

Abstract
RhBG, a human member of the Amt/Mep/Rh/superfamily of ammonium transporters, has been shown to facilitate NH3 transport and to be anchored to the basolateral plasma membrane of kidney epithelial cells, via ankyrin-G. We showed here that triple alanine substitution of the 419FLD421 sequence, which links the cytoplasmic C-terminal domain of RhBG to ankyrin-G, not only disrupted the interaction of RhBG with the spectrin-based skeleton but also delayed its cell surface expression, decreased its plasma membrane stability, and abolished its NH3 transport function in epithelial cell lines. Similarly, we demonstrated that both anchoring to the membrane skeleton and ammonium transport activity are regulated by the phosphorylation status of the C-terminal tail of RhBG. Tyrosine 429, which belongs to the previously reported YED basolateral targeting signal of RhBG, was demonstrated to be phosphorylated in vitro using purified Src and Syk kinases and ex vivo by analyzing the effect of pervanadate treatment on wild-type RhBG or Y429A mutants. Then, we showed that Y429D and Y429E mutations, mimicking constitutive phosphorylation, abolished NH3 transport and enhanced Triton X-100 solubilization of RhBG from the cell membrane. In contrast, the nonphosphorylated/nonphosphorylatable Y429A and Y429F mutants behaved the same as wild-type RhBG. Conversely, Y/A or Y/F but not Y/E or Y/D mutations of residue 429 abolished the exclusive basolateral localization of RhBG in polarized epithelial cells. All these results led to a model in which targeting and ammonium transport function of RhBG are regulated by both phosphorylation and membrane skeleton binding of the C-terminal cytoplasmic domain.
The protein homologues Rh, RhAG, RhBG, and RhCG are the four members of the human Rh2 (Rhesus) family. They share a common predicted secondary structure with twelve transmembrane domains and both N and C termini located in the cytoplasm, a structure reminiscent of many membrane transporters (1). Rh and RhAG are erythroid-specific membrane proteins and represent the “core” of the Rh membrane complex (2–4). The nonerythroid RhBG and RhCG proteins exhibit a polarized expression, basolateral and apical, respectively, in epithelial cells from organs specialized in ammonia production and excretion such as kidney, liver, and intestine (5–7). Phylogenetic studies (1, 8, 9) and experimental evidence (10–18) have shown that these proteins belong to the Amt/Mep/Rh protein superfamily of ammonium transporters. Moreover, both in human and mouse red blood cells (16) and in recombinant kidney epithelial cells (18), we showed by fast kinetic studies based on stopped-flow spectrometry analysis that Rh glycoproteins (RhAG, RhBG, and RhCG) facilitate NH3 movement, rather than NH+4, across the membrane and therefore represent the first examples of gas channels in mammals. By contrast, the nonglycosylated erythroid RhD and RhCE proteins are not able to transport NH3 (16). Supporting these findings, crystallographic structure determination and transport experiments demonstrated that Escherichia coli AmtB is a channel that conducts uncharged NH3 (19, 20). Based on the three-dimensional structure of AmtB, homology modeling emphasizing critical residues involved in the NH3 channel of the Rh protein family members has been proposed (21, 22). More recently, the structure of a bacterial homologue (from Nitrosomonas europaea) of Rh glycoproteins was solved, showing a homotrimeric conformation as for AmtB (23, 24). Altogether, these data provide a basis to further undertake the study of the structure/function relationship of Rh proteins.
The cytosolic domains of transmembrane proteins are known to be involved in their sorting (25, 26) and, in some cases of adhesion molecules and ion transporters, anchoring to the underlying spectrin-based skeleton through the membrane adaptor ankyrins (27, 28). We previously demonstrated that the Rh complex constitutes a major interaction site between the phospholipid bilayer and the erythrocyte membrane skeleton through direct binding of the cytoplasmic C-terminal tails of Rh and RhAG with ankyrin-R (29). This finding led to the proposal of an erythroid model of the Rh/AE1 (type 1 anion exchanger) macrocomplex described by Bruce et al. (30), in which Rh complex proteins and AE1 could associate either directly or indirectly through their common interaction with ankyrin-R (29). Moreover, we recently reported that the targeting and anchoring of human RhBG to the basolateral plasma membrane of epithelial kidney cells require a cis-tyrosine-based signal and an ankyrin-G-binding motif, respectively, both located in the cytoplasmic C-terminal tail (31) (Fig. 1).
FIGURE 1.

Amino acid sequence of the cytoplasmic C terminus of human RhBG (RhBG-Cter). The FLD ankyrin-G interaction domain and the YED targeting signal were previously identified (31). Terminal DTQA sequence conforms to a possible type I PDZ-binding motif. Potential phosphorylation sites are denoted by asterisks. TM-12, polytopic structure of RhBG with 12 transmembrane domains upstream the cytoplasmic C terminus.
Although the structure of the cytosolic C termini of AmtB and Rh glycoproteins has not been resolved, this domain did not appear to be a central constituent of the gas channel. However, several mutational studies on both fungal (9, 13, 32, 33) and plant (34) Amt proteins provided evidence of the importance of the C-terminal tail for transport function. Moreover, the recent structure of an Rh homologue from N. europaea revealed the presence of a cytoplasmic C-terminal helix that could regulate channel opening by interaction with a protein partner (23). Alternatively, the crystal structure of the archeal Amt-1 strongly suggested that the C terminus interacts physically with cytosolic loops of the protein (35), and recent reports on the plant AtAMT1;1 (36) and AtAMT1;2 (37) proposed that phosphorylation of conserved sites in the C terminus controls this interaction and consequently the ammonium transport function of the protein.
The cytoplasmic C-terminal tail of human RhBG contains several potential phosphorylation sites, Ser422, Ser426, Tyr429, and Thr456, as determined using in silico prediction programs (Fig. 1). Moreover, the extreme four C-terminal residues (DTQA), in which Thr456 is included, resemble a canonical type I PDZ-binding domain (X-Ser/Thr-X-Φ) known to regulate the polarized distribution of numerous membrane proteins in neurons and epithelial cells (38, 39) and also to modulate the function of ion channels and receptors (40–43).
In this study, we confirmed the importance of the C terminus of RhBG for targeting and showed its crucial part in NH3 transport function. We investigated the role of phosphorylation and anchorage to the plasma membrane skeleton on surface expression and transport activity of RhBG. We propose a model of regulation of RhBG targeting and function by the cytoplasmic C-terminal tail.
EXPERIMENTAL PROCEDURES
Materials—Primers used in PCR and mutagenesis experiments were from MWG Biotech (Ebersberg, Germany). The QuikChange XL site-directed mutagenesis kit and Escherichia coli BL21 and TKB1 strains were provided by Stratagene (La Jolla, CA). The pGEX-5X-3 vector, the protein A-Sepharose CL4B beads, and the glutathione-Sepharose 4B beads were purchased from Amersham Biosciences. Complete protease inhibitor mixture was supplied by Roche Applied Science. Purified Src and Syk kinases were provided by Cell Signaling Technology (Danvers, MA), and sodium orthovanadate was purchased from Calbiochem (Darmstadt, Germany).
Wild-type and Mutant RhBG and RhBG-Cter Expression Vectors—The mutated human RhBG cDNAs S422A, S426A, T456A, S422D, S426D, Y429D, T456D, Y429E, Y429F, and 454Stop were obtained by in vitro mutagenesis from the pcDNA3-RhBG vector previously described (31), according to the supplier's instructions (Stratagene). The PCR-amplified cDNA fragment encoding the C-terminal tail of RhBG (RhBG-Cter, residues 416–458, starting from the ATG codon, Fig. 1) was inserted between the EcoRI and XhoI sites of the pGEX-5X-3 vector, in-frame with the DNA coding for the GST protein. The mutant form of RhBG-Cter Y429A was derived from pGEX-5X-3-RhBG-Cter by in vitro mutagenesis. All the inserts were sequenced using an ABI-PRISM 310 genetic analyzer (Applied Biosystems, Foster City, CA). The pCEP4-RhBG vector, containing the full-length cDNA for RhBG and the hygromycin resistance gene as selection marker, was described previously (18).
Antibodies—Rabbit polyclonal antiserum raised against human RhBG-Cter has been described previously (6). This antibody was affinity-purified using the SulfoLink kit from Pierce (Rockford, IL). A mouse polyclonal antibody was raised against the extracellular loops of RhBG by immunization with recombinant mIMCD-3 cells expressing human RhBG at their surface. The immunization schedule was as follows (Kernov, St. Etienne-en-Cogles, France): BALB/C SSC mice were immunized by three intraperitoneal injections of 2 × 106 cells, the first injection with Freund's complete adjuvant, the following injections with Freund's incomplete adjuvant. Ascites fluids were screened and checked for the specificity of the antibody by enzyme-linked immunosorbent assay and flow cytometry on both HEK293-RhBG transfectants and parental HEK293 cells; ascites A-08 was selected for further studies. The specificity of this mouse polyclonal antibody for RhBG protein was further demonstrated by flow cytometry (Table 1), immunofluorescence (Fig. 2B), and immunoprecipitation studies (below under “Experimental Procedures” and Fig. 7). Peroxidase-conjugated anti-rabbit and anti-mouse IgG were provided by P.A.R.I.S. (Compiègne, France). Mouse anti-human ZO-1 was purchased from Zymed Laboratories Inc. (San Francisco, CA). The monoclonal anti-phosphotyrosine antibodies P-Tyr-100 and P-Tyr-102 were provided by Cell Signaling Technology and 4G10 by Upstate (Charlottesville, VA). Alexa Fluor anti-rabbit and anti-mouse IgG were from Invitrogen. Erk1+Erk2 rabbit polyclonal antibody was provided by Abcam (Cambridge, UK).
TABLE 1.
Cell surface expression and NH3 transport activity of C terminus mutants of RhBG
| Cell clones | Expressiona | Alkalinization rate constants | Transport efficiencyb |
|---|---|---|---|
| % | k(s–1) | % | |
| WTc | 0 | 0.22 ± 0.068 (14)d | 0 |
| RhBG | 100 | 1.38 ± 0.215 (14) | 100 |
| Emptye | 0 | 0.24 ± 0.025 (3) | 2 |
| F419A/L420A/D421A | 102 | 0.31 ± 0.087 (3) | 7 |
| S422A | 102 | 1.05 ± 0.081 (4) | 70 |
| S422D | 70 | 0.84 ± 0.078 (4) | 76 |
| S426A | 105 | 1.03 ± 0.072 (4) | 66 |
| S426D | 68 | 0.84 ± 0.065 (4) | 78 |
| Y429A | 113 | 1.31 ± 0.278 (3) | 83 |
| Y429F | 98 | 1.32 ± 0.110 (4) | 97 |
| Y429D | 115 | 0.28 ± 0.034 (3) | 5 |
| Y429E | 65 | 0.25 ± 0.028 (3) | 3 |
| 454Stop | 71 | 0.88 ± 0.059 (3) | 79 |
| T456A | 108 | 1.17 ± 0.221 (4) | 76 |
| T456D | 104 | 1.19 ± 0.255 (4) | 81 |
Relative values to MFI for RhBG
Relative values of alkalinization rate constants corrected for membrane expression of RhBG and after subtraction of the passive diffusion constant (WT)
Parental HEK293 cells
n values are in parentheses
HEK293 cells transfected with an empty pcDNA3 vector
FIGURE 2.
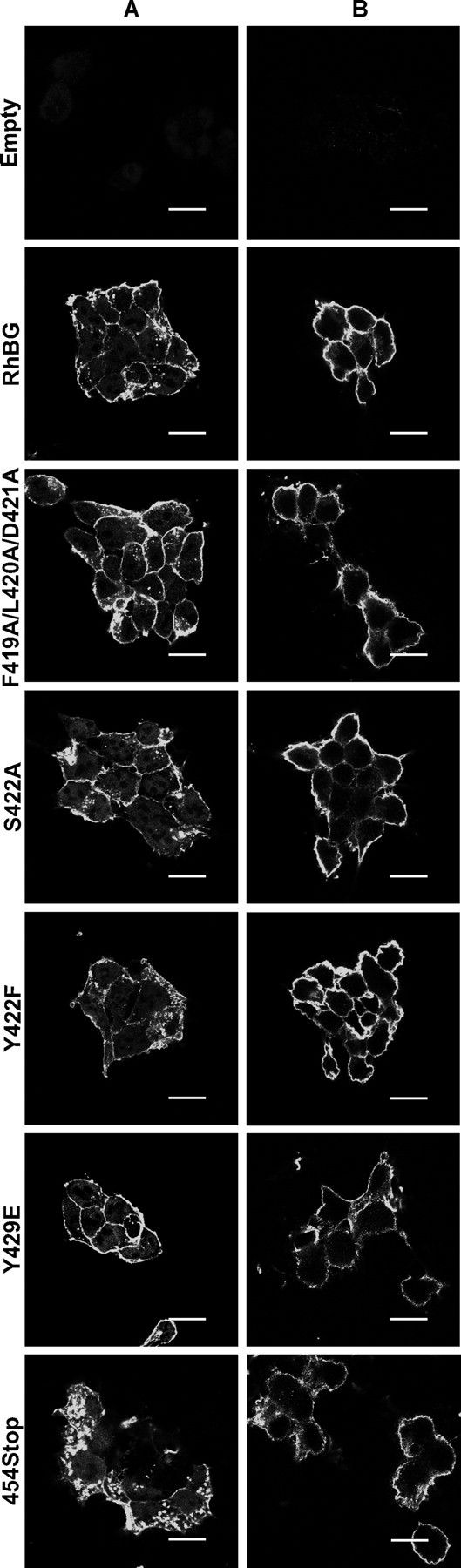
Immunofluorescence microscopy analysis of the expression of the RhBG C terminus mutants in HEK293 cells. HEK293 clones transfected with an empty pcDNA3 vector (Empty), or expressing wild-type (RhBG), or mutant RhBG were cultured on poly-l-lysine coverslips and fixed in 4% paraformaldehyde. A, cells were permeabilized in 1% SDS before staining with rabbit anti-RhBG-Cter and Alexa Fluor 488 goat anti-rabbit IgG. B, cells were directly labeled with murine ascites A-08 and Alexa Fluor 488 goat anti-mouse IgG. S422D, S426A, S426D, Y429A, Y429D, T456A, and T456D (not shown) exhibited similar plasma membrane and internal staining in A, and exclusive surface labeling in B. Scale bar, 15 μm.
FIGURE 7.

Delivery and turnover of RhBG, Y429F, Y429E, and F419A/L420A/D421A at the surface of HEK293 cells. HEK293 cells expressing RhBG mutants of Tyr429 or ankyrin-G binding site were pulse-chase-labeled ([35S]methionine/[35S]cysteine), and total membrane proteins were biotinylated and immunoprecipitated. A, newly delivered RhBG proteins at the cell surface were monitored by autoradiography of biotinylated proteins. Total amounts of RhBG proteins expressed at the plasma membrane at each time of chase were determined by Western blot using anti-RhBG A-08. B, the curves show the ratio of radiolabeled biotinylated RhBG/total biotinylated RhBG at the membrane reflecting the turnover of newly delivered native or mutant RhBG at the cell surface as a function of time. The curves represent mean values from three experiments. ♦, RhBG; ▪ Y429F; ⋄, Y429E; ▵, F419A/L420A/D421A.
Cell Culture, Transfection, Flow Cytometry, and Confocal Microscopy—Madin-Darby canine kidney (MDCK) cells, human embryonic kidney (HEK) 293 cells, and murine inner medullary collecting duct (mIMCD-3) cells were supplied by the American Type Culture Collection (Manassas, VA). MDCK cells were grown in Dulbecco's modified Eagle's medium/Glutamax I (Invitrogen), HEK293 and mIMCD-3 cells were grown in Iscove's modified Dulbecco's medium (Invitrogen), and all media were supplemented with 10% fetal calf serum (Dutscher, Brumath, France). Stable MDCK, HEK293, and mIMCD-3 cells expressing native or mutated RhBG proteins were obtained after transfection with the relevant expression vectors using Lipofectin reagent (Invitrogen) and selection in culture medium supplemented with 0.6 mg/ml (MDCK) or 0.8 mg/ml (HEK293) neomycin (Geneticin, Invitrogen), or 0.4 mg/ml hygromycin (Invitrogen) for mIMCD-3 cells. RhBG-positive cells were detected by flow-cytometry using the rabbit anti-RhBG-Cter polyclonal antibody after permeabilization of the cells, as described previously (31), or using the murine anti-RhBG ascites A-08 (1:200) without prior permeabilization and then cloned by limiting dilutions. At least three clones of each transfectant were grown and subsequently analyzed. Subconfluent (HEK293) or confluent (MDCK) monolayers of transfectants were cultured on poly-l-lysine coverslips or polycarbonate membranes, respectively, immunostained with the appropriate primary and Alexa Fluor secondary antibodies, and examined by confocal microscopy as described previously (31).
Stopped-flow Analysis—The ammonium transport function of wild-type RhBG and mutants was determined by stopped-flow spectrofluorometry analysis. Intracellular pH variation, using BCECF-AM (Sigma-Aldrich) as a pH-sensitive fluorescent probe, was measured in parental and transfected HEK293 cells exposed to inwardly directed 20 mmol of ammonium (NH+4) gradients at pH 7.2, as extensively described previously (18, 44). When indicated, pervanadate was added at 0.1 mm to culture medium 30 min before cell trypsinization and during each step before analysis with the stopped-flow instrument (SFM3, Bio-Logic, Grenoble, France).
Electrophoresis and Western Blot Analysis—SDS-PAGE was performed using 12.5% polyacrylamide gels according to Laemmli (45). Western blots were performed on nitrocellulose membranes, which then were stained with Ponceau Red (0.1%) and/or incubated with relevant primary antibodies followed with the appropriate peroxidase-conjugated secondary antibody (1:1000). Immunoblots were visualized using the ECL Plus Western blotting Detection System (Amersham Biosciences).
In Vitro Phosphorylation Assays—The GST fusion proteins expressed in E. coli BL21 and TKB1 were purified by elution from glutathione-Sepharose 4B beads (150 mm NaCl, 50 mm Tris-HCl, pH 8, 20 mm glutathione) and quantified by absorption at 280 nm. For in vitro phosphorylation with purified kinases, 15 μg of GST-RhBG-Cter fusion protein produced in BL21 cells and bound to glutathione-Sepharose 4B beads were mixed with 100 ng of Src or Syk kinase in the reaction buffer: 60 mm HEPES, pH 7.5, 5 mm MgCl2, 5 mm MnCl2, 1 mm DTT, 3 μm sodium orthovanadate, and incubated with 10 μCi of [γ-32P]ATP (200 μm) for 30 min at 37 °C. Beads were washed six times with phosphate-buffered saline (PBS) containing 0.05% Triton X-100 and then resuspended and boiled for 5 min in 1× Laemmli buffer. Samples were subjected to 12.5% SDS-PAGE and transferred to nitrocellulose membrane. Radioactive bands corresponding to GST-RhBG-Cter were visualized using a Bio Imaging Analyser BAS-1800 II (Fujifilm-Raytest, Courbevoie, France).
Protein Extraction From HEK293 or MDCK Cells and Immunoprecipitation—Wild-type or transfected cells were lysed for 1 h at 4 °C in lysis buffer (150 mm NaCl, 20 mm Tris-HCl, pH 8, 5 mm EDTA) containing complete protease inhibitor mixture and usually 1% Triton X-100, or variable amounts when indicated. Lysates were centrifuged at 15,000 × g for 15 min at 4 °C. Aliquots of lysates were mixed with 5× loading buffer (1.25 m sucrose, 20% SDS, 250 mm Tris-HCl, pH 6.8, 25% β-mercaptoethanol, 1% bromphenol blue) before electrophoresis. For RhBG immunoprecipitation, lysates were submitted to preclearing by incubation with protein A-Sepharose CL4B beads supplemented with 2% goat serum for 2 h at 4 °C and centrifuged at 1,000 × g for 5 min at 4 °C. Supernatants were incubated with 1 μg of purified rabbit anti-RhBG-Cter and protein A-Sepharose CL4B beads for 2 h at 4 °C. Beads were then washed three times with lysis buffer, and immunocomplexes were eluted in Laemmli buffer for 1 h at room temperature and mixed with loading buffer. Samples were electrophoresed in 12.5% SDS-PAGE gels and immunoblotted using murine ascites A-08 (1:5,000), which is directed against the extracellular loops of RhBG. The recognition of RhBG protein, immunoprecipitated by the anti-RhBG-Cter, actually demonstrated the specificity of ascites A-08 for RhBG.
Membrane Targeting Assays of RhBG Protein—Delivery of newly synthesized RhBG proteins to the membrane and their turnover were determined by performing a pulse-labeled biotin targeting assay. HEK293 cells expressing native or mutated RhBG protein were grown to confluency on 60-mm Petri dishes. Cells were washed twice with Dulbecco's modified Eagle's medium minus methionine and cysteine and then incubated for 30 min in the same medium supplemented with 10% fetal calf serum. Newly synthesized proteins were labeled by adding 150 μCi of [35S]methionine/[35S]cysteine (Invitrogen) in the culture medium for 10 min at 37 °C. Cells were washed and incubated in nonradioactive medium for 0.5, 1.5, 3.5, 6, 10, 13, and 17 h at 37 °C. After each time, cells were washed twice with cold PBS and incubated with 0.5 mg/ml nonpenetrating Sulfo-NHS-LC-biotin (Pierce) diluted in biotinylation buffer (10 mm Hepes, 150 mm NaCl, 0.2 mm CaCl2, 0.2 mm MgCl2, pH 7.5) for 30 min at 4 °C. After removal of biotin, cells were incubated with 10 mm glycine for 10 min at 4 °C and washed twice with cold PBS. Cell lysis and immunoprecipitation of RhBG proteins were performed as described above. The immunocomplexes were washed twice with lysis buffer, and one-fourth (200 μl) was eluted in Laemmli buffer for 1 h at room temperature (total RhBG proteins). The remaining three-fourths (600 μl) were eluted with 10% SDS for 30 min at room temperature and diluted in 500 μl of cold lysis buffer. The supernatant was incubated for 3 h at 4 °C with immobilized monomeric avidin (Pierce) to isolate membrane RhBG proteins. Beads were washed twice with lysis buffer, and proteins were eluted in Laemmli buffer for 1 h at room temperature. Eluates from both steps (total and membrane RhBG proteins) were electrophoresed by SDS-PAGE followed by immunoblotting using murine anti-RhBG A-08. Samples of radioactive biotinylated RhBG proteins were analyzed using the Bio Imaging Analyser to quantify the newly delivered membrane proteins as a function of time.
Domain-selective Biotinylation—Confluent MDCK monolayers of RhBG transfectants were grown on polycarbonate membranes for 7 days. Control cells were cultured in parallel under the same conditions and analyzed by confocal microscopy to check that they have formed a polarized monolayer. Cells were metabolically labeled with [35S]methionine/[35S]cysteine overnight at 37 °C and washed with cold PBS before incubating either the apical or the basolateral side with biotin as described above; the nonbiotinylated surface was incubated with PBS only. Filters were washed three times and excised. Cell lysis, immunoprecipitation, incubation with avidin, and analysis of biotinylated surface RhBG proteins were performed as described above.
RESULTS
Steady-state Cell Surface Expression of C Terminus Mutants of RhBG—As predicted in silico, the cytoplasmic C-terminal tail of RhBG (RhBG-Cter) contains four potential phosphorylation sites, Ser422, Ser426, Tyr429, and Thr456, and a potential type I PDZ-binding domain DTQA, including Thr456 at the extreme C terminus (Fig. 1). From pcDNA3-RhBG expression vector, we generated mutants encoding proteins that contain one alanine or one aspartic acid in place of either serine, tyrosine, and threonine residue, changes that block or mimic phosphorylation, respectively; Tyr429 was also replaced by phenylalanine or glutamic acid, which have the same effect as alanine and aspartic acid, respectively, and a mutant lacking the last four amino acids of the C terminus, 454Stop, was designed to remove the PDZ-binding sequence. The F419A/L420A/D421A triple mutant disrupting the ankyrin-G-binding site (Fig. 1) has been previously described (31). Human epithelial kidney cells HEK293 were stably transfected with pcDNA3 vectors bearing the different mutant constructs described above, and cell clones were obtained for each transfection. Flow cytometry of permeabilized cells using the previously described anti-RhBG-Cter rabbit polyclonal antibody (6) indicated that parental HEK293 or cells transfected with an empty pcDNA3 vector lacked endogenous expression of RhBG, whereas all different RhBG mutants were significantly expressed as compared with wild-type RhBG (mean fluorescence intensities (MFIs) = 78–180%, not shown). However, fluorescence microscopic analysis (Fig. 2A) revealed variable intracellular accumulation of RhBG in addition to a strong plasma membrane staining. For example, the 454Stop mutant exhibited a strong internal staining compared with wild-type RhBG-expressing cells that could account for the overexpression of the mutant protein (MFI = 180% of RhBG). Analysis of the impact of the different mutations on NH3 transport activity of RhBG required to compare the expression level only of cell surface proteins. For this purpose, we developed a new murine ascites A-08 directed against extracellular loops of RhBG (see “Experimental Procedures”). Flow cytometry analysis of intact cells using the A-08 ascites revealed cell surface expression levels of the mutated RhBG comprised between 65 and 113% as compared with wild-type RhBG (Table 1). Accordingly, fluorescence microscopic analysis using the A-08 ascites revealed strong staining of the plasma membrane of all nonpermeabilized cell clones (Fig. 2B). The absence of membrane labeling of cells transfected with an empty pcDNA3 vector (Empty) confirmed that RhBG protein was not expressed endogenously.
NH3 Transport Activity of RhBG Mutated on Potential Phosphorylation Sites—Because phosphorylation of transmembrane proteins is known to mediate regulation of their function, the NH3 transport capacity of RhBG mutated on potential phosphorylation sites was investigated. Stable recombinant HEK293 cells were loaded with the intracellular pH-sensitive probe BCECF-AM and analyzed by stopped-flow spectrofluorometry, as described previously (18). The time course of the fluorescence increase (corresponding to intracellular pH increase) of HEK293 cells exposed to inwardly directed 20 mmol of NH+4 gradients at pH 7.2 was recorded (Fig. 3) and allowed the calculation of alkalinization rate constants, which reflect the incidence of the different curve slopes. As shown in Table 1, the alkalinization process was ∼6–7 times faster in HEK293 cells expressing RhBG than in nontransfected parental (WT) cells or cells transfected with an empty pcDNA3 vector (rate constants were 1.38 ± 0.215 s-1 versus 0.22 ± 0.068 s-1 or 0.24 ± 0.025 s-1, respectively). Cells expressing S422A, S422D, S426A, S426D, Y429A, Y429F, T456A, and T456D mutants, exhibited slightly lower or similar rate constants compared with cells expressing RhBG. In contrast, cells expressing Y429D and Y429E mutants displayed drastically decreased alkalinization rate constants (0.28 ± 0.034 s-1 and 0.25 ± 0.028 s-1, respectively). The rate constants were corrected taking into account the relative surface expression level of each mutant protein as compared with RhBG and by subtracting the passive diffusion constant of parental cells (WT). The relative values reported in Table 1 indicated that all mutations of Ser422, Ser426, and Thr456 as well as mutations of Tyr429 into alanine or phenylalanine had little effect on the alkalinization rate (transport efficiencies between 66 and 97%, as compared with RhBG). Of note, the 454Stop mutation removing the potential PDZ-binding domain (which contains Thr456) did not significantly alter the NH3 transport capacity of RhBG and resulted in a transport efficiency identical to T456A and T456D mutants (79% versus 76 and 81%, respectively). Conversely, the Y429D and Y429E mutations reduced the rate of alkalinization to 5 and 3%, respectively, as compared with wild-type RhBG. These results indicate that, among the four potential phosphorylation sites present in the C terminus of RhBG, only mutations of Tyr429 into aspartic acid or glutamic acid (both mimicking constitutive phosphorylation) dramatically impaired the NH3 transport capacity of the protein. Thus, we focused our next study on this residue which appeared to be critical for RhBG function.
FIGURE 3.

Typical time course of fluorescence changes in HEK293 cells expressing RhBG C terminus mutants and exposed to ammonium gradients. Cells loaded with the fluorescent pH-sensitive probe BCECF-AM were exposed to an iso-osmotic inwardly directed 20 mEq NH+4 gradient at 15 °C. pHi-dependent fluorescence changes were monitored using a stopped-flow spectrofluorometer. Data from seven to nine time courses were averaged and fitted to a mono-exponential function using the simplex procedure of Biokine software (Bio-Logic), from which alkalinization rate constants (k, s-1) were calculated. WT, parental HEK293 cells. S422D, S426A, S426D, Y429F, 454Stop, T456A, and T456D (not shown) exhibited time courses similar to S422A and Y429A. The Y429D profile (not shown) resembled those of Y429E and F419A/L420A/D421A. All experiments were repeated at least three or four times to obtain the k values in Table 1.
Phosphorylation at Tyr429—We investigated whether the cytoplasmic C terminus of RhBG (Cter) can be phosphorylated at Tyr429. Recombinant fusion proteins, GST-Cter and GST-Cter Y429A, were first produced in TKB1 bacterial cells, which carry a tyrosine kinase gene whose expression allows the production of tyrosine-phosphorylated proteins, and analyzed by SDS-PAGE and immunoblotting. As shown in Fig. 4A (top panel), GST-Cter, but not GST alone, strongly reacted with the anti-phosphotyrosine P-Tyr-100 antibody. Similar results were obtained with two other anti-phosphotyrosine antibodies, P-Tyr-102 and 4G10 (not shown). The antibody labeling was abolished for the Y429A mutant, indicating that phosphorylation of the C terminus was specific of the tyrosine residue. We verified that similar amounts of fusion proteins were loaded on gels by Ponceau Red staining (Fig. 4A, bottom panel). Because Tyr429 is the target amino acid of a highly consensus sequence for both Src and Syk kinases, according to the Group-based Phosphorylation Scoring method, the same GST fusion proteins were then produced in BL21 cells and incubated with [γ-32P]ATP and purified Src or Syk. As shown in Fig. 4B (top panel), GST-Cter was clearly a substrate for both kinases in vitro, as opposed to GST alone. The absence of phosphorylation of the Y429A mutant demonstrated the specificity of enzyme reactions. Ponceau Red staining confirmed that equal amounts of proteins were used in all assays (Fig. 4B, bottom panel). To explore the impact of tyrosine phosphorylation on the activity of RhBG ex vivo, HEK293 cells expressing either wild-type or Y429A mutant RhBG were treated with an inhibitor of protein-tyrosine phosphatases, pervanadate, before monitoring their NH3 transport capacity. As shown in Fig. 4C, pervanadate-treated RhBG-expressing cells exhibited a 60% reduction of transport as compared with untreated cells. In contrast, pervanadate had no effect on NH3 transport when Tyr429 was mutated to alanine. The alteration by pervanadate of NH3 transport in RhBG-expressing cells corroborates the impairment of activity obtained with Y429D/E mutants (Table 1). Indeed, an inhibition of protein-tyrosine phosphatases mimics an activation of phosphorylation at tyrosine residue(s). Hence, the lack of transport inhibition by pervanadate observed for the Y429A mutant together with the deleterious effect of Y429D/E mutations demonstrated that specific phosphorylation of Tyr429 results in negative regulation of NH3 transport.
FIGURE 4.

Phosphorylation of RhBG at Tyr429. A, top panel, purified recombinant GST, GST-Cter, and GST-Cter Y429A were produced in E. coli TKB1. GST protein phosphorylation was analyzed by Western blot using anti-phosphotyrosine P-Tyr-100 antibody. Bottom panel, Ponceau Red staining of the different GST constructs. B, top panel, GST proteins were phosphorylated in the presence of [γ-32P]ATP by purified Src or Syk kinases. Phosphorylation was analyzed by autoradiography (32P). Bottom panel, Ponceau Red staining. C, effect of pervanadate on NH3 transport efficiency in HEK293 cells expressing RhBG or Y429A. Transport efficiency was expressed as relative values of alkalinization rate constants corrected for membrane expression of native RhBG, like in Table 1.
Membrane Targeting and Anchoring of RhBG Mutated on the Tyr429 Phosphorylation Site—We have previously identified Tyr429 as part of the YED basolateral sorting signal (Fig. 1) (31). To assess the influence of tyrosine phosphorylation on the polarized expression of RhBG, MDCK cells were stably transfected with the Y429A, Y429F, Y429D, or Y429E mutants. Confluent monolayers of clones expressing the different recombinant proteins were grown on permeable filters, allowed to polarize, and analyzed by confocal microscopy (Fig. 5A). Wild-type RhBG was localized at the basolateral membrane (XY horizontal and XZ sections, green), as compared with ZO-1 staining (red) representative of the tight junctions. The Y429A (not shown) and Y429F mutants exhibited a nonpolarized expression pattern at both apical and basolateral domains of the cells. This result is in accordance with our previous mapping, where each single Y429A, E430A, and D431A mutant of the targeting determinant was expressed at both domains of polarized MDCK cells (31). In contrast, Y429D (not shown) and Y429E mutants were properly localized on the basolateral side of the cells. Biotinylation and immunoprecipitation experiments on polarized monolayers of the same clones (Fig. 5B) revealed that RhBG (migrating with an apparent molecular mass of 50 kDa) and the Y429E mutant were expressed almost exclusively on the basolateral surface (89 and 91%, respectively). Expression of the Y429F mutant was nonpolar but remained predominant on the basolateral side of the cells (31 and 69% of the protein at the apical and basolateral surfaces, respectively), possibly because the Glu430 and Asp431 residues from the YED basolateral targeting signal were not altered. Indeed, we previously showed that the three residues of this motif are necessary and have additive effects on the targeting machinery (31). Y429D and Y429A mutants (not shown) exhibited a distribution similar to Y429E and Y429F, respectively. We also analyzed the expression of the S422A, S422D, S426A, S426D, T456A, T456D, and 454Stop C terminus mutants in polarized MDCK cells. All of them displayed a normal basolateral targeting of RhBG (not shown). These results indicated that replacement of Tyr429 from the YED motif by a negatively charged residue (Asp or Glu) mimicking constitutive phosphorylation resulted in normal polarity of RhBG expression in MDCK cells, whereas blocking phosphorylation by substitution of tyrosine with either small (alanine) or aromatic (phenylalanine) hydrophobic amino acids resulted in nonpolarized expression of RhBG.
FIGURE 5.

Targeting of Tyr429 mutants of RhBG in polarized MDCK cells. A, confocal microscopy sections. MDCK clones expressing wild-type (RhBG) or mutant RhBG were filter-grown for 7 days and processed for indirect immunofluorescence microscopy. Cells were fixed and permeabilized before adding rabbit anti-RhBG-Cter and mouse anti-ZO-1 antibodies. ZO-1 (zonula occludens) is a marker for tight junctions, which delimit apical and basolateral domains. Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat antimouse IgG were used as second antibodies, respectively. Top panels, XY horizontal middle sections (en face views) showing RhBG labeling in native and mutant clones. Bottom panels, XZ sections (side views) showing both RhBG (green) and ZO-1 (red) stainings. Y429A and Y429D (not shown) exhibited the same RhBG distribution as Y429F and Y429E, respectively. Scale bar, 15 μm. B, cell surface distribution detected by domain-selective biotinylation. Filter-grown MDCK cells were metabolically labeled with [35S]methionine/[35S]cysteine and surface biotinylated from either the apical (Ap) or the basolateral (Bl) side. After lysis, total RhBG proteins were immunoprecipitated using anti-RhBG-Cter, and biotinylated proteins were isolated with avidin beads. The biotinylated RhBG proteins were subjected to SDS-PAGE, electrotransferred to nitrocellulose membranes, and autoradiographed. Y429A (Ap 35%/Bl 65%) and Y429D (Ap 8%/Bl 92%) (not shown) displayed a distribution similar to Y429F and Y429E, respectively. Percentage values are averages from two independent experiments.
We have previously shown that binding to ankyrin-G through the 419FLD421 motif is necessary for membrane anchoring of RhBG to the basolateral domain of polarized MDCK cells (31). Because of the proximity of this motif with Tyr429, we investigated a potential involvement of this Tyr-phosphorylation site on the attachment of RhBG to the spectrin-based skeleton. HEK293 or polarized MDCK cells expressing wild-type RhBG or Tyr429 mutants were lysed in the presence of variable amounts of Triton X-100, and solubilized proteins were analyzed by electrophoresis and Western blot (Fig. 6, A and B, respectively). The F419A/L420A/D421A mutant that disrupts binding to ankyrin-G (and most probably the linkage to the underlying skeleton) was also analyzed as a control. RhBG migrates in SDS-PAGE as two bands with an estimated apparent molecular mass of 50–55 kDa (top panels), representing the N-glycosylated forms of the protein (6, 46). The amounts of solubilized RhBG protein in each lane were normalized to the amounts of the endogenous cytoplasmic Erk1 and Erk2 proteins (42–44 kDa, bottom panels). In HEK293 cells (Fig. 6A), wild-type RhBG and the Y429F mutant were progressively extracted from the skeleton with increasing concentrations of detergent to reach a peak at 0.9 and 1% Triton X-100, respectively. By contrast, Y429E and F419A/L420A/D421A mutants were equally solubilized from 0.2% to 1% Triton X-100. In polarized MDCK cell lines (Fig. 6B), both RhBG and Y429F were maximally extracted at the highest detergent concentration, and Y429E and F419A/L420A/D421A were readily solubilized in low amounts of Triton X-100 (0.3 and 0.2%). These data show that Y429E is solubilized like F419A/L420A/D421A in both HEK293 cells and polarized MDCK cells and suggest that phosphorylation at Tyr429 alters anchorage of RhBG to the spectrin-based skeleton. Of note, the removal of the potential PDZ-binding domain (454Stop mutant) did not modify the extractability of RhBG protein (not shown).
FIGURE 6.

Triton X-100 extractability of RhBG, Y429F, Y429E, and F419A/L420A/D421A from HEK293 and polarized MDCK cells. HEK293 (A) or polarized MDCK (B) cells expressing wild-type (RhBG) or mutant RhBG were lysed in the presence of increasing concentrations (%) of Triton X-100 as indicated. Total extracted proteins were analyzed by Western blot. Top panels, RhBG proteins (50–55 kDa) revealed using anti-RhBG A-08. Bottom panels, endogenous cytoplasmic Erk1 and Erk2 proteins (42–44 kDa).
Delivery and Turnover of Tyr429 RhBG Mutants at the Surface of HEK293 Cells—To further investigate whether phosphorylation at Tyr429 could play a role in other steps of RhBG surface expression, the delivery and turnover of RhBG proteins at the membrane of HEK293 cells was monitored using a pulse-chase experiment combined with biotinylation of surface proteins. All cell clones investigated expressed equivalent amounts of wild-type or mutant proteins, as determined by flow cytometry. The amounts of radiolabeled biotinylated RhBG proteins were normalized to the total biotinylated RhBG proteins detected by Western blot (Fig. 7A). Newly delivered RhBG proteins at the cell surface, after 10 min of radiolabeling, reached a peak after 1.5 h of chase (Fig. 7, A (Autoradiography) and B). RhBG protein displayed a great stability, because only one-third of the proteins was internalized after extended times of chase (17 h). Y429F and Y429E mutants exhibited a profile similar to the native protein. Therefore, the phosphorylation state of RhBG at position 429 had no effect on the delivery and turnover of RhBG at the surface of HEK293 cells. We intended to provide corroborating data from polarized MDCK epithelial cells. However, these cells expressed five times less RhBG proteins at the plasma membrane than HEK293 cells, as determined by flow cytometry (MFI = 170 versus 850, respectively). Moreover, polarized MDCK cells were grown on polycarbonate filters whose area is only 1 cm2, compared with the 28 cm2 of Petri dishes on which HEK293 cells were cultured for membrane targeting experiments. For these reasons, pulse-chase labeling of MDCK cells did not allow us to visualize the delivery and turnover of RhBG proteins at their surface.
NH3 Transport Activity and Membrane Delivery of RhBG Mutated on the Ankyrin-G-binding Site—Because the F419A/L420A/D421A mutant was clearly not tightly linked to the spectrin-based skeleton of both HEK293 and MDCK cells (see Fig. 6), we assessed its capacity to transport NH3 in the same experimental conditions as described above. The time course of intracellular pH increase (Fig. 3) and the deduced alkalinization rate constant (Table 1) showed a drastic inhibition of NH3 transport in F419A/L420A/D421A mutant compared with wild-type RhBG-expressing cells (0.31 ± 0.087 s-1 versus 1.38 ± 0.215 s-1). The relative value of the constant corrected for membrane expression gave a transport efficiency of 7%, as compared with native RhBG, which is equivalent to the transport efficiencies of Y429D and Y429E (5 and 3%). An intact ankyrin-G-binding motif is therefore necessary for a normal transport function of RhBG in HEK293 cells. Like for the Tyr429 mutants, we next studied the membrane delivery of the F419A/L420A/D421A mutant in HEK293 cells. As shown in Fig. 7, newly synthesized F419A/L420A/D421A proteins reached the plasma membrane more slowly than RhBG and Y429F/Y429E mutant proteins (6 versus 1.5 h). Moreover, F419A/L420A/D421A proteins were less stable, because their half-life at the cell surface was reached after ∼15 h of chase. Therefore, in contrast to the mutation of the Tyr429 phosphorylation site, an inhibition of interaction with ankyrin-G modified both surface delivery and stability of RhBG.
DISCUSSION
In this study, we demonstrated the functional importance of the cytoplasmic C terminus of RhBG. We showed that Tyr429 belonging to the YED basolateral targeting signal is involved in the regulation of NH3 transport capacity of RhBG. This residue is phosphorylatable, and the phosphorylated state of RhBG negatively regulates both its transport activity and its linkage to the spectrin-based skeleton in epithelial cell lines. Similarly, disruption of ankyrin-G binding via the FLD motif abolishes NH3 transport and is correlated with a dramatic decrease of anchorage to the skeleton. In contrast, phosphorylation at Tyr429 is necessary for the basolateral targeting of RhBG in polarized MDCK cells.
Both HEK293 and MDCK systems have been suitable for measurement of NH3 transport by stopped-flow spectrofluorometry analysis, ascertaining that transport data were not cell line-dependent but directly correlated to RhBG expression (18). However RhBG-transfected HEK293 cells displayed higher surface expression levels and transport activities than RhBG-transfected MDCK cells and, therefore, represented a more accurate model for the comparison of different mutants in the present study.
From the four potential phosphorylation sites located in the C terminus, only Tyr429 had a dramatic negative effect on NH3 transport when mutated in aspartic acid or glutamic acid, mimicking constitutive phosphorylation. All the other mutants displayed very moderate reduction of activity, between 3 and 34% compared with native RhBG. Moreover, this mild decrease of transport capacity was equivalent whether Ser422, Ser426, and Thr456 were changed into alanine or aspartic acid, strongly suggesting that phosphorylation of these residues, if occurs, would not modulate RhBG function. Therefore, we concentrated our work on Tyr429. We showed that Tyr429 was phosphorylatable both in TKB1 bacterial cells and in vitro by Src and Syk kinases. Tyrosine-based sorting motifs are very similar to SH2 (Src-homology 2) binding domains specific of numerous kinases, especially the Src and Syk families. Tyrosine-based signals have actually been shown to play a role in both trafficking events and, when tyrosine residue is phosphorylated, intracellular signaling events (47, 48). Regulation of membrane channel activity by tyrosine phosphorylation is well documented. For example, phosphorylation by Src kinase can either activate TRPC6 (49) and TRPC4 (50) or inhibit voltage-dependent calcium channels (51) and ROMK channels (52). Pervanadate treatment of RhBG-expressing HEK293 cells resulted in a 60% inhibition of NH3 transport. This limited reduction of activity, compared with the 95 and 97% inhibition for the Y429D and Y429E mutants, is probably due to a balance of phosphorylated/nonphosphorylated RhBG molecules at the surface of cells treated with pervanadate, whereas all of the Y429D/E molecules mimicked phosphorylated proteins. It is noteworthy that there are four other internal tyrosines located in the first, third, and fifth intracellular loops of the protein (1, 46). The lack of transport inhibition in the Y429A mutant incubated with pervanadate thus implies that Tyr429 is the only tyrosine residue of RhBG to be involved in the regulation of NH3 transport via pervanadate-sensitive phosphorylation. Moreover, these data indirectly showed that RhBG is actually phosphorylated ex vivo at Tyr429.
The F419A/L420A/D421A mutant of RhBG abolishing ankyrin-G binding also exhibited a dramatically reduced (93%) NH3 transport activity in HEK293 cells. The study of a mutation associated with Brugada syndrome, where a single amino acid change in a cytoplasmic loop of the cardiac sodium channel Nav1.5 (E1053K) disrupts its interaction with ankyrin-G, showed altered channel properties when this mutant was expressed in HEK293 cells (53). Likewise, ankyrin-G was shown to modulate gating properties of the neuronal sodium channel Nav1.6 in tsA201 cells, which are derived from HEK293 cells (54). We report here the first evidence that a mutation disrupting the interaction with ankyrin results in a nonfunctional gas channel in epithelial kidney cells. Recently, Lowe et al. (55) demonstrated that in cardiomyocytes lacking ankyrin-G, the few residual Nav1.5 channels that reached the plasma membrane had normal biophysical properties, pointing out the importance of studying ankyrin-membrane protein interactions in the physiological context of a native cell. Although we studied the function of human RhBG in human kidney epithelial cells, new measurements of NH3 transport in cell lines originating from connecting tubule or collecting duct, where RhBG is normally expressed (6, 56), should clarify this issue.
Both Y429D/E and F419A/L420A/D421A mutants were nonfunctional, although they were expressed at the surface of HEK293 cells. However, they behaved differently in membrane targeting assays. Although Y429E displayed the same profile of membrane delivery and turnover as native RhBG, F419A/L420A/D421A proteins reached the surface within a delay of 4.5 h (6 versus 1.5 h), and were internalized more rapidly. These data suggest that ankyrin-G may be involved in both targeting and turnover of RhBG at the plasma membrane of HEK293 cells, and an inhibition of interaction of the two proteins may slow down surface delivery of RhBG and decrease its stability. A number of reports described the requirement of ankyrin-G (53, 55, 57–59) or ankyrin-B (60–64) in the targeting and stability of their membrane protein partners, and we previously showed that expression of RhBG at the basolateral membrane of polarized MDCK cells was impaired when interaction with ankyrin-G was inhibited (31).
In nonpolarized cells, an ankyrin-binding mutant of RhBG can be expressed at the plasma membrane (this study and Ref. 31), however the mutant protein is not tightly linked to the skeleton. Different signals or a different hierarchy of signals present on the RhBG molecule could be involved in nonpolarized and polarized cells, as proposed by Toye et al. (65) to explain different localizations of renal AE1 (kAE1) in the two states of MDCK cells. Y429E and Y429F were normally delivered to the HEK293 cell surface, most probably because they were still interacting with ankyrin-G. However, only Y429F was linked to the skeleton as strongly as native RhBG, whereas Y429E was solubilized using the same low concentrations of Triton X-100 as for F419A/L420A/D421A. This result was unexpected, because membrane delivery experiments showed that targeting and turnover time courses of Y429E and F419A/L420A/D421A were clearly dissociated. We propose that the state of phosphorylation at Tyr429 does not influence membrane delivery of RhBG, but when the protein is phosphorylated, tethering to the underlying skeleton is altered, leading to NH3 transport inhibition, as for a mutant that cannot bind to ankyrin-G (F419A/L420A/D421A).
In polarized MDCK cells, Y429A/F mutants were expressed on both apical and basolateral domains, because the YED basolateral sorting motif (31) was altered, and Y429D/E mutants were exclusively targeted to the basolateral domain, like native RhBG. Moreover, F419A/L420A/D421A mutant was retained intracellularly (31). Solubilization experiments revealed the same patterns as for HEK293 cells: Y429F was tightly linked to the skeleton as RhBG, whereas Y429E and F419A/L420A/D421A were not anchored to it. These data corroborate our above proposal of a plasma membrane expression of RhBG that is independent of Tyr429 phosphorylation, except that for a correct basolateral localization in polarized cells, RhBG has to be phosphorylated on this residue. Moreover, like in HEK293, phosphorylated RhBG is not or poorly linked to the skeleton of polarized MDCK cells. Previous reports have shown that phosphorylation of the tyrosine in the FIGQY ankyrin-binding motif of the L1CAM family of proteins prevented the interaction of the two proteins (66, 67). Another report demonstrated that the association of sodium channel β1 subunits and ankyrin was mediated through a 16-amino acid sequence of the intracellular domain of β1 and that phosphorylation of a tyrosine residue located at an extremity of this segment abolished ankyrin recruitment (68). Tyr429 of RhBG is separated by 7 amino acids from the FLD ankyrin-binding domain (Fig. 1); however, its phosphorylation state could modulate the interaction of RhBG with ankyrin-G by a mechanism not elucidated yet.
The 454Stop mutant did not exhibit significantly altered NH3 transport capacity and was properly targeted to the membrane and anchored to the spectrin-based skeleton. A more detailed analysis should precise whether the C-terminal DTQA motif actually binds a PDZ protein and whether it could be involved in a specific targeting pathway. Indeed, the PDZ-binding domain of kAE1 has been proposed to function in a direct route of targeting to the plasma membrane, whereas the tyrosine-based motif may function as a basolateral/endosomal recycling signal (69).
Considering the overall data presented here, we propose a model of regulation of RhBG targeting and function by the cytoplasmic C terminus. In nonpolarized cells, RhBG is optimally delivered to the plasma membrane through its recruitment by ankyrin-G, whatever the phosphorylation state of Tyr429. Once at the membrane, RhBG is dephosphorylated to be anchored to the underlying spectrin-based skeleton via ankyrin-G and to become active in NH3 transport. In polarized epithelial cells, i.e. in more physiological conditions, phosphorylation of Tyr429 from the YED sorting signal is necessary for targeting RhBG to the basolateral membrane. Like in nonpolarized cells, dephosphorylation of the tyrosine residue allows tethering of the protein to the skeleton through ankyrin-G and most probably activation of channel function. A similar involvement of phosphorylation of the C-terminal tyrosine from the targeting motif has been proposed for the basolateral localization of kAE1 (69). However, there is up to now no evidence of an interaction of this protein with ankyrin-G in the kidney, although erythrocytic AE1 has been shown to bind ankyrin-R (27). Rh/RhAG/AE1/ankyrin-R are supposed to form a macrocomplex in red blood cells (29, 30), and we expect the existence of a similar RhBG/AE1/ankyrin-G macrocomplex in kidney epithelial cells (31, 70). Future investigations will hopefully answer this question and provide new insights on the possibility that ankyrins are likely to contribute to higher order organization of multiple channels and transporters, as it has been previously predicted for ankyrin-G in axon initial segments (57) and ankyrin-B in cardiomyocytes (71). Moreover, the C-terminal α-helix structure of the Rh homologue from N. europaea implying that its interaction with a protein partner could regulate channel opening (23) suggests that binding of ankyrin-G to the C terminus of RhBG might lead to NH3 transport activation. Further studies are needed to determine whether phosphorylation directly modulates the intrinsic NH3 transport capacity of RhBG and/or acts through interaction with ankyrin-G.
Acknowledgments
We are grateful to Drs. Wassim El Nemer and Emilie Gauthier for helpful advices on membrane targeting and in vitro phosphorylation assays, respectively. We acknowledge Pierre Gane and Julien Picot for skillful technical assistance in flow cytometric analysis.
This investigation was supported in part by the Institut National de la Transfusion Sanguine, INSERM, and the Université Paris Diderot-Paris 7. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Rh, Rhesus; Amt/Mep, ammonia transporters/methylammonium-ammonium permease; RhBG-Cter, cytoplasmic C terminus of RhBG; kAE1, kidney type 1 anion exchanger; PDZ, PSD-95 Discslarge ZO-1; ZO-1, zonula occludens-1; HEK, human embryonic kidney; MDCK, Madin-Darby canine kidney; mIMCD, murine inner medullary collecting duct; GST, glutathione S-transferase; BCECF-AM, 2′,7′-bis-(2-carboxyethyl)-5(6)-carboxyfluoresceinacetoxymethyl ester; MFI, mean fluorescence intensity; PBS, phosphate-buffered saline.
References
- 1.Huang, C. H., and Liu, P. Z. (2001) Blood Cells Mol. Dis. 2790 -101 [DOI] [PubMed] [Google Scholar]
- 2.Avent, N. D., and Reid, M. E. (2000) Blood 95375 -387 [PubMed] [Google Scholar]
- 3.Cartron, J. P. (1999) Baillieres Best Pract. Res. Clin. Haematol. 12655 -689 [DOI] [PubMed] [Google Scholar]
- 4.Van Kim, C. L., Colin, Y., and Cartron, J. P. (2006) Blood Rev. 2093 -110 [DOI] [PubMed] [Google Scholar]
- 5.Handlogten, M. E., Hong, S. P., Zhang, L., Vander, A. W., Steinbaum, M. L., Campbell-Thompson, M., and Weiner, I. D. (2005) Am. J. Physiol. 288G1036 -G1047 [DOI] [PubMed] [Google Scholar]
- 6.Quentin, F., Eladari, D., Cheval, L., Lopez, C., Goossens, D., Colin, Y., Cartron, J. P., Paillard, M., and Chambrey, R. (2003) J. Am. Soc. Nephrol. 14545 -554 [DOI] [PubMed] [Google Scholar]
- 7.Weiner, I. D., and Verlander, J. W. (2003) Acta Physiol. Scand. 179331 -338 [DOI] [PubMed] [Google Scholar]
- 8.Huang, C. H., and Peng, J. (2005) Proc. Natl. Acad. Sci. U. S. A. 10215512 -15517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marini, A. M., Urrestarazu, A., Beauwens, R., and Andre, B. (1997) Trends Biochem. Sci. 22 460-461 [DOI] [PubMed] [Google Scholar]
- 10.Bakouh, N., Benjelloun, F., Hulin, P., Brouillard, F., Edelman, A., Cherif-Zahar, B., and Planelles, G. (2004) J. Biol. Chem. 27915975 -15983 [DOI] [PubMed] [Google Scholar]
- 11.Benjelloun, F., Bakouh, N., Fritsch, J., Hulin, P., Lipecka, J., Edelman, A., Planelles, G., Thomas, S. R., and Cherif-Zahar, B. (2005) Pflugers Arch. 450155 -167 [DOI] [PubMed] [Google Scholar]
- 12.Ludewig, U. (2004) J. Physiol. 559751 -759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marini, A. M., Matassi, G., Raynal, V., Andre, B., Cartron, J. P., and Cherif-Zahar, B. (2000) Nat. Genet. 26 341-344 [DOI] [PubMed] [Google Scholar]
- 14.Mayer, M., Schaaf, G., Mouro, I., Lopez, C., Colin, Y., Neumann, P., Cartron, J. P., and Ludewig, U. (2006) J. Gen. Physiol. 127133 -144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakhoul, N. L., Dejong, H., Abdulnour-Nakhoul, S. M., Boulpaep, E. L., Hering-Smith, K., and Hamm, L. L. (2005) Am. J. Physiol. 288F170 -F181 [DOI] [PubMed] [Google Scholar]
- 16.Ripoche, P., Bertrand, O., Gane, P., Birkenmeier, C., Colin, Y., and Cartron, J. P. (2004) Proc. Natl. Acad. Sci. U. S. A. 10117222 -17227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Westhoff, C. M., Ferreri-Jacobia, M., Mak, D. O., and Foskett, J. K. (2002) J. Biol. Chem. 27712499 -12502 [DOI] [PubMed] [Google Scholar]
- 18.Zidi-Yahiaoui, N., Mouro-Chanteloup, I., D'Ambrosio, A. M., Lopez, C., Gane, P., Le van Kim, C., Cartron, J. P., Colin, Y., and Ripoche, P. (2005) Biochem. J. 391 33-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khademi, S., O'Connell, J., 3rd, Remis, J., Robles-Colmenares, Y., Miercke, L. J., and Stroud, R. M. (2004) Science 3051587 -1594 [DOI] [PubMed] [Google Scholar]
- 20.Zheng, L., Kostrewa, D., Berneche, S., Winkler, F. K., and Li, X. D. (2004) Proc. Natl. Acad. Sci. U. S. A. 10117090 -17095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callebaut, I., Dulin, F., Bertrand, O., Ripoche, P., Mouro, I., Colin, Y., Mornon, J. P., and Cartron, J. P. (2006) Transfus. Clin. Biol. 13 70-84 [DOI] [PubMed] [Google Scholar]
- 22.Conroy, M. J., Bullough, P. A., Merrick, M., and Avent, N. D. (2005) Br. J. Haematol. 131543 -551 [DOI] [PubMed] [Google Scholar]
- 23.Li, X., Jayachandran, S., Nguyen, H. H., and Chan, M. K. (2007) Proc. Natl. Acad. Sci. U. S. A. 10419279 -19284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lupo, D., Li, X. D., Durand, A., Tomizaki, T., Cherif-Zahar, B., Matassi, G., Merrick, M., and Winkler, F. K. (2007) Proc. Natl. Acad. Sci. U. S. A. 10419303 -19308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonifacino, J. S., and Dell'Angelica, E. C. (1999) J. Cell Biol. 145923 -926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellman, I. (1996) Annu. Rev. Cell Dev. Biol. 12575 -625 [DOI] [PubMed] [Google Scholar]
- 27.Bennett, V., and Baines, A. J. (2001) Physiol. Rev. 811353 -1392 [DOI] [PubMed] [Google Scholar]
- 28.Mohler, P. J., and Bennett, V. (2005) Front. Biosci. 102832 -2840 [DOI] [PubMed] [Google Scholar]
- 29.Nicolas, V., Le Van Kim, C., Gane, P., Birkenmeier, C., Cartron, J. P., Colin, Y., and Mouro-Chanteloup, I. (2003) J. Biol. Chem. 27825526 -25533 [DOI] [PubMed] [Google Scholar]
- 30.Bruce, L. J., Beckmann, R., Ribeiro, M. L., Peters, L. L., Chasis, J. A., Delaunay, J., Mohandas, N., Anstee, D. J., and Tanner, M. J. (2003) Blood 1014180 -4188 [DOI] [PubMed] [Google Scholar]
- 31.Lopez, C., Metral, S., Eladari, D., Drevensek, S., Gane, P., Chambrey, R., Bennett, V., Cartron, J. P., Le Van Kim, C., and Colin, Y. (2005) J. Biol. Chem. 2808221 -8228 [DOI] [PubMed] [Google Scholar]
- 32.Biswas, K., and Morschhauser, J. (2005) Mol. Microbiol. 56649 -669 [DOI] [PubMed] [Google Scholar]
- 33.Monahan, B. J., Unkles, S. E., Tsing, I. T., Kinghorn, J. R., Hynes, M. J., and Davis, M. A. (2002) Fungal Genet. Biol. 3635 -46 [DOI] [PubMed] [Google Scholar]
- 34.Ludewig, U., Wilken, S., Wu, B., Jost, W., Obrdlik, P., El Bakkoury, M., Marini, A. M., Andre, B., Hamacher, T., Boles, E., von Wiren, N., and Frommer, W. B. (2003) J. Biol. Chem. 27845603 -45610 [DOI] [PubMed] [Google Scholar]
- 35.Andrade, S. L., Dickmanns, A., Ficner, R., and Einsle, O. (2005) Proc. Natl. Acad. Sci. U. S. A. 10214994 -14999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loque, D., Lalonde, S., Looger, L. L., von Wiren, N., and Frommer, W. B. (2007) Nature 446195 -198 [DOI] [PubMed] [Google Scholar]
- 37.Neuhauser, B., Dynowski, M., Mayer, M., and Ludewig, U. (2007) Plant Physiol. 1431651 -1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheng, M., and Sala, C. (2001) Annu. Rev. Neurosci. 241 -29 [DOI] [PubMed] [Google Scholar]
- 39.Songyang, Z., Fanning, A. S., Fu, C., Xu, J., Marfatia, S. M., Chishti, A. H., Crompton, A., Chan, A. C., Anderson, J. M., and Cantley, L. C. (1997) Science 275 73-77 [DOI] [PubMed] [Google Scholar]
- 40.Hall, R. A., Premont, R. T., Chow, C. W., Blitzer, J. T., Pitcher, J. A., Claing, A., Stoffel, R. H., Barak, L. S., Shenolikar, S., Weinman, E. J., Grinstein, S., and Lefkowitz, R. J. (1998) Nature 392626 -630 [DOI] [PubMed] [Google Scholar]
- 41.Raghuram, V., Mak, D. O., and Foskett, J. K. (2001) Proc. Natl. Acad. Sci. U. S. A. 981300 -1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang, S., Yue, H., Derin, R. B., Guggino, W. B., and Li, M. (2000) Cell 103169 -179 [DOI] [PubMed] [Google Scholar]
- 43.Xia, Z., Gray, J. A., Compton-Toth, B. A., and Roth, B. L. (2003) J. Biol. Chem. 27821901 -21908 [DOI] [PubMed] [Google Scholar]
- 44.Zidi-Yahiaoui, N., Ripoche, P., Le Van Kim, C., Gane, P., D'Ambrosio, A. M., Cartron, J. P., Colin, Y., and Mouro-Chanteloup, I. (2006) Transfus. Clin. Biol. 13 128-131 [DOI] [PubMed] [Google Scholar]
- 45.Laemmli, U. K. (1970) Nature 227680 -685 [DOI] [PubMed] [Google Scholar]
- 46.Liu, Z., Peng, J., Mo, R., Hui, C., and Huang, C. H. (2001) J. Biol. Chem. 2761424 -1433 [DOI] [PubMed] [Google Scholar]
- 47.Stephens, D. J., and Banting, G. (1997) FEBS Lett. 41627 -29 [DOI] [PubMed] [Google Scholar]
- 48.Wu, H., Windmiller, D. A., Wang, L., and Backer, J. M. (2003) J. Biol. Chem. 27840425 -40428 [DOI] [PubMed] [Google Scholar]
- 49.Hisatsune, C., Kuroda, Y., Nakamura, K., Inoue, T., Nakamura, T., Michikawa, T., Mizutani, A., and Mikoshiba, K. (2004) J. Biol. Chem. 27918887 -18894 [DOI] [PubMed] [Google Scholar]
- 50.Odell, A. F., Scott, J. L., and Van Helden, D. F. (2005) J. Biol. Chem. 28037974 -37987 [DOI] [PubMed] [Google Scholar]
- 51.Vela, J., Perez-Millan, M. I., Becu-Villalobos, D., and Diaz-Torga, G. (2007) Am. J. Physiol. 293C951 -C959 [DOI] [PubMed] [Google Scholar]
- 52.Lin, D., Kamsteeg, E. J., Zhang, Y., Jin, Y., Sterling, H., Yue, P., Roos, M., Duffield, A., Spencer, J., Caplan, M., and Wang, W. H. (2008) J. Biol. Chem. 2837674 -7681 [DOI] [PubMed] [Google Scholar]
- 53.Mohler, P. J., Rivolta, I., Napolitano, C., LeMaillet, G., Lambert, S., Priori, S. G., and Bennett, V. (2004) Proc. Natl. Acad. Sci. U. S. A. 10117533 -17538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shirahata, E., Iwasaki, H., Takagi, M., Lin, C., Bennett, V., Okamura, Y., and Hayasaka, K. (2006) J. Neurophysiol. 961347 -1357 [DOI] [PubMed] [Google Scholar]
- 55.Lowe, J. S., Palygin, O., Bhasin, N., Hund, T. J., Boyden, P. A., Shibata, E., Anderson, M. E., and Mohler, P. J. (2008) J. Cell Biol. 180173 -186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Verlander, J. W., Miller, R. T., Frank, A. E., Royaux, I. E., Kim, Y. H., and Weiner, I. D. (2003) Am. J. Physiol. 284F323 -F337 [DOI] [PubMed] [Google Scholar]
- 57.Jenkins, S. M., and Bennett, V. (2001) J. Cell Biol. 155739 -746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan, Z., Kao, T., Horvath, Z., Lemos, J., Sul, J. Y., Cranstoun, S. D., Bennett, V., Scherer, S. S., and Cooper, E. C. (2006) J. Neurosci. 262599 -2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou, D., Lambert, S., Malen, P. L., Carpenter, S., Boland, L. M., and Bennett, V. (1998) J. Cell Biol. 1431295 -1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cunha, S. R., Bhasin, N., and Mohler, P. J. (2007) J. Biol. Chem. 2824875 -4883 [DOI] [PubMed] [Google Scholar]
- 61.Mohler, P. J., Davis, J. Q., Davis, L. H., Hoffman, J. A., Michaely, P., and Bennett, V. (2004) J. Biol. Chem. 27912980 -12987 [DOI] [PubMed] [Google Scholar]
- 62.Mohler, P. J., Gramolini, A. O., and Bennett, V. (2002) J. Biol. Chem. 27710599 -10607 [DOI] [PubMed] [Google Scholar]
- 63.Scotland, P., Zhou, D., Benveniste, H., and Bennett, V. (1998) J. Cell Biol. 1431305 -1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tuvia, S., Buhusi, M., Davis, L., Reedy, M., and Bennett, V. (1999) J. Cell Biol. 147995 -1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toye, A. M., Banting, G., and Tanner, M. J. (2004) J. Cell Sci. 1171399 -1410 [DOI] [PubMed] [Google Scholar]
- 66.Garver, T. D., Ren, Q., Tuvia, S., and Bennett, V. (1997) J. Cell Biol. 137703 -714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tuvia, S., Garver, T. D., and Bennett, V. (1997) Proc. Natl. Acad. Sci. U. S. A. 9412957 -12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malhotra, J. D., Koopmann, M. C., Kazen-Gillespie, K. A., Fettman, N., Hortsch, M., and Isom, L. L. (2002) J. Biol. Chem. 27726681 -26688 [DOI] [PubMed] [Google Scholar]
- 69.Toye, A. M. (2005) Biochem. Soc. Symp. 7247 -63 [DOI] [PubMed] [Google Scholar]
- 70.Nicolas, V., Mouro-Chanteloup, I., Lopez, C., Gane, P., Gimm, A., Mohandas, N., Cartron, J. P., Le Van Kim, C., and Colin, Y. (2006) Transfus. Clin. Biol. 13 23-28 [DOI] [PubMed] [Google Scholar]
- 71.Mohler, P. J., Davis, J. Q., and Bennett, V. (2005) PLoS Biol. 3e423. [DOI] [PMC free article] [PubMed] [Google Scholar]