
FIGURE 4.
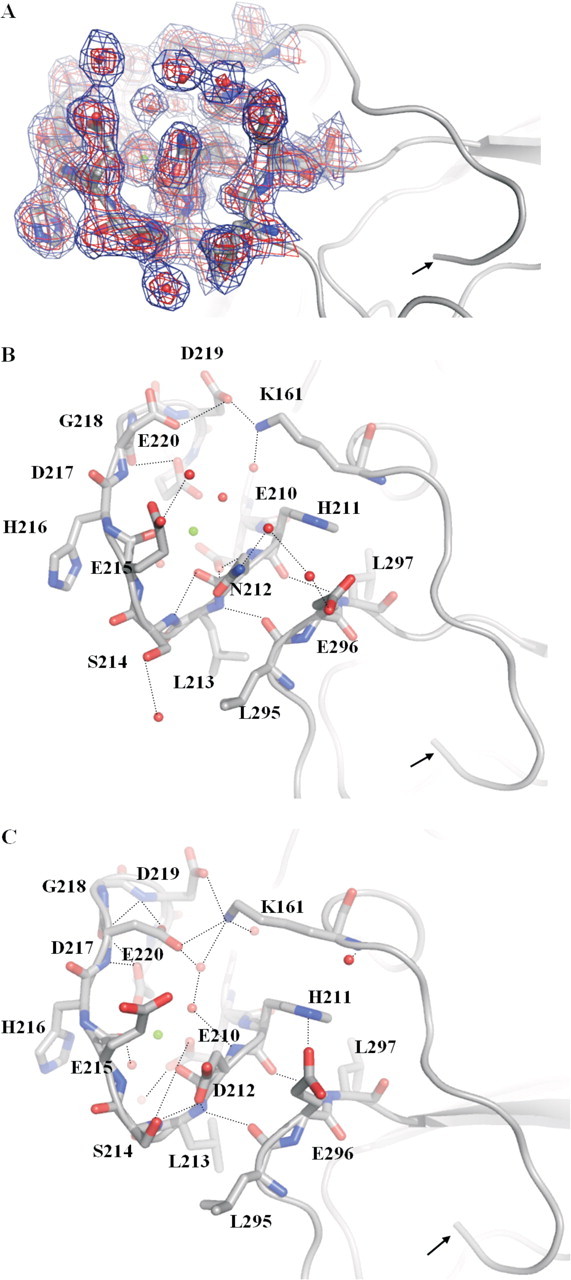
Highlight of residues in the Ca2+-binding loop of the protease domain in the structures of FVIIaD212N (A and B; 2.7 Å) and wild-type FVIIa (C; 2.0 Å; PDB code 1DAN). The N terminus is indicated with an arrow and hydrogen bonds shown by dotted lines.2Fo – Fc electron density maps are shown at 1σ (blue) and 2σ (red). The mutant structure shows changes of hydrogen bond networks in the Ca2+-binding loop (Ca2+ shown as a green sphere), for example, surrounding mutated residue Asn212 and Ser214 and Glu296. A hydrogen bond is abolished between Asn212 and Ser214 because of a side chain movement of Ser214 in the mutant structure. A hydrogen bond network is introduced between Asn212, Glu296, and two strongly defined water molecules. In the wild-type structure, Asp212 and Glu296 are not in an electrostatically optimal configuration because of charge repulsion and the conformations of the two residues are slightly changed in the mutant structure. A distinct side chain movement of Asp217 can be observed as well. In turn, a hydrogen bond between Lys161 and Asp217 is lost, whereas bonding to Asp219 is strengthened: Asp219 to Lys161 is 2.6 Å in the mutant structure (see B) versus 3.9 Å in the wild-type structure (see C). Root mean square displacements (Cα) of the Ca2+-binding loop of the mutant structure versus the wild structure were 0.74 Å compared with an overall of 0.65 Å for the heavy chains.