
Figure 2.
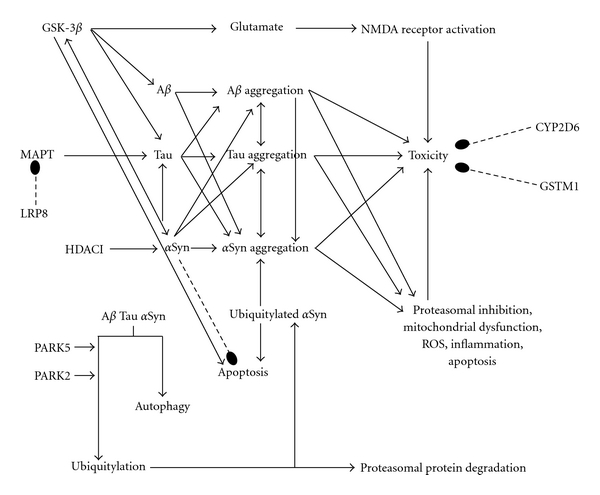
Interactions of neuroprotective and neurodegenerative pathways emphasizing pathogenic proteins and toxins. Relations terminating in an arrowhead indicate facilitation, those with double arrowheads indicate mutual facilitation, whereas dashed lines terminating in a bulb indicate inhibition. The enzyme glycogen synthase kinase 3 beta (GSK-3β) activates glutamatergic excitotoxicity mediated through the N-methyl-D-aspartate (NMDA) receptor. GSK-3β also drives production of alpha-synuclein (αSyn), the pathogenic proteins beta-amyloid (Aβ) and tau, and apoptosis. Whereas αSyn appears to be neuroprotective and inhibits apoptosis, mono-ubiquitylated αSyn promotes αSyn aggregation and apoptosis. On the other hand, αSyn can also increase GSK-3β and tau concentrations, in turn increasing aggregated αSyn, Aβ, and tau itself. Tau can further increase concentrations of αSyn. Aggregated αSyn, Aβ, and tau inhibit the proteasome and induce cellular toxicity, reactive oxygen species (ROS), mitochondrial dysfunction, apoptosis, and inflammation, leading to neuronal demise. The three proteins promote the formation of each other, as do their aggregated forms. The LRP8 gene product stabilizes microtubule associated protein tau (MAPT), the gene that produces tau protein, and dysfunctional LRP8 leads to excessive MAPT expression, increasing tau and driving pathogenic protein aggregation. Pathogenic proteins are disposed of through autophagy and the ubiquitin-proteasomal system, wherein proteins targeted for destruction are polyubiquitylated, a process that appears to be regulated by PARK5 (UCHL1) and PARK2 (parkin). Interference with autophagy or ubiquitylation prevents disposal of proteins, leading to their accumulation and their subsequent inhibition of the proteasome. GSTM1 and CYP2D6 gene products promote solvent detoxification, and deficiencies in these proteins permit toxicity. GSTM1 is particularly important in the context of CYP2D6 dysfunction.