

Abstract
Cutinase, which exists in both fungi and bacteria, catalyzes the cleavage of the ester bonds of cutin. Fungal cutinases have been extensively studied, however, reports on bacterial cutinases have been limited due to the lack of knowledge concerning the identity of their open reading frames. In the present study, the cutinase from Thermobifida fusca was induced by cutin and purified to homogeneity by following p-nitrophenyl butyrate hydrolyzing activity. Peptide mass fingerprinting analysis of the wild-type enzyme matched two proteins, Tfu_0883 and Tfu_0882, which are 93% identical in sequence. Both proteins were cloned and overexpressed in their mature form. Recombinant Tfu_0883 and Tfu_0882 display very similar enzymatic properties and were confirmed to be cutinases by their capability to hydrolyze the ester bonds of cutin. Comparative characterization of Fusarium solani pisi and T. fusca cutinases indicated that they have similar substrate specificity and catalytic properties except that the T. fusca enzymes are thermally more stable. Homology modeling revealed that T. fusca cutinases adopt an α/β-hydrolase fold that exhibits both similarities and variations from the fungal cutinase structure. A serine hydrolase catalytic mechanism involving a Ser170-His248-Asp216 (Tfu_0883 numbering) catalytic triad was supported by active site-directed inhibition studies and mutational analyses. This is the first report of cutinase encoding genes from bacterial sources.
Cutin is a major component of the plant cuticle. It is an insoluble lipid-polyester formed primarily from C16 and C18 hydroxy and epoxy fatty acids (Fig. 1) (1–3). Although the precise composition of cutin varies among species, all cutins contain hydroxy fatty acids as the base units (4).
FIGURE 1.

Structure of cutin.
Cutinases are inducible extracellular enzymes secreted by microorganisms that are capable of degrading plant cell walls. They catalyze the cleavage of ester bonds of cutin, resulting in the release of cutin monomers (5–8). Cutinases have been found in both fungi and bacteria; however, studies have focused on fungal cutinases, including Fusarium solani pisi (9), Monilinia fructicola (7, 10), Botrytis cinerea (11), and Aspergillus oryzae (12). Cutinase from F. solani pisi, for example, has been extensively investigated by biochemical and structural approaches (13, 14). X-ray crystallography studies of F. solani pisi cutinase revealed that it belongs to the α/β-hydrolase fold superfamily, and contains the characteristic GXSXG motif as well as a Ser-His-Asp catalytic triad (9). Interestingly, fungal cutinases hydrolyze not only cutin, but also insoluble triglycerides and soluble esters such as p-nitrophenyl butyrate (pNPB).3 Thus, fungal cutinases are considered as being intermediate between lipases and esterases (8, 9, 14). Recently, it was found that F. solani pisi cutinase can hydrolyze and thus improve the surface properties of synthetic fibers such as polyethyleneterephthalate fibers in an environmentally friendly way (15). Such versatile hydrolytic activities of cutinases suggest that they may have promising applications in chemical, textile, and other industries (16–18).
In contrast to fungal cutinases, little is known about cutinases from bacterial sources. There are a few reports of bacterial cutinases from Streptomyces scabies (19), Thermobifida fusca (20), and Pseudomonas putida (6, 21); however, these studies were limited to screening of enzyme-producing strains and initial characterization on crude enzyme preparations. Most critically, no cutinase open reading frame has been identified in bacteria, thus it is impossible to obtain sufficient amounts of pure recombinant enzymes for in depth studies.
In the present study, for the first time, the gene responsible for the expression of bacterial cutinase was identified. Two cutin-induced wild-type pNPB hydrolases were isolated from T. fusca. Peptide mass fingerprinting and data base search of these hydrolases revealed that they are encoded by open reading frames, Tfu_0882 and Tfu_0883, respectively. The two enzymes are 93% identical in amino acid sequence. The mature forms of both enzymes were cloned, expressed, and purified to homogeneity, and were confirmed to function as cutinase. The biochemical properties of T. fusca cutinases were investigated and compared with their F. solani pisi counterpart. Molecular modeling, inhibition studies, and mutational analysis shed light on the mechanism and key residues in catalysis.
EXPERIMENTAL PROCEDURES
Materials—A Bacillus subtilis strain harboring the plasmid pBSMuL3 containing the F. solani pisi cutinase was kindly provided by Dr. Thorsten Eggert. The EZ-10 Spin Column Plasmid Mini-Preps kit, agarose gel DNA purification kit, restriction enzymes, and T4 DNA ligase were obtained from TakaRa Biotechnology Co. Ltd. The plasmids pMD18T-simple and pET20b(+) were obtained from Novagen. Sodium taurodeoxycholate, phenylmethylsulfonyl fluoride (PMSF), bis-(trimethylsilyl)trifluoroacetamide, pNPB, and Pseudomonas sp. MIS38 lipase (PsL) were obtained from Sigma. Other chemicals were obtained from Sinopharm Chemical Reagent Co. Ltd. Phenyl-Sepharose FF and DEAE-Sepharose FF resins were obtained from Amersham Biosciences. Cellulase and pectinase were obtained from Wuxi Boli Biotechnologies Co. Ltd. DNA primers were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. DNA sequencing was performed by Shanghai Generay Biotechnology Co. Ltd.
Culture of T. fusca—The T. fusca strain is from laboratory stock. The cells were grown at 50 °C with shaking at 200 rpm. The seed medium (pH 8.0) contained 20 g/liter soluble starch, 10 g/liter beef extract, 5 g/liter yeast extract, 2 g/liter K2HPO4, and 5 g/liter NaCl. The fermentation medium (pH 8.0) contained 7.5 g/liter sodium acetate, 5 g/liter peptone, 7.5 g/liter yeast extract, 2 g/liter K2HPO4, 5 g/liter NaCl, and 1 g/liter cutin.
Isolation and Purification of a Cutin-induced pNPB Hydrolase from T. fusca—Cells were removed from the cutin-induced culture by centrifugation (40,000 × g, 20 min, 4 °C). Solid (NH4)2SO4 was slowly added to the supernatant to a final concentration of 70% (w/v). The precipitated protein was collected by centrifugation (40,000 × g, 30 min, 4 °C), immediately dissolved in buffer A (20 mm Tris-HCl, pH 8.0), and dialyzed against 2 liters of buffer A at 4 °C overnight. Solid (NH4)2SO4 was added to the dialyzed sample to a final concentration of 20% (w/v). The sample was filtered (0.22 μm) and loaded onto a phenyl HP-Sepharose FF column pre-equilibrated with 20% (NH4)2SO4 in buffer A. A reverse gradient from 20 to 0% (NH4)2SO4 in buffer A was applied at a flow rate of 1.0 ml/min over a 60-min period. The fractions containing pNPB hydrolase activity, which was assayed as described below, were pooled and dialyzed against 1 liter of buffer A overnight. The dialyzed sample was applied to a DEAE-Sepharose FF column pre-equilibrated with buffer A. The column was eluted at a flow rate of 0.8 ml/min using a linear gradient of 0 to 1 m NaCl in buffer A over 100 min. The fractions containing pNPB hydrolase activity were pooled and dialyzed against 1 liter of buffer A overnight. The final preparation was concentrated by ultrafiltration (Centriprep YM-10 concentrator) to 1 mg/ml, and stored at –80 °C.
Peptide Mass Fingerprinting Analysis and N-terminal Amino Acid Sequencing—The protein band corresponding to the cutin-induced pNPB hydrolase was excised from the SDS-PAGE gel and subjected to peptide mass fingerprinting analysis and N-terminal amino acid sequencing. Peptide mass finger-printing analysis was performed by the Proteomics solution I system and the N-terminal residues were sequenced with Edman method by Applied Biosystems model 492cLC protein sequencer. Both analyses were performed by Shanghai Gene Core BioTechnologies Co. Ltd.
Cloning, Expression, and Purification of Tfu_0882 and Tfu_0883—The genes encoding the mature forms of Tfu_0882 (NCBI accession number YP_288943) and Tfu_0883 (NCBI accession number YP_288944) were amplified from T. fusca genomic DNA by standard polymerase chain reaction methodologies using Taq DNA polymerase. Because the N- and C-terminal nucleotide sequences of the two genes are identical, the same primers were utilized. The forward primer was GGAATACCATATGTCCATGGCCAACCCCTACGAGCGCGG (NcoI underlined), and the reverse primer was CATCTCGAGAGAATTCGGGAACGGGCAGGTGGAGCG (EcoRI underlined). The amplification product was isolated and ligated into the vector pMD18T-simple. The ligation mixture was used to transform chemically competent Escherichia coli JM109 cells. The plasmid isolated from these transformants was verified by restriction analysis and the gene sequence was confirmed by DNA sequencing. The plasmids with the correct sequences of Tfu_0882 and Tfu_0883 were named Tfu_0882-pMD18T and Tfu_0883-pMD18T, respectively. Because both genes contain one internal NcoI site, site-directed mutagenesis was performed to eliminate these sites while keeping the encoded amino acid unchanged. The resulting plasmids were digested with NcoI and EcoRI and ligated into the similarly restriction-digested expression vector pET20b(+). The ligation mixture was used to transform chemically competent E. coli JM109 cells. The plasmid isolated from these transformants was verified by restriction analysis and the gene sequence was confirmed by DNA sequencing. The plasmid with the correct sequence, pET20b-Tfu_0882 or pET20b-Tfu_0883, was used to transform chemically competent E. coli BL21 Rossetta (DE3) PlysS cells.
The E. coli Rossetta (DE3) pLysS cells harboring pET20b-Tfu_0883 were grown in TB medium containing ampicillin (100 mg/liter) and chloramphenicol (50 mg/liter) at 37 °C with shaking at 200 rpm. Isopropyl β-d-thiogalactoside was added to a final concentration of 0.4 mm when the culture reached an A600 of 1.5. The culture supernatant was collected by centrifugation (40,000 × g, 20 min, 4 °C) 16 h after induction.
The culture supernatant was concentrated and dialyzed against 2 liters of buffer B (20 mm sodium phosphate, 0.5 m NaCl, 20 mm imidazole, pH 7.4) overnight and applied to a nickel affinity column pre-equilibrated with buffer B. The column was eluted at a flow rate of 1.0 ml/min using a linear gradient from 0 to 480 mm imidazole in buffer B over 100 min. The fractions containing pNPB hydrolase activity were pooled and dialyzed against 2 liters of buffer C (20 mm Tris-HCl, pH 8.0) at 4 °C overnight. The purified enzyme was aliquoted, and stored at –80 °C. The expression and purification of recombinant Tfu_0882 were performed using the same procedures as described above for Tfu_0883.
Purification of Recombinant F. solani pisi Cutinase—The B. subtilis harboring the plasmid pBSMuL3 expressing F. solani pisi cutinase was grown in TB medium containing kanamycin (50 mg/liter) at 37 °C with shaking at 200 rpm overnight. The culture supernatant was collected by centrifugation (40,000 × g, 20 min, 4 °C). Solid (NH4)2SO4 was added to the culture supernatant to a final concentration of 70% (w/v). The precipitated proteins were collected by centrifugation (40,000 × g,30 min, 4 °C), dissolved in buffer D (20 mm Tris-HCl, pH 7.5), and dialyzed against 2 liters of buffer D at 4 °C overnight. Solid (NH4)2SO4 was added to the dialyzed sample to a final concentration of 20% (w/v). The sample was filtered (0.22 μm) and loaded onto a phenyl HP-Sepharose FF column pre-equilibrated with 20% (NH4)2SO4 in buffer D. A reverse gradient from 20 to 0% (NH4)2SO4 in buffer D was applied at a flow rate of 1.0 ml/min over a 60-min period. The fractions containing pNPB hydrolase activity, which was assayed as described below, were pooled, dialyzed against 1 liter of buffer E (20 mm Tris-HCl, pH 7.0) overnight, and loaded onto a CM-Sepharose column pre-equilibrated with buffer E. The column was eluted at a flow rate of 0.5 ml/min using a linear gradient of 0 to 1 m NaCl in buffer E over 30 min. The fractions containing pNPB hydrolase activity were pooled and dialyzed against 500 ml of buffer E at 4 °C overnight. The purified enzyme was aliquoted, flash frozen, and stored at –80 °C.
Enzyme Assays—Esterase activity was determined by a continuous spectrophotometric assay using pNPB as the substrate (22). One unit of enzyme activity is defined as the production of 1 μmol of p-nitrophenol per minute. The standard assay was measured in a final volume of 1 ml containing pNPB (1 mm), the enzyme, and the assay buffer (20 mm Tris-HCl, 10 mm NaCl, and 50 mm sodium taurodeoxycholate, pH 8.0) at 20 °C. The reaction was initiated by the addition of pNPB. The hydrolysis of pNPB was spectrophotometrically monitored for the formation of the p-nitrophenol at 405 nm.
Cutinase activity was determined as previously reported with the following modification (20). Apple cutin, prepared from mature apples as described previously (2), served as the substrate. For a typical assay, a certain amount of enzyme and 100 mg of apple cutin were added into a 25 mm potassium phosphate buffer (pH 8.0) in a final volume of 10 ml. The tube was shaken (125 rpm) for 18 h in a water bath preset at the desired temperatures. At the end of reaction, the remaining cutin was removed by centrifugation. The resulting supernatant was acidified with acetic acid and the released cutin monomers were extracted with chloroform. The organic soluble material was collected and dried under a stream of nitrogen. The dried cutin monomers were converted to their corresponding methyl esters and then silylated with bis-(trimethylsilyl)trifluoroacetamide (23). The silylated methyl esters were dissolved in hexane and analyzed by Finnigan GC/MS 4610B on a 122-7032 DBWAX 30M capillary column with temperature programmed as following: 125 °C for 5 min, 4 °C/min to 250 °C, and 250 °C for 15 min.
Lipase activity was measured as previously reported (24) with the following modification. Triolein served as the substrate. The reaction solution contained 2.5 ml of 25 mm potassium phosphate buffer (pH 8) and 2 ml of emulsified triolein (3% PVA:triolein = 3:1). The reaction was initiated by adding the enzyme to the reaction solution and quenched by adding 7.5 ml of ethanol. The released fatty acids were quantified via titration by 0.05 n NaOH. One unit of lipase activity was defined as the release of 1 μmol of fatty acid per min.
Temperature and pH Dependence of Cutinase—The temperature dependence of enzyme activity was determined between 20 and 70 °C. For esterase activity, the reaction was performed in a buffer containing 20 mm Tris-HCl, 10 mm NaCl, and 50 mm sodium taurodeoxycholate at pH 8.0 using pNPB as the substrate. Because the pH of Tris buffer is temperature dependent, the buffers were adjusted to pH 8.0 at the desired temperatures. The enzyme activity was measured by preincubating the buffer at a desired temperature for 2 min to allow it to reach the final pH of 8.0. For lipase activity, the reaction was performed in 25 mm potassium phosphate (pH 8.0) using triolein as the substrate. The pH dependence of enzyme activity was determined between pH 6.0 and 9.0 using either 25 mm potassium phosphate buffer (pH 6.0–7.0) or 20 mm Tris-HCl buffer (pH 7.0–9.0).
Thermostability of Cutinase—The thermostability of the enzyme was determined by incubating the enzyme in 20 mm Tris-HCl (pH 8.0) at 40 or 60 °C. At various times, aliquots of enzymes were removed and assayed for residual activity using pNPB as the substrate.
Evaluation of Potential Synergistic Effect between Tfu_0882 and Tfu_0883—To evaluate the possibility of a synergism between the two T. fusca cutinases in cutin degradation, Tfu_0882 (0.08 μmol) and Tfu_0883 (0.08 μmol) were incubated, either individually or as a 1:1 mixture, with 1% (w/v) apple cutin in 25 mm potassium phosphate buffer (pH 8.0) at 60 °C. At various times, aliquots were removed and assayed for fatty acids by titration with 0.02 n NaOH. The fatty acid monomers released after 18 h of incubation were identified by GC/MS as described above.
Inactivation of Cutinase—Inactivation of the enzyme was performed by incubating the enzyme (2 μm) with 1 mm PMSF in 20 mm Tris-HCl (pH 7.0) at 37 °C for varying amounts of time. The inactivation reactions were stopped by 200-fold dilution into ice-cold standard esterase assay buffer as described above. The residual enzyme activity was determined immediately by standard esterase assay using pNPB as the substrate. Inactivation was shown not to proceed during the assay under these conditions in control experiments (data not shown). The concentration dependence of PMSF inhibition was performed similarly utilizing an inactivation time of 10 min.
Generation of Cutinase Mutants—Cutinase mutations were generated by the standard QuikChange mutagenesis methodology using complementary primers. The S188A mutant of Tfu_0882 was generated using pET20b-Tfu_0882 as template; S170A mutant of Tfu_0883 was generated using pET20b-Tfu_0883 as template. Mutated sequences were verified by DNA sequencing. The mutants were expressed and purified using the same procedures as described for the original recombinant T. fusca cutinases. The esterase activity of the purified mutants was assayed using pNPB as substrate.
Miscellaneous Methods—Optical spectroscopy was performed using the UV-2450 ultraviolet-visible spectrophotometer from Shimadzu. SDS-PAGE was performed under reducing conditions on a 12% polyacrylamide gel. The gel was visualized with 0.25% Coomassie Brilliant Blue R-250 stain. Protein concentration was determined by the Bradford method.
RESULTS
Purification of a Cutin-induced pNPB Hydrolase from T. fusca—A major hurdle to the isolation of wild-type bacterial cutinase has been the difficulty in monitoring cutin hydrolysis. We set out to isolate this enzyme using pNPB as the substrate based on the following rationale: cutinase is inducible by cutin (1), is excreted to the culture media, and can efficiently hydrolyze pNPB (14). Indeed, we found that pNPB hydrolase activity in T. fusca culture supernatant reached about 19 units/ml in the presence of cutin, whereas no pNPB hydrolase activity was detectable in the absence of cutin. Notably, analysis of proteins in the culture supernatant clearly indicated that a polypeptide of 29 kDa was the major protein induced by cutin (supplemental Fig. S1), suggesting that it might be a candidate for cutinase protein.
Subsequently, by monitoring the hydrolysis of pNPB, a pNPB hydrolase was purified from cutin-induced culture supernatant of T. fusca through ammonium sulfate precipitation, hydrophobic interaction (phenyl Sepharose), and anion exchange (DEAE-Sepharose) chromatography. The purified enzyme exhibited a high specific activity of 398.6 units/mg for pNPB (supplemental Table S1). As expected, SDS-PAGE analysis demonstrated that this pNPB hydrolase corresponded to the 29-kDa protein induced by cutin (supplemental Fig. S1).
Cutin-induced pNPB Hydrolase Displays Cutin-hydrolyzing Activity—To determine whether this T. fusca pNPB hydrolase also displayed cutinase activity, the protein was assayed for its ability of degrade cutin. For comparison, recombinant cutinase from F. solani pisi was purified and assayed under the same condition. As shown in supplemental Table S1, both enzymes exhibited activity for cutin hydrolysis, yielding comparable fatty acid monomers. For example, the proportion of C16 and C18 family fatty acids monomers released after enzymatic reaction were 48.85% for T. fusca pNPB hydrolase and 53.13% for F. solani pisi cutinase. Additionally, the proportion of hydroxy fatty acid is 16.66% for T. fusca pNPB hydrolase and 15.27% for F. solani pisi cutinase (supplemental Table S1). These results indicated that the cutin-induced pNPB hydrolase from T. fusca functions as a cutinase.
Identification of T. fusca Cutinase Encoding Gene—To identify the gene encoding T. fusca cutinase, the 29-kDa band corresponding to cutin-induced pNPB hydrolase was excised from the SDS-PAGE gel (supplemental Fig. S1) and subjected to trypsin digestion. The resulting degradation peptides were analyzed by mass spectrometry. The peptide mass data were used to query the Mascot proteomics data base (www.matrixscience.com), resulting in two significant matches, Tfu_0883 and Tfu_0882 (Table 1). Of the matched peptides, 9 peptides were shared by both proteins, 4 were unique for Tfu_0883, and 1 was unique for Tfu_0882 (Table 2). Furthermore, N-terminal amino acid sequencing of the 29-kDa protein band resulted only a single sequence of ANPYERGPN, which corresponded to residues 59–67 of Tfu_0882 and residues 41–49 of Tfu_0883. These results strongly suggested that residues 1–58 of Tfu_0882 and residues 1–40 of Tfu_0883 are signal peptides. With their signal peptides excluded, both Tfu_0882 and Tfu_0883 are 269 amino acids in length and share a high sequence identity of 93%. It is to be noted that both proteins were annotated as triacylglycerol lipase in the genomic data base and contain the consensus sequence GXSXG, a characteristic of the α/β-hydrolase fold superfamily (25).
TABLE 1.
Sequence comparison of Tfu_0882 and Tfu_0883
TABLE 2.
Summary of matrix-assisted laser desorption ionization peptide mass data identifying Tfu_0883 and Tfu_0882
| Peptide sequence | Sequence length | MWobserved | MWexpect | Comments |
|---|---|---|---|---|
| GPNPTDALLEASSGPFSVSEENVSR | 25 | 2560.2356 | 2559.2283 | Unique for Tfu_0883 |
| LSASGFGGGTIYYPR | 15 | 1545.7740 | 1544.7667 | |
| ENNTYGAVAISPGYTGTEASIAWLGER | 27 | 2827.4700 | 2826.4628 | |
| AEQLNAALNHMINR | 14 | 1594.8105 | 1593.8032 | |
| IASHGFVVITIDTNTTLDQPDSR | 23 | 2500.2482 | 2499.2410 | Unique for Tfu_0882 |
| LAVMGHSMGGGGTLR | 15 | 1443.7270 | 1442.7197 | Shared by Tfu_0882 and Tfu_0883 |
| LASQRPDLK | 9 | 1027.6167 | 1026.6094 | |
| AAIPLTPWHLNK | 12 | 1360.7967 | 1359.7894 | |
| AYLELDGATHFAPNIPNK | 18 | 1971.0039 | 1969.9966 | |
| YSVAWLK | 7 | 866.5405 | 865.5333 | |
| YSVAWLKR | 8 | 1022.6554 | 1021.6481 | |
| RFVDNDTR | 8 | 1022.4574 | 1021.4501 | |
| FVDNDTR | 7 | 866.4168 | 865.4095 | |
| DGLFGEVEEYR | 11 | 1313.6225 | 1312.6152 |
Cloning, Expression, and Purification of Tfu_0882 and Tfu_0883—The genes encoding the mature forms of Tfu_0882 and Tfu_0883 were expressed for further characterization. The vector pET-20b(+), chosen for expression, contains a C-terminal His6 tag and signal peptide PelB, which allows the heterologously expressed proteins to be secreted. The pNPB hydrolyzing activity in the culture supernatant of transformed E. coli cells harboring plasmid pET20b-Tfu_0883 was 180 units/ml, which was 782-fold higher than that of un-transformed E. coli cells and 9.5-fold higher than that of cutin-induced T. fusca cells. The pNPB hydrolyzing activity in the culture supernatant of transformed E. coli cells harboring pET20b-Tfu_0882 plasmid was 69 units/ml, which was 300-fold higher than that of un-transformed E. coli cells and 3.6-fold higher than that of cutin-induced T. fusca cells. The recombinant enzymes were purified to homogeneity in a single step by nickel affinity chromatography (supplemental Fig. S2) and exhibited a specific activity of 458 units/mg for Tfu_0883 and 223 units/mg for Tfu_0882.
Catalytic Properties of Recombinant Tfu_0882 and Tfu_0883—Because the fungal cutinase from F. solani pisi can hydrolyze both insoluble triglycerides and soluble esters (3), Tfu_0882 and Tfu_0883 were first characterized for the hydrolysis of triolein and pNPB, and compared with their fungal counterpart. As shown in Fig. 2, all three enzymes effectively hydrolyzed triolein and pNPB. Tfu_0882 and Tfu_0883 exhibited an optimal temperature at 60 °C for both triolein and pNPB, whereas F. solani pisi cutinase displayed an optimal temperature at 30 °C for triolein and 40 °C for pNPB (Fig. 2). Furthermore, all three enzymes exhibited a pH optimum of about 8.0 for the two substrates (Fig. 3).
FIGURE 2.

Temperature optimum of cutinase. ▵ and ▴, Tfu_0882 using pNPB and triolein as substrates, respectively; ⋄ and ♦, Tfu_0883 using pNPB and triolein as substrates, respectively; □ and ▪, F. solani pisi cutinase using pNPB and triolein as substrates, respectively. Error bars correspond to the standard deviation of three determinations.
FIGURE 3.

pH optimum of cutinase. ▵ and ▴, Tfu_0882 using pNPB and triolein as substrates, respectively; ⋄ and ♦, Tfu_0883 using pNPB and triolein as substrates, respectively; □ and ▪, F. solani pisi cutinase using pNPB and triolein as substrates, respectively. Enzyme activity was measured at 40 °C for F. solani pisi cutinase and 60 °C for Tfu_0882 and Tfu_0883, in either 25 mm potassium phosphate buffers (pH 6.0–7.0) or 20 mm Tris-HCl buffers (pH 7.0–9.0). Error bars correspond to the standard deviation of three determinations.
The thermostability of Tfu_0882, Tfu_0883, and F. solani pisi cutinase were evaluated at both 40 and 60 °C (Fig. 4). Interestingly, the activity of Tfu_0882 and Tfu_0883 increased initially during incubation at both temperatures, whereas F. solani pisi cutinase exhibited a similar initial increase at 40 °C, but a simple exponential decay at 60 °C. T. fusca cutinases exhibited superior thermostability, with residual activities of over 80% after 160 h at 40 °C or over 50% after 40 h at 60 °C. In contrast, F. solani pisi cutinase was significantly less stable, with 50% of activity retained after 85 h at 40 °C or after 5 min at 60 °C.
FIGURE 4.
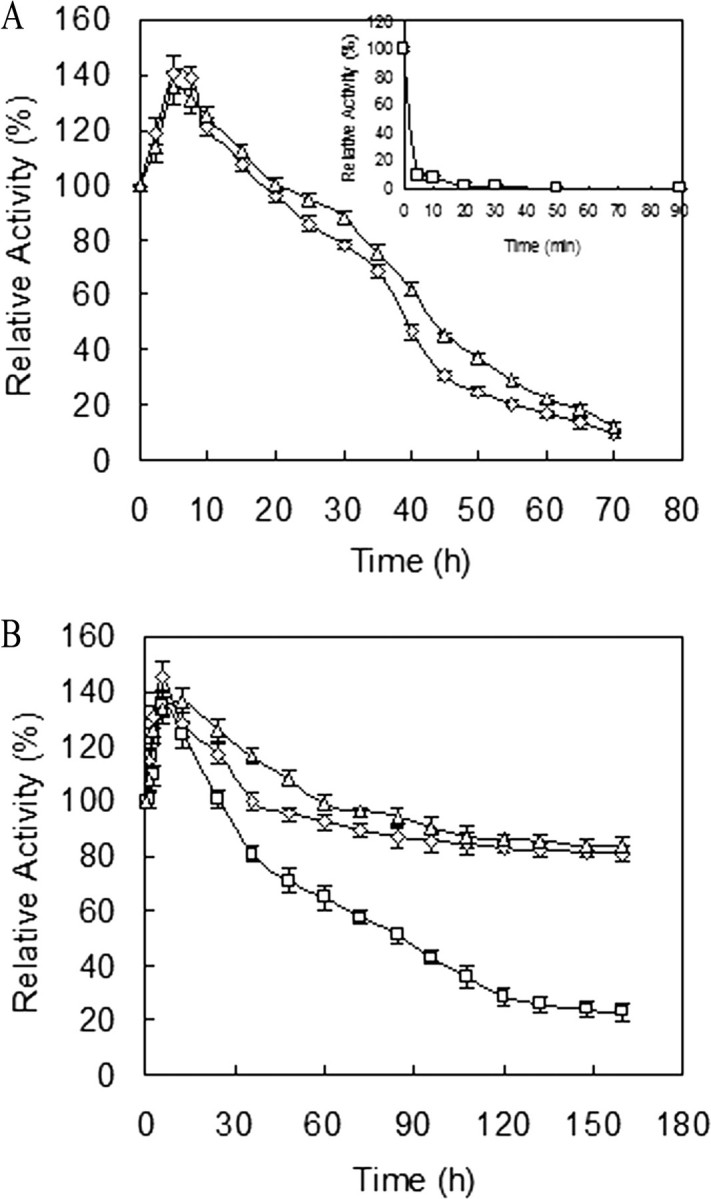
Thermostability of cutinase. The enzyme was incubated in 20 mm Tris-HCl (pH 8.0) at 60 °C (A) or 40 °C (B). ▵, ⋄, and □ represent the relative activity of Tfu_0882, Tfu_0883, and F. solani pisi cutinase, respectively. At various intervals, aliquots of the samples were removed and assayed for residual activity at their optimal temperature and pH. Error bars correspond to the standard deviation of three determinations.
Subsequently, to determine whether Tfu_0882 and Tfu_0883 can indeed hydrolyze cutin, both enzymes and the fungal cutinase were evaluated under their individual optimal temperature and pH. As shown in Table 3, the C16 and C18 family fatty acid monomers released after enzymatic reaction were 50.14% for Tfu_0883, 46.03% for Tfu_0882, and 57.54% for F. solani pisi cutinase. The hydroxy fatty acids that are specific in cutin were 28.10% for Tfu_0883, 25.16% for Tfu_0882, and 23.66% for F. solani pisi cutinase. These results confirmed that both Tfu_0882 and Tfu_0883 are efficient cutin hydrolases.
TABLE 3.
Monomeric products released from cutin hydrolysis by recombinant cutinases and a bacterial lipase
Cutin hydrolysis by F. solani pisi cutinase, PsL, and T. fusca cutinases were incubated in 25 mm potassium phosphate buffer (pH 8.0) at temperatures of 40, 40, and 60 °C, respectively, for 18 h. The results are the averages of triplicate assays.
| Cutin hydrolysis products | F. solani cutinase Area (%) | T. fusca cutinase (Tfu_0882) Area (%) | T. fusca cutinase (Tfu_0883) Area (%) | P. sp. MIS38 lipase Area (%) |
|---|---|---|---|---|
| Hexadecanoic acid | 15.94 ± 2.62 | 9.27 ± 2.07 | 10.96 ± 2.89 | 24.89 ± 3.71 |
| 9-Hexadecenoic acid | 0.13 ± 0.02 | 0.06 ± 0.02 | 0.09 ± 0.03 | 0.77 ± 0.12 |
| Octadecanoic acid | 14.99 ± 1.71 | 9.24 ± 3.01 | 8.77 ± 2.69 | 21.61 ± 4.09 |
| 9-Octadecenoic acid | 1.74 ± 0.61 | 0.98 ± 0.36 | 1.16 ± 0.79 | 1.39 ± 0.67 |
| 9,12-Octadecadienoic acid | 1.08 ± 0.23 | 1.32 ± 0.37 | 1.06 ± 0.23 | 1.33 ± 0.54 |
| 16-Hydroxyhexadecanoic acid | 1.65 ± 0.31 | 5.66 ± 1.14 | 4.74 ± 0.54 | NDa |
| 10,16-Dihydroxyhexadecanoic acid | 10.18 ± 1.12 | 8.29 ± 2.35 | 9.15 ± 0.97 | ND |
| 18-Hydroxyoctadeca-9-enoic acid | 1.90 ± 0.34 | 1.03 ± 0.32 | 1.52 ± 0.12 | ND |
| 18-Hydroxyoctadeca-9,12-dienoic acid | 7.98 ± 0.98 | 9.19 ± 2.25 | 10.91 ± 1.07 | ND |
| 9,10,18-Trihydroxyoctadecanoic acid | 1.95 ± 0.42 | 0.99 ± 0.17 | 1.78 ± 0.35 | ND |
ND, not detectable.
A question arises as to whether there is a synergistic effect between the two T. fusca enzymes in the breakdown of cutin. When Tfu_0882, Tfu_0883, or a mixture containing the same amounts of both enzymes were incubated with cutin under the same conditions, the amount of free fatty acids released by the enzyme mixtures was almost identical to the hypothetical sum of values from individual enzymes (Fig. 5) throughout the monitored time points during the reaction. In addition, GC/MS analysis of the 18-h reaction samples revealed no appreciable difference in the types and proportions of fatty acid monomers released by Tfu_0882, Tfu_0883, or the 1:1 mixture of them (data not shown). Thus, it seems likely there is no synergism between Tfu_0882 and Tfu_0883 in cutin degradation.
FIGURE 5.

Evaluation of possible synergistic effects between Tfu_0882 and Tfu_0883 in cutin degradation. Tfu_0882 (0.08 μmol) and Tfu_0883 (0.08 μmol) were together or individually incubated with 1% (w/v) apple cutin in 25 mm potassium phosphate buffer (pH 8) at 60 °C. The released fatty acids were quantified by titration with 0.02 n NaOH. ♦, Tfu_0883 alone; ▴, Tfu_0882 alone; •, Tfu_0882 and Tfu_0883; □, hypothetical sum of Tfu_0883 alone (♦) plus Tfu_0882 alone (▴). Error bars correspond to the standard deviation of three determinations.
Homology Modeling of T. fusca Cutinase—Because Tfu_0882 and Tfu_0883 are highly similar in sequence, only one sequence, Tfu_0883, was randomly chosen for sequence and structure analysis. A BLAST search of the NCBI data base with Tfu_0883 revealed that most matches are annotated as triacylglycerol lipase (Table 4). The highest similarity (63%) was found with Streptomyces exfoliates lipase (SeL) whose x-ray structure has been solved at 1.9 Å (26). To better understand the mechanism of bacterial cutinase, a homology model of Tfu_0883 based on SeL structure was constructed by the SWISS-MODEL homology modeling web server (27). The predicted structure exhibits an α/β-hydrolase fold consisting of a central twisted β-sheet flanked on both sides by α-helices (Fig. 6A). In addition, similar to SeL, Tfu_0883 contains an additional C-terminal extension. This model also revealed a potential Ser170-His248-Asp216 catalytic triad in which the nucleophilic Ser170 is located in a sharp turn called the “nucleophile elbow,” which is commonly observed in the α/β-hydrolases (25). The “oxyanion hole” is predicted to be formed by the backbone amides of Met171 and Tyr100. Notably, similar to fungal cutinase, the enzyme does not contain a lid insertion that is commonly observed in lipases (24), resulting in a nucleophilic serine that is exposed to the solvent (Fig. 6B).
TABLE 4.
Tfu_0883-like sequences from other bacterial sources
| Sources | NCBI accession No. | Current definition | Identities to Tfu_0883 | Sequence length |
|---|---|---|---|---|
| % | ||||
| Streptomyces exfoliatus | 1JFR_A | Lipase | 63 | 262a |
| Streptomyces coelicolor A3(2) | NP_625018 | Lipase | 63 | 310 |
| Streptomyces albus | AAA53485 | Lipase precursor | 62 | 304 |
| Streptomyces ambofaciens | CAJ88461 | Putative secreted lipase | 59 | 334 |
| Kineococcus radiotolerans | YP_001363557 | Triacylglycerol lipase | 57 | 301 |
| Acidovorax delafieldii | BAB86909 | Poly(tetramethylene succinate) depolymerase | 48 | 304 |
Signal peptide not included.
FIGURE 6.
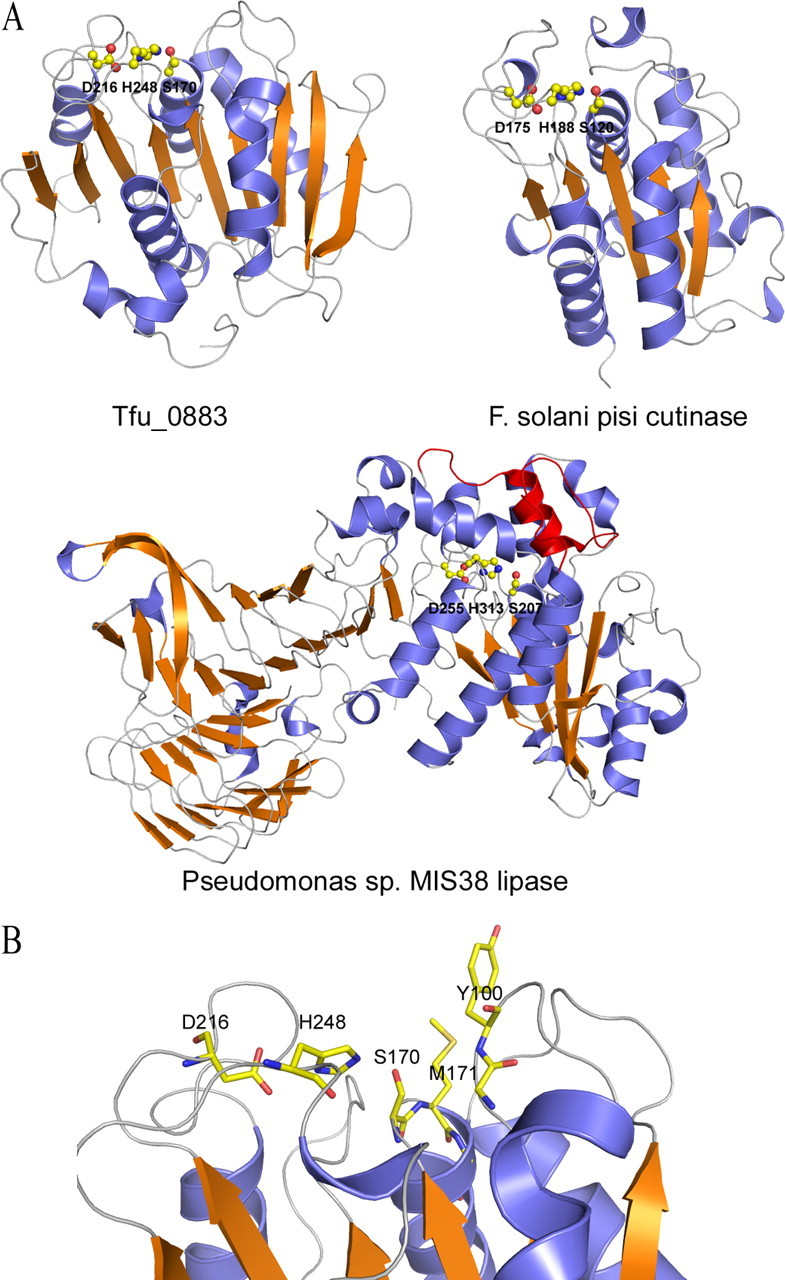
Homology modeling of T. fusca cutinase. A, ribbon diagram of a predicted Tfu_0883 model compared with the structures of a fungal cutinase and a true bacterial lipase (Pseudomonas sp. MIS38 lipase). The catalytic triad residues are shown as ball-and-sticks. The lid domain of Pseudomonas sp. MIS38 lipase is shown in red. The Tfu_0883 model was predicted by SWISS-MODEL, an automated comparative protein modeling server (27). The template was SeL (PDB code 1jfr), which shares 63% sequence identity with Tfu_0883. B, the active site residues of T. fusca cutinase. The catalytic triad is formed by Ser170, His248, and Asp216. The oxyanion hole is formed by the main chain amides of Met171 and Tyr100.
Inhibition and Mutational Analysis of T. fusca Cutinase—To determine whether the enzyme indeed utilizes a serine-based catalytic triad mechanism, we tested the effect of PMSF, a classic mechanism-based serine hydrolase inhibitor, on enzyme activity, and investigated the role of the putative nucleophilic serine on catalysis. As shown in Fig. 7, the activities of the two cutinases were essentially abolished by PMSF (1 mm) after 17.5 min of incubation for Tfu_0882 and 15 min of incubation for Tfu_0883, with a half-life of ∼7 min for Tfu_0882 and 6 min for Tfu_0883, thereby indicating irreversible modification of the catalytic serine for both enzymes. When the enzymes were preincubated with PMSF for 10 min, 50% inhibition of Tfu_0882 and Tfu_0883 were observed at about 0.50 and 0.45 mm PMSF, respectively. Subsequently, mutation of the putative catalytic serine (Ser188 in Tfu_0882 and Ser170 in Tfu_0883) to alanine completely eliminated their activities.
FIGURE 7.
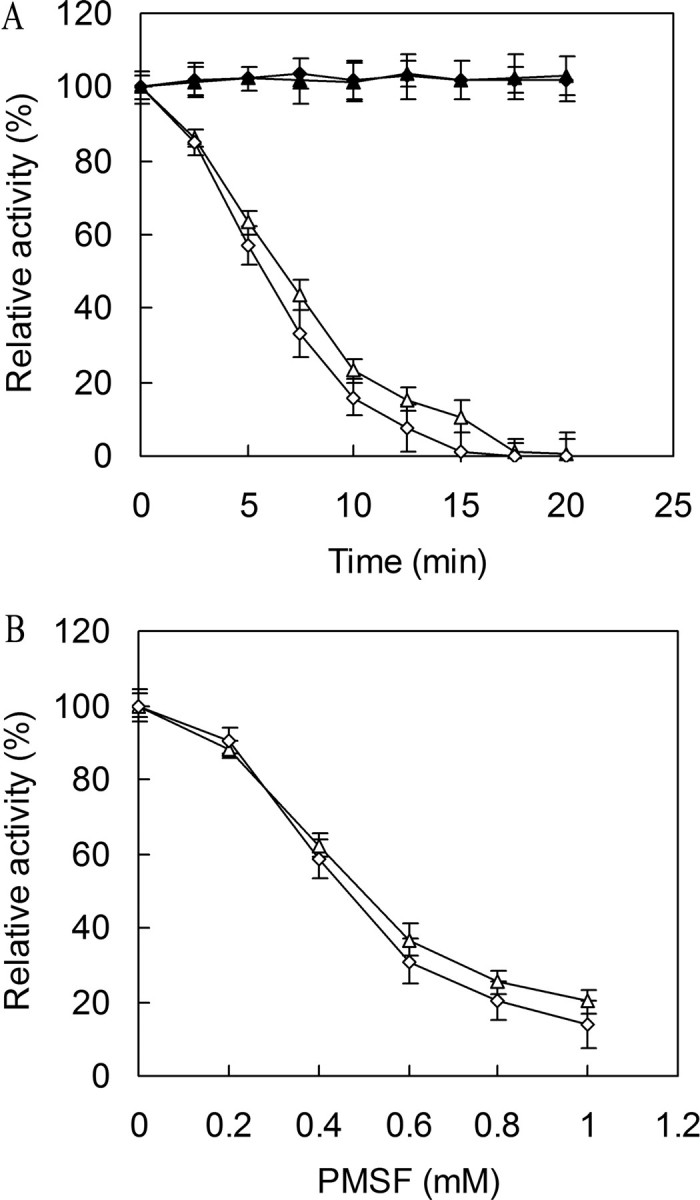
Inactivation of cutinase by PMSF. A, time course of cutinase inactivation by PMSF. The enzymes were incubated with 1 mm PMSF (▵, Tfu_0882; ⋄, Tfu_0883) or without PMSF (▴, Tfu_0882; ♦, Tfu_0883) at 37 °C for various times and assayed for residual activity. B, concentration dependence of cutinase inactivation by PMSF. The enzymes (▵, Tfu_0882; ⋄, Tfu_0883) were incubated with various concentrations of PMSF at 37 °C for 10 min and assayed for residual activity. Error bars correspond to the standard deviation of three determinations.
DISCUSSION
Although fungal cutinase has been extensively characterized for decades, the molecular identity of bacterial cutinase has remained a mystery. In the present study, several lines of evidence indicate that Tfu_0882 and Tfu_0883 from the actinomycete T. fusca function as cutinases: (i) a 29-kDa protein was secreted into the T. fusca culture upon cutin induction and exhibited cutinase activity (supplemental Fig. S1); (ii) peptide mass fingerprinting analysis and N-terminal amino acid sequencing of the 29-kDa protein matched two proteins, Tfu_0882 and Tfu_0883, which are 93% identical in sequence; (iii) both Tfu_0882 and Tfu_0883 are able to hydrolyze cutin, resulting in monomeric products typical of fungal cutinase.
Given their high sequence identity, it is not surprising that Tfu_0882 and Tfu_0883 have similar physical properties and were thus unresolved during our purification of the wild-type enzyme. Catalytically, both Tfu_0882 and Tfu_0883 are relatively versatile in that they can utilize both insoluble triglycerides (triolein) and soluble esters (pNPB) as substrate in addition to cutin. Moreover, the two enzymes share similar temperature and pH dependence profiles and thermostability. We therefore suggest that they may be defined as cutinase isoenzymes in T. fusca. Although Tfu_0882 and Tfu_0883 are sequential in the genome, likely due to a gene duplication event, they do not appear to be in an operon as determined by the operon prediction tool VIMSS (www.microbesonline.org/operons) (28). Moreover, there is no synergism between the two enzymes in cutin degradation (Fig. 5). Thus further studies are needed to understand why there are two sequential genes for T. fusca cutinase.
Sequence analysis suggested that T. fusca cutinases belong to the α/β-hydrolase fold superfamily (25). Enzymes in this superfamily exhibit a wide variety of hydrolytic activities (29), however, they all adopt a conserved three-dimensional fold and are believed to have evolved from a common ancestor (25). As expected, the homology model of Tfu_0883 displays a canonical α/β-hydrolase fold with a Ser170-His248-Asp216 triad and a preformed oxyanion hole (Fig. 6B), suggesting a classic serine hydrolase mechanism involving two tetrahedral transition states and an acyl-enzyme intermediate (30). Indeed this mechanism was supported by PMSF-mediated irreversible inhibition of Tfu_0882 and Tfu_0883 (Fig. 7) as well as site-directed mutagenesis of the catalytic serine (to alanine) in both enzymes.
A notable feature of the T. fusca cutinase model is that the enzyme does not contain a lid insertion commonly observed in true lipases (31–34), and exposes its nucleophilic serine to the solvent (Fig. 6B). In lipases, the lid insertion is reported to be involved in the interfacial activation in which it undergoes conformational change in response to adsorption at the oil-water interface (24). The absence of such a lid insertion suggests that the T. fusca cutinases should belong to a class of α/β-hydrolases different from the classic lipases. On the other hand, the open active site of T. fusca cutinase readily explains the ability of the enzyme to accommodate large substrates like cutin. Further studies, especially x-ray crystallography studies of ligand-bound cutinase may reveal the structural basis of substrate recognition.
Comparative biochemical characterization of bacterial and fungal cutinases indicated that they have similar substrate specificity and catalytic properties except that T. fusca cutinases demonstrate remarkably greater thermostability. This unique feature may render T. fusca cutinases practically more amenable for industrial applications.
Although both T. fusca and F. solani pisi cutinases belong to the α/β-hydrolases superfamily and contain an open active site, the bacterial enzymes have significantly longer sequences and demonstrate no sequence similarity to the fungal enzyme. Moreover, the fungal cutinase contains neither the two N-terminal β strands of the canonical α/β-hydrolase fold nor the unique C-terminal extension of Tfu_0883. Thus, the bacterial and fungal enzymes must have undergone extensive evolutionary differentiation, and are suggested to be classified into prokaryotic and eukaryotic cutinase subfamilies, respectively.
Previously, Tfu_0883 was shown to be a hydrolase capable of degrading aliphatic-aromatic copolyesters by the Muller group (35). In their studies, an extracellular hydrolase (TfH) responsible for the degradation of aliphatic-aromatic copolyesters was purified from the culture broth of T. fusca and determined for amino acid sequence. The sequence of their enzyme is identical to that of Tfu_0883. Although cutin-degrading activity was observed for TfH, they claimed that classification of this enzyme as a cutinase is “generally questionable” because a true lipase Pseudomonas sp. MIS38 lipase (PsL) was also shown to hydrolyze their cutin preparation and release fatty acids (determined by a titration assay).
To clarify this ambiguity, we subjected PsL to cutinase assay by using GC/MS to detect the released fatty acid monomers. It turned out that this enzyme exhibited no cutinase activity because C16 and C18 hydroxyl fatty acids, the characteristic cutin components, were absent in the enzymatic reaction products (Table 3). The fatty acids found in the enzymatic reaction could be the hydrolyzed products of lipids that coexisted in the cutin preparation. In fact, we have found that it is particularly hard to obtain cutin that is free of lipid contamination, which could also be the case in the study by Muller and co-workers (35). Furthermore, the catalytic residues of PsL are completely buried under the lid surface, which is different from the active sites in TfH (Tfu_0883) and the fungal cutinase (Fig. 6). Therefore, these analyses indicated that TfH (Tfu_0883) differs from PsL in its cutin-hydrolyzing activity and should be classified as a cutinase.
In conclusion, the present study has definitively established that the Tfu_0882 and Tfu_0883 from the bacteria T. fusca are cutinases. Although their catalytic properties and mechanisms are similar to the fungal cutinases, sequence and structural differences place the T. fusca enzymes into a different subfamily of cutinases from the fungal enzymes. Given their potential applications in biotechnology, the elucidation of the bacterial cutinase encoding genes provides an important breakthrough that should contribute significantly to further investigation into cutinases.
Supplementary Material
Acknowledgments
We thank other members of the group for helpful discussions.
This work was supported by Program for New Century Excellent Talents in University Grant NCET-06-0486, National Science Fund for Distinguished Young Scholars of China Grant 20625619, 111 Project Grant 11-2-06, and Research Program of State Key Laboratory of Food Science and Technology Grant SKLF-MB-200802. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Table S1.
Footnotes
The abbreviations used are: pNPB, p-nitrophenyl butyrate; SeL, Streptomyces exfoliates lipase; PMSF, phenylmethylsulfonyl fluoride; PsL, Pseudomonas sp. MIS38 lipase; GC/MS, gas chromatography/mass spectrometry.
References
- 1.Fett, W. F., Gerard, H. C., Moreau, R. A., Osman, S. F., and Jones, L. E. (1992) Appl. Environ. Microb. 58 2123–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walton, T. J., and Kolattukudy, P. E. (1972) Biochemistry 11 1885–1896 [DOI] [PubMed] [Google Scholar]
- 3.Purdy, R. E., and Kolattukudy, P. E. (1973) Arch. Biochem. Biophys. 159 61–69 [DOI] [PubMed] [Google Scholar]
- 4.Gerard, H. C., Osman, S. F., Fett, W. F., and Moreau, R. A. (1992) Phytochem. Analysis 3 139–144 [Google Scholar]
- 5.Ferreira, B. S., Calado, C. R., van Keulen, F., Fonseca, L. P., Cabral, J. M., and da Fonseca, M. M. (2003) Appl. Microbiol. Biotechnol. 61 69–76 [DOI] [PubMed] [Google Scholar]
- 6.Sebastian, J., and Kolattukudy, P. E. (1988) Arch. Biochem. Biophys. 263 77–85 [DOI] [PubMed] [Google Scholar]
- 7.Wang, G. Y., Michailides, T. J., Hammock, B. D., Lee, Y. M., and Bostock, R. M. (2002) Fungal Genet. Biol. 35 261–276 [DOI] [PubMed] [Google Scholar]
- 8.Carvalho, C. M., Aires-Barros, M. R., and Cabral, J. M., (1999) Biotechnol. Bioeng. 66 17–34 [DOI] [PubMed] [Google Scholar]
- 9.Egmond, M. R., and de Vlieg, J. (2000) Biochimie (Paris) 82 1015–1021 [DOI] [PubMed] [Google Scholar]
- 10.Wang, G. Y., Michailides, T. J., Hammock, B. D., Lee, Y. M., and Bostock, R. M. (2000) Arch. Biochem. Biophys. 382 31–38 [DOI] [PubMed] [Google Scholar]
- 11.Gindro, K., and Pezet, R. (1999) FEMS Microbiol. Lett. 171 239–243 [DOI] [PubMed] [Google Scholar]
- 12.Maeda, H., Yamagata, Y., Abe, K., Hasegawa, F., Machida, M., Ishioka, R., Gomi, K., and Nakajima, T. (2005) Appl. Microbiol. Biotechnol. 67 778–788 [DOI] [PubMed] [Google Scholar]
- 13.Cunha, M. T., Costa, M. J., Calado, C. R., Fonseca, L. P., Aires-Barros, M. R., and Cabral, J. M. (2003) J. Biotechnol. 100 55–64 [DOI] [PubMed] [Google Scholar]
- 14.Longhi, S., and Cambillau, C. (1999) Biochim. Biophys. Acta 1441 185–196 [DOI] [PubMed] [Google Scholar]
- 15.Araujo, R., Silva, C., O'Neill, A., Micaelo, N., Guebitz, G., Soare, C. M., Casal, M., and Cavaco-Paulo, A. (2007) J. Biotechnol. 128 849–857 [DOI] [PubMed] [Google Scholar]
- 16.Alisch-Mark, M., Herrmann, A., and Zimmermann, W. (2006) Biotechnol. Lett. 28 681–685 [DOI] [PubMed] [Google Scholar]
- 17.Vertommen, M. A., Nierstrasz, V. A., Veer, M., and Warmoeskerken, M. M. (2005) J. Biotechnol. 120 376–386 [DOI] [PubMed] [Google Scholar]
- 18.Degani, O., Gepstein, S., and Dosoretz, C. G. (2002) Appl. Biochem. Biotechnol. 102–103 277–289 [DOI] [PubMed] [Google Scholar]
- 19.Lin, T. S., and Kolattukudy, P. E. (1980) Plant Pathol. 17 1–15 [Google Scholar]
- 20.Fett, W. F., Wijey, C., Moreau, R. A., and Osman, S. F. (1999) J. Appl. Microbiol. 86 561–568 [DOI] [PubMed] [Google Scholar]
- 21.Sebastian, J., Chandra, A. K., and Kolattukudy, P. E. (1987) J. Bacteriol. 169 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fett, W. F., Gerard, H. C., Moreau, R. A., Osman, S. F., and Jones, L. E. (1992) Curr. Microbiol. 25 165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Osman, S. F., Irwin, P., Fett, W. F., Connor, J. V., and Parris, N. (1999) J. Agric. Food Chem. 47 799–802 [DOI] [PubMed] [Google Scholar]
- 24.Jaeger, K. E., Ransac, S., Dijkstra, B. W., Colson, C., van Heuvel, M., and Misset, O. (1994) FEMS Microbiol. Rev. 15 29–63 [DOI] [PubMed] [Google Scholar]
- 25.Nardini, M., and Dijkstra, B. W. (1999) Curr. Opin. Struct. Biol. 9 732–737 [DOI] [PubMed] [Google Scholar]
- 26.Wei, Y., Swenson, L., Castro, C., Derewenda, U., Minor, W., Arai, H., Aoki, J., Inoue, K., Servin-Gonzalez, L., and Derewenda, Z. S. (1998) Structure 6 511–519 [DOI] [PubMed] [Google Scholar]
- 27.Schwede, T., Kopp, J., Guex, N., and Peitsch, M. C. (2003) Nucleic Acids Res. 31 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price, M. N., Huang, K. H., Alm, E. J., and Arkin, A. P. (2005) Nucleic Acids Res. 33 880–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmquist, M. (2000) Curr. Protein Pept. Sci. 1 209–235 [DOI] [PubMed] [Google Scholar]
- 30.Dodson, G., and Wlodawer, A. (1998) Trends Biochem. Sci. 23 347–352 [DOI] [PubMed] [Google Scholar]
- 31.Angkawidjaja, C., You, D. J., Matsumura, H., Kuwahara, K., Koga, Y., Takano, K., and Kanaya, S. (2007) FEBS Lett. 581 5060–5064 [DOI] [PubMed] [Google Scholar]
- 32.Brzozowski, A. M., Savage, H., Verma, C. S., Turkenburg, J. P., Lawson, D. M., Svendsen, A., and Patkar, S. (2000) Biochemistry 39 15071–15082 [DOI] [PubMed] [Google Scholar]
- 33.Brzozowski, A. M., Derewenda, U., Derewenda, Z. S., Dodson, G. G., Lawson, D. M., Turkenburg, J. P., Bjorkling, F., Huge-Jensen, B., Patkar, S. A., and Thim, L. (1991) Nature 351 491–494 [DOI] [PubMed] [Google Scholar]
- 34.Lang, D., Hofmann, B., Haalck, L., Hecht, H. J., Spener, F., Schmid, R. D., and Schomburg, D. (1996) J. Mol. Biol. 259 704–717 [DOI] [PubMed] [Google Scholar]
- 35.Kleeberg, I., Welzel, K., Vandenheuvel, J., Muller, R. J., and Deckwer, W. D. (2005) Biomacromolecules 6 262–270 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.