fuscachelins, peptide siderophores from the moderate thermophile Thermobifida fusca. Proc Natl Acad Sci U S A. 2008;105:15311–15316. doi: 10.1073/pnas.0805451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ichikawa N, Oguchi A, Ikeda H, Ishikawa J, Kitani S, Watanabe Y, Nakamura S, Katano Y, Kishi E, Sasagawa M, Ankai A, Fukui S, Hashimoto Y, Kamata S, Otoguro M, Tanikawa S, Nihira T, Horinouchi S, Ohnishi Y, Hayakawa M, Kuzuyama T, Arisawa A, Nomoto F, Miura H, Takahashi Y, Fujita N. Genome sequence of Kitasatospora setae NBRC 14216T: an evolutionary snapshot of the family Streptomycetaceae. DNA Res. 2010;17:393–406. doi: 10.1093/dnares/dsq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamb DC, Ikeda H, Nelson DR, Ishikawa J, Skaug T, Jackson C, Omura S, Waterman MR, Kelly SL. Cytochrome p450 complement (CYPome) of the avermectin-producer Streptomyces avermitilis and comparison to that of Streptomyces coelicolor A3(2) Biochem Biophys Res Commun. 2003;307:610–619. doi: 10.1016/s0006-291x(03)01231-2. [DOI] [PubMed] [Google Scholar]
- 32.Pfennig F, Schauwecker F, Keller U. Molecular characterization of the genes of actinomycin synthetase I and of a 4-methyl-3-hydroxyanthranilic acid carrier protein involved in the assembly of the acylpeptide chain of actinomycin in Streptomyces. J Biol Chem. 1999;274:12508–12516. doi: 10.1074/jbc.274.18.12508. [DOI] [PubMed] [Google Scholar]
- 33.Blodgett JA, Oh DC, Cao S, Currie CR, Kolter R, Clardy J. Common biosynthetic origins for polycyclic tetramate macrolactams from phylogenetically diverse bacteria. Proc Natl Acad Sci U S A. 2010;107:11692–11697. doi: 10.1073/pnas.1001513107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Umezawa H, Aoyagi T, Ogawa K, Obata T, Iinuma H, Naganawa H, Hamada M, Takeuchi T. Foroxymithine, a new inhibitor of angiotensin-converting enzyme, produced by actinomycetes. J Antibiot (Tokyo) 1985;38:1813–1815. doi: 10.7164/antibiotics.38.1813. [DOI] [PubMed] [Google Scholar]
- 35.Lazos O, Tosin M, Slusarczyk AL, Boakes S, Cortes J, Sidebottom PJ, Leadlay PF. Biosynthesis of the putative siderophore erythrochelin requires unprecedented crosstalk between separate nonribosomal peptide gene clusters. Chem Biol. 2010;17:160–173. doi: 10.1016/j.chembiol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Owaku M, Majima T, Matsugami M, Goto M, Nakajima T, Ito T, Namako K, Nozawa A, Miki T. In Patent Abstracts of Japan. Japan: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.