

Abstract
The meso-diaminopimelate decarboxylase (DAPDC, EC 4.1.1.20) catalyzes the final step of l-lysine biosynthesis in bacteria and is regarded as a target for the discovery of antibiotics. Here we report the 2.3Å crystal structure of DAPDC from Helicobacter pylori (HpDAPDC). The structure, in which the product l-lysine forms a Schiff base with the cofactor pyridoxal 5′-phosphate, provides structural insight into the substrate specificity and catalytic mechanism of the enzyme, and implies that the carboxyl to be cleaved locates at the si face of the cofactor. To our knowledge, this might be the first reported external aldimine of DAPDC. Moreover, the active site loop of HpDAPDC is in a “down” conformation and shields the ligand from solvent. Mutations of Ile148 from the loop greatly impaired the catalytic efficiency. Combining the structural analysis of the I148L mutant, we hypothesize that HpDAPDC adopts an induced-fit catalytic mechanism in which this loop cycles through “down” and “up” conformations to stabilize intermediates and release product, respectively. Our work is expected to provide clues for designing specific inhibitors of DAPDC.
l-Lysine is an essential amino acid in the bacterial life cycle. In addition to its fundamental role in protein synthesis, l-lysine is also a basic building block of bacterial cell wall peptidoglycan. The biosynthesis of l-lysine in bacteria involves a multistep pathway (1, 2) transforming l-aspartate to a common precursor meso-diaminopimelate (DAP).4 DAP is then decarboxylated at the d-stereocenter by meso-diaminopimelate decarboxylase (DAPDC) encoded by lysA gene to give the final product l-lysine. Thus, DAPDC acts as a key enzyme in this pathway and is of great importance to bacterial growth and survival. De Lancastre et al. (3) have demonstrated that abolishing the function of DAPDC in methicillin-resistant Staphylococcus aureus by transposon insertion could reduce the level of the methicillin resistance of methicillin-resistant S. aureus by nearly 100-fold; and Gokulan et al. (13) have established that the lysA gene of Mycobacterium tuberculosis was essential for in vivo bacterial viability in immunocompromised mice. In contrast, instead of synthesizing l-lysine de novo, the human body obtains it from food, and no counterpart of DAPDC is found in mammals. Therefore, DAPDC could be a potential target for developing novel antibiotics.
DAPDC is a group IV pyridoxal 5′-phosphate (PLP)-dependent enzyme (also known as alanine racemase (ALR) family) (5). Except ALR, all enzymes of this family are basic amino acid decarboxylases (6-8), including DAPDC, ornithine decarboxylase (ODC), arginine decarboxylase (ADC), decarboxylases with dual specificity for l-lysine and l-ornithine (l/ODC), and carboxynorspermidine decarboxylases. Reactions catalyzed by representative enzymes are shown in Fig. 1a. Crystal structures of these group IV PLP enzymes (except carboxynorspermidine decarboxylases) (6, 9-12) reveal that although the family members do not share high sequence identities (only 5% between DAPDC and ALR), they adopt a similar fold consisting of an N-terminal classical α/β-barrel and a C-terminal β-strand domain, and all exist as an obligate homodimer with two active sites lining the dimer interface.
FIGURE 1.

Reactions catalyzed by representative group IV PLP-dependent decarboxylases (a) and overall structure of HpDAPDC (b-d). b, monomer structure. The active site loop is colored in green. c, dimer structure. Different protomers are colored white and magenta, respectively. The active site loops and the ligands are shown as ribbon and sticks, respectively. d, electron density around PLP and the bound l-lysine.
To date, the crystal structures of DAPDC from Methanococcus jannaschii (MjDAPD, PDB codes 1TUF and 1TWI) and M. tuberculosis (MtDAPDC, PDB codes 1HKV, 1HKW, and 2O0T) (4, 13) have been reported, and several other DAPDC structures (DAPDC from Aquifex aeolicus (AaDAPDC, PDB code 2P3E) and Escherichia coli (EcDAPDC, PDB codes: 1KO0 and 1KNW) (14)) were released in the Protein Data Bank. Some detailed description of cofactor and substrate binding was supplied in the reported structures. However, because of the inconsistent binding patterns of the substrate analogues (lysine or azelaic acid), the complete set of substrate-recognizing residues of DAPDC still remains uncertain.
Here we report the 2.3 Å crystal structure of DAPDC from Helicobacter pylori (HpDAPDC), a severe clinical pathogen causing peptic ulcer and gastric cancer (15-17). In the structure, the product l-lysine is covalently bonded to cofactor PLP, and the resultant external aldimine is believed to closely mimic the catalytic intermediate formed between DAP and PLP during the decarboxylation reaction. Detailed information about substrate binding is then derived and carefully compared with DAPDCs from other species as well as other group IV PLP enzymes (ALR, ODC, and ADC). In addition, structural analysis also implied the putative catalytic mechanism of DAPDC, especially the position and recognition of the carboxyl group to be cleaved. Moreover, a loop covering the active site of HpDAPDC is in a “down” conformation and shields the ligand from the solvent. Mutations of Ile148 from this loop greatly impaired the catalytic efficiency, especially the kcat value of the enzyme. Combining the crystal structure of the I148L mutant and the results from previous studies, we propose that HpDAPDC adopts an induced-fit mechanism for catalysis in which the active site loop cycles through down and “up” conformations to stabilize intermediates and release product, respectively. Our work is expected to gain insight into the essential catalytic mechanism of HpDAPDC and provide clues for designing specific inhibitors.
EXPERIMENTAL PROCEDURES
Cloning of lysA Gene from H. pylori—Based on the genome sequences of H. pylori strains 26695 and J99 (GenBank™ accession numbers NC_000915 and NC_000921), two PCR primers, 5′-TGTATGTGAATGCGGGGGTGG-3′ (sense) and 5′-GGGGCTTTCTTGTTTGATGAG-3′ (antisense), were designed to amplify the corresponding region, including the lysA gene from the genome of H. pylori strain SS1 (obtained from Shanghai Institute of Digestive Disease). According to the sequencing result of the amplified DNA segment, another pair of primers, 5′-TGGGCATATGTTTAATTATGAAGAGC-3′ (sense) and 5′-TTTCTCGAGTCAAACCCCTTTTAAGCC-3′ (antisense), was synthesized to amplify lysA gene from the genome again. The PCR product was cloned into the NdeI and XhoI sites of pET-28a (Novagen) to produce N-terminal His-tagged protein. The gene sequence has been submitted to GenBank™ with accession number EU056336.
Cloning of lysA Gene from E. coli—Similar to the cloning procedures of pET28a-HpDAPDC, the lysA gene of E. coli was cloned from the genome of E. coli strain JM109 based on the sequence of strain K12 (GenBank™ accession number NC_000913). The gene was amplified by PCR with the primers 5′-ATATGGATCCATGCCACATTCACTGTTCAG-3′ (sense) and 5′-ATATCTCGAGTTAAAGCAATTCCAGCG-3′ (antisense) and cloned into the BamHI and XhoI sites of pET28a.
Site-directed Mutagenesis—Six single point mutants of HpDAPDC (I148L, I148F, I148A, I148K, I148D, and I148G) were constructed with Quikchange site-directed mutagenesis kit (Stratagene) following the instruction manual and verified by sequencing. The PCR primers containing the desired single mutations (in boldface) are as follows (only sense primers are shown): 5′-CTAAAACGCACCCCTATCTTTCTACCGGCTTGAAAG-3′ (I148L), 5′-GACGCTAAAACGCACCCCTATTTTTCTACCGGCTTGAAAG-3′ (I148F), 5′-CTAAAACGCACCCCTATGCTTCTACCGGCTTGAAAG-3′ (I148A), 5′-CTAAAACGCACCCCTATAAGTCTACCGGCTTGAAAG-3′ (I148K), 5′-CTAAAACGCACCCCTATGATTCTACCGGCTTGAAAG-3′ (I148D), and 5′-CTAAAACGCACCCCTATGGTTCTACCGGCTTGAAAG-3′ (I148G).
Expression and Purification of HpDAPDC and EcDAPDC—The recombinant plasmid pET28a-HpDAPDC was transformed into E. coli strain BL21 (DE3) for expression. The transformed cells were grown in LB media supplemented with 50 μg/ml kanamycin at 37 °C. When A600 reached 0.6, the culture was induced with 0.4 mm isopropyl β-d-thiogalactopyranoside and incubated at 25 °C for an additional 5 h. The cells were harvested by centrifugation at 6000 × g for 15 min at 4 °C and resuspended in buffer A (20 mm MES, pH 6.0, 500 mm NaCl, 200 μm PLP, and 10 mm imidazole). The mixture was sonicated on ice for 30 min and centrifuged at 16,000 × g for 45 min at 4 °C. The supernatant was loaded onto a column filled with nickel-nitrilotriacetic acid resin (Qiagen) pre-equilibrated in buffer A. The column was washed with 100 ml of buffer B (20 mm MES, pH 6.0, 500 mm NaCl, and 20 mm imidazole) and then eluted with 10 ml of buffer C (20 mm MES, pH 6.0, 500 mm NaCl, and 500 mm imidazole). The eluent was dialyzed against buffer D (20 mm MES, pH 6.0, 200 μm PLP, and 150 mm NaCl) for subsequent enzymatic assay and crystallization. Same protocols were used for the expression and purification of EcDAPDC and HpDAPDC mutants. Protein concentration was determined by Bradford assay using bovine serum albumin as the standard.
Enzymatic Assay—The enzymatic activities of HpDAPDC and EcDAPDC were measured by using DAPDC/saccharopine dehydrogenase (EC 1.5.1.7)-coupled spectrophotometric assay system as reported before (18). In brief, 100 μl of reaction buffer containing 100 mm Tris, pH 8.0, 10 mm α-ketoglutarate, 0.31 mg/ml NADH (A340 = 1.0), 10 μm saccharopine dehydrogenase, 10 μm PLP, and DAPDC (50-8000 nm, depending on the catalytic efficiency of the enzyme) was added to the 96-well plate, and then 100 μl of DAP solution at desired concentration was supplemented to start the reaction. The initial velocity of the reaction was reflected by the rate of NADH consumption monitored by a Benchmark Plus Microplate spectrophotometer from Bio-Rad. The kinetic constants were obtained by Lineweaver-Burk double-reciprocal plots.
Crystallization—All crystallization experiments were performed by hanging drop vapor diffusion method at 4 °C. Initial crystallization trials of apo-HpDAPDC failed. Thus, 10 mg/ml HpDAPDC solution was incubated with 5 mm l-lysine on ice overnight, and then 1 μl of the complex was mixed with 1 μl of the mother liquor containing 100 mm Bicine, pH 8.0, and 1.6 m ammonium sulfate, and equilibrated with 500 μl of the same solution. Crystals grew to 0.1 × 0.1 × 0.2 mm after 1 week. The crystals of I148L were obtained in the same condition as the wild type.
Data Collection and Structure Determination—The crystals of HpDAPDC were cryoprotected in the mother liquor supplemented with 30% glycerol and flash-frozen in liquid nitrogen. Diffraction data were collected in-house at -180 °C from a Rigaku rotating-anode x-ray generator (wavelength 1.5418 Å) operated at 100 mA and 100 kV. Images were recorded by a Rigaku R-Axis IV++ image plate with an oscillation step of 1°. The data from wild-type HpDAPDC crystal was processed by CrystalClear software from Rigaku. The data from I148L crystal was indexed and integrated by Mosflm (19) and scaled by Scala (20). Statistics of the diffraction data are summarized in Table 1.
TABLE 1.
Statistics of diffraction data and structure refinement
| Wild type | I148L | |
|---|---|---|
| Data collection | ||
| Space group | P3221 | P3221 |
| Cell dimensions | ||
| a, b, c (Å) | 90, 90, 120 | 90, 90, 120 |
| α, β, γ (°) | 79.2, 79.2, 134.3 | 79.1, 79.1, 134.8 |
| Resolution (Å) | 20.0-2.30 (2.38-2.30)a | 15.0-2.40 (2.49-2.40) |
| Rsym or Rmergeb | 0.128 (0.390) | 0.140 (0.373) |
| I/σI | 5.1 (1.9) | 4.9 (1.9) |
| Completeness (%) | 97.0 (97.0) | 98.2 (98.2) |
|
Redundancy
|
7.1 (7.2)
|
7.3 (7.5)
|
| Refinement | ||
| Resolution (Å) | 15.0-2.30 | 15.0-2.40 |
| No. of reflections | 21,499 | 19,161 |
| Rwork/Rfreec | 0.206/0.267 | 0.211/0.257 |
| No. of atoms | 3254 | 3254 |
| Protein | 3102 | 3102 |
| Ligand/ion | 31 | 31 |
| Water | 121 | 121 |
| B-factors (Å2) | 27.23 | 24.37 |
| Protein | 27.27 | 24.41 |
| Ligand/ion | 22.97 | 19.13 |
| Water | 27.20 | 24.57 |
| R.m.s.d. | ||
| Bond lengths (Å) | 0.007 | 0.007 |
|
Bond angles (°)
|
1.282
|
1.304
|
| Ramachandran plot (%) | ||
| Most favored regions | 90.8 | 88.2 |
| Allowed regions | 9.2 | 11.8 |
| Generously allowed regions | 0 | 0 |
| Disallowed regions | 0 | 0 |
Values in parentheses are for highest resolution shell.
Rsym or Rmerge = ∑h∑i|Ihi - 〈Ih〉|/∑h∑iIhi, where Ihi and <Ih> are the i-th and mean measurement of the intensity of reflection h, respectively.
Rwork/Rfree = ∑h|Fo.h - Fc.h|/∑hFo.h, where Fo.h and Fc.h are the observed and calculated structure factor amplitudes, respectively.
The structure of HpDAPDC was solved by molecular replacement with Molrep (21) using the monomer structure of MjDAPDC (PDB code 1TUF, chain A) as the search model. Structure refinement was carried by CNS (22) using standard protocols (simulated annealing, energy minimization, and B-factor refinement). Model building was performed in O (23) and Coot (24). The quality of the final model was checked by PROCHECK (25). The statistics of structure refinement are summarized in Table 1. The coordinates of the wild type and I148L HpDAPDC have been deposited in Protein Data Bank with accession numbers of 2QGH and 3C5Q, respectively. Structural superpositions were calculated with LSQKAB (26) from the CCP4 software suite (27). All structure figures were prepared by PyMOL (28).
RESULTS AND DISCUSSION
Overall Structure—As a classic group IV PLP enzyme, HpDAPDC consists of an N-terminal 8-fold α/β-barrel and a C-terminal β-strand domain (Fig. 1b). A dimer of HpDAPDC could be reconstituted by crystallographic 2-fold symmetry (Fig. 1c). The two protomers adhere to each other in a “head-to-tail” manner burying about 19% (3533 Å2) of their surface area, confirming that the functional unit of DAPDC is an obligate dimer (13). The PLP molecule together with the covalently bonded l-lysine are positioned unambiguously in the well defined electron density map (Fig. 1d), indicating the location of the active site. Two catalytic pockets line at the dimer interface, each formed by a cavity from the N-terminal domain of one protomer and a protruded surface from the C-terminal domain of the other protomer.
The overall folding of HpDAPDC is similar to other known DAPDC structures with small r.m.s.d. values superposing either monomeric or dimeric forms (Table 2). However, the N terminus of HpDAPDC is substantially shorter and lacks the secondary structure elements usually found at this terminus (4, 13). Besides, the active pocket of HpDAPDC is covered by a loop and completely shielded from the solvent (Fig. 1c). Structural superposition of HpDAPDC with other typical group IV PLP enzymes (ornithine decarboxylase from Trypanosoma brucei (TbODC) (11, 29-31), arginine decarboxylase from Paramecium bursaria chlorella virus (cvADC) (12), and alanine racemase from Bacillus stearothermophilus (BsALR) (9, 32)) reveals that the HpDAPDC is more structurally similar to ADC and ODC than to ALR (Table 2), especially in the strand pattern and orientation of the C-terminal domain.
TABLE 2.
r.m.s.d. values (Å) from superposing various structures with HpDAPDC
The protomer and dimer of HpDAPDC contain 394 and 788 residues, respectively.
| Protomer | Aligned C-α | Dimer | Aligned C-α | |
|---|---|---|---|---|
| AeDAPDC (2P3Ea, 398/801b) | 1.21 | 370 | 1.34 | 735 |
| EcDAPDC (1KO0, 419/838) | 1.55 | 364 | 1.64 | 728 |
| MtDAPDC (1HKV, 447/894) | 1.64 | 374 | 1.64 | 743 |
| MjDAPDC (1TUF, 434/868) | 1.32 | 376 | 1.37 | 751 |
| TbODC (1F3T, 378/759) | 1.98 | 339 | 2.04 | 679 |
| CvADC (2NVA, 369/738) | 1.83 | 342 | 1.91 | 681 |
| BsALR (1BD0, 381/761) | 3.12 | 252 | 4.87 | 525 |
| I148L (3C5Q, 394/788) | 0.15 | 394 | 0.16 | 788 |
PDB code of the structure.
Number of residues in protomer and dimer.
PLP-binding Site—The cofactor PLP binds in a deep cavity at the mouth of the N-terminal α/β-barrel (Fig. 1b) with nearly all contacting residues from the parental protomer (Fig. 2a). The pyridoxal ring is stacked in a shallow crevice, and the phosphate moiety is inserted in a highly negatively charged hole (Fig. 2c, upper panel). The si face of the pyridoxal ring is packed against His185, which is conserved among all group IV PLP enzymes (alignment not shown). The decarboxylation of DAP is believed to happen at this side of PLP (13). The phenolate oxygen (O-3) on the PLP ring is recognized by the highly conserved residues Arg135 and Cys329. The interaction between the PLP pyridinium nitrogen and the two acidic residues Asp65 and Glu259 is thought to keep PLP protonated to enhance its “electron sink” nature (7). Upon the formation of the l-lysine-PLP external aldimine, Lys46 that originally makes Schiff base with the cofactor (internal aldimine) now points away from PLP and is stabilized by Asp65. A similar ion pair between a broken Schiff base lysine and an asparagine residue is also observed in ALR (Lys39-Asp313) (9, 33), which is believed to be a naturally occurring conformation during catalysis.
FIGURE 2.

Active site structure of HpDAPDC. a, PLP-binding site of HpDAPDC. Oxygen, nitrogen, phosphorus, and sulfur atoms are colored red, blue, orange, and dark yellow, respectively, and hydrogen bonds are presented as dotted lines (same below). The carbons of ligand and protein are colored cyan and yellow, respectively. b, substrate-binding site of HpDAPDC. The color scheme is the same as a except that the residues from another protomer (with letter “B” in their names) are colored green. c, electrostatic potential distribution on the active site inner walls, colored red to blue representing -20 e/kT to +20 e/kT. The potential is calculated by Adaptive Poisson-Boltzmann Solver (42). d, active pocket viewed by looking into the interior of HpDAPDC dimer. The substrate DAP is modeled into the pocket (lower panel) by being superposed onto the bound l-lysine (upper panel). The white circles denote the space to accommodate the d-stereocenter carboxyl of DAP.
The phosphate group of PLP makes numerous hydrogen bonds, either directly or via water molecules, with residues from conserved motifs, including HIGS (residues 185-188), the glycine-rich region (residues 224-226), EPGRS (residues 259-263), and GAY (residues 356-358) motifs (Fig. 2a). Notably, two PLP-binding residues (Arg262 and Tyr358) are also involved in recognizing the l-stereocenter of the substrate.
Substrate-binding Site—The external aldimine conformation of HpDAPDC provides valuable structural information on the substrate binding and the catalytic mechanism of DAPDC.
General Description—The substrate DAP is a meso compound with two stereocenters, namely the l-stereocenter and the d-stereocenter (l-center and d-center for short). DAPDC specifically cleaves the d-center carboxyl to give the product l-lysine. This specificity comes from both the recognition of the d-center and the proper anchoring of the l-center in the enzyme. Therefore, we will dissect the substrate DAP into three parts, the aliphatic chain and the two stereocenters, and describe their interactions with the enzyme separately.
The aliphatic chain of the l-lysine is sandwiched between the side chains of Ile148 and Tyr358 (Fig. 2b), which constitute the “ceiling” and “floor” of the active pocket, respectively. The ceiling, Ile148, is in a small 310 helix α9 from the active site loop and is nonconserved among DAPDCs. The floor, Tyr358, is highly conserved and plays a dual role in both substrate recognition and PLP binding. Besides the sandwich structure, the alkyl moiety of the side-chain of Glu330B (“B” denotes the residue from another protomer, same below) also makes hydrophobic interaction with the aliphatic chain of l-lysine (Fig. 2b).
The l-center of the l-lysine is wrapped by several charged and polar residues electrostatically complementary with the carboxyl and amine groups of the substrate. In detail, Arg262 and Arg298 line beside the l-center carboxyl with their side-chain nitrogens salt-bridging to the carboxyl oxygens (Fig. 2b). These two basic residues constitute a positively charged inner wall at one side of the active pocket (Fig. 2c, upper panel). The other side of the pocket, however, is mainly negative (Fig. 2c, lower panel), composed of Tyr302, Tyr366B, and Glu330B, which hydrogen bond to the l-center amine of the substrate (Fig. 2b). This side of the pocket is less charged than the opposite side (Fig. 2c), because part of the side-chain of Glu330B is hydrophobically interacting with the aliphatic chain of l-lysine. All residues mentioned above are absolutely conserved among all known DAPDC sequences, indicating their significance in substrate binding. Notably, Arg298 and Tyr302 come from a small helix α14 referred to as the “specific element” of group IV PLP-dependent decarboxylases. The position of this helix varies across enzymes to accommodate substrates of different sizes (6, 12). Besides this specificity element, we believe the two oppositely charged inner walls of the active site of the DAPDC also determine the specificity of the enzyme for DAP, because other group IV PLP-dependent decarboxylases carry only negative charge at the corresponding area. Thus, this distinct property of DAPDC might offer a chance to design specific DAPDC inhibitors.
The d-center carboxyl of DAP is very likely to locate at the si face (the face toward solvent) of PLP in DAPDC, because superposition of DAP onto the bound l-lysine unambiguously places its d-center carboxyl into the only room left in the active pocket, which is above the si face of PLP (Fig. 2d). Several previous studies also support this conclusion. First, the Dunathan stereoelectronic hypothesis states that the bond to be broken by PLP-dependent enzymes lies orthogonal to the π system of the cofactor (34). Second, the crystal structure of MtDAPDC complexed with l-lysine suggests that the decarboxylation occurs at the si face of PLP (13). Third, mechanistic and structural studies of TbODC indicate that the carboxyl-leaving group of l-ornithine is buried on the re face of PLP (35), whereas the CO2 of d-ornithine locates at the si face (31). Attempt to reverse the orientation of DAP results in both electrostatic repulsion and steric hindrance, indicating that the substrate specificity of DAPDC is determined by both the shape and charge property of the active pocket. Based on these observations, the substrate DAP is modeled into the active site of HpDAPDC to provide mechanistic insights.
Putative Mechanism of the Recognition and Decarboxylation of the d-Center Carboxyl—In the modeled HpDAPDC-DAP structure, the d-center carboxyl of DAP could be recognized by several residues via water molecules, including the absolutely conserved PLP-binding residues Arg136 (36) and His185 and the active site loop residues Ile148 and Thr150 (Fig. 3a). Although the water molecule “Wx” is not included in the submitted coordinate because of its relatively weak but detectable Fo - Fc signal, this molecule could probably be stabilized in the presence of the d-center carboxyl). Interestingly, superposition of HpDAPDC-DAP with the BsALR-l-Ala-P (substrate analogue) complex structure (32) reveals that the substrate carboxyl groups (or the phosphonate analogue), the highly conserved arginine residues involved in carboxyl recognition, and the catalytic active lysine residues are all overlaid perfectly well (Fig. 3a), indicating that these two enzymes, although with distinctive functions and overall structures, might share some common features in substrate recognition and catalytic mechanism.
FIGURE 3.

Comparison of the substrate-binding sites among group IV PLP-dependent decarboxylase. a, modeled HpDAPDC-DAP structure (cyan) is superposed with the BsALR-l-Ala-P structure (yellow). The active site loop of HpDAPDC is drawn as ribbon. Residues with red names are of particular interest and described in detail in the text (same below). b, substrate-binding residues of various DAPDCs are superposed. Only the residues of HpDAPDC are named. The numbers in parentheses indicate corresponding residues from other species. c and d, superposition of HpDAPDC (cyan), TbODC (magenta), and cvADC (yellow). The two figures are viewed toward the l-stereocenter amine (c) and carboxyl (d) of the bound l-lysine in HpDADPC, respectively. The specificity element helices are shown as ribbon.
A putative decarboxylation mechanism of DAPDC could also be derived. Two residues lie within 4.5 Å from the d-center C-α of DAP, one is the original Schiff base residue Lys46, and the other is the highly conserved Cys329 at the re side of PLP (Fig. 3a). It has been reported that in eukaryotic ornithine decarboxylases (ODC), the analogues of these two residues, Lys69 and Cys360, play multiple roles in catalysis (30, 37). Notably, Cys360 is the target of α-difluoromethylornithine, a drug treating African sleeping sickness (11, 31). Thus, it is plausible that Lys46 and Cys329 would have similar catalytic functions in HpDAPDC. In detail, Lys46 might catalyze the formation and hydrolysis of the Schiff base, as well as carboxyl cleavage (37). Cys326, with its sulfhydryl group only 4.3 Å away from the d-center C-α, might be involved in carbanion protonation, Schiff base formation, and decarboxylation (30).
Comparison of the Substrate-binding Sites among Group IV PLP-dependent Decarboxylases—Although the group IV PLP-dependent decarboxylases all work on basic amino acids with similar mechanisms, they have evolved distinctive specificity determinants to recognize their own substrates.
Before comparison, we overlaid all known DAPDC structures and confirmed that the substrate-binding residues from HpDAPDC are representative of DAPDCs (Fig. 3b), which is consistent with the sequence alignment (Fig. 4). Thus, we will take HpDAPDC, TbODC (PDB code 1F3T) (30), and cvADC (PDB code 2NVA) (12) to carry out the comparison. Notably each structure contains a product-PLP external aldimine.
FIGURE 4.

Structure-based sequence alignment of DAPDCs, TbODC and cvADC. PDB codes are included in parentheses beside the protein names. Secondary structures of HpDAPDC are indicated above the alignment. Functional residues are shaded and labeled according to the footnote below the alignment. The italic letters in the boxed region indicate the crystallographically unobservable residues of AaDAPDC. The alignment was produced by STRAP (43) using the combinatorial extension (CE) method (44).
Because of the different length and chirality of the substrates (Fig. 1a), the inner walls of the active pockets are substantially different among the three enzymes. Accordingly, we will “cut” the active pocket of HpDAPDC into halves, namely the “DAP-amine side” and the “DAP-carboxyl side,” and compare them with the corresponding regions of ODC and ADC separately.
As shown in Fig. 3c, at the DAP-amine side, the arginine residues recognizing PLP and probably the d-center carboxyl (Arg135/DAPDC, Arg154/ODC, and Arg133/ADC), the catalytic active cysteine residues (Cys329B/DAPDC, Cys360B/ODC, and Cys324B/ADC), and the acidic residues lining beside the substrate (Glu330B/DAPDC, Asp361B/ODC, and Asp325B/ADC) are all structurally conserved, indicating the similar catalytic mechanism shared by these decarboxylases. The difference occurs at the PLP distal site. In detail, the glutamate residue (Glu330B) of DAPDC is substituted by aspartate residues in ODC and ADC (Asp361B/ODC and Asp325B/ADC), probably to interact with the more PLP-proximal nitrogen atoms of ornithine and arginine. Moreover, Tyr366B of DAPDC shift toward PLP compared with Tyr323B/ODC and Tyr287B/ADC, probably to avoid electrostatic repulsion with the l-center carboxyl of DAP. In addition, the two tyrosine residues of ODC and ADC are stabilized by forming hydrophobic interaction with nearby residues (Phe397B, Tyr331B of ODC and Phe361B, Leu357, and Phe295 of ADC). Such stabilization effect is absent in HpDAPDC. Finally, as has been reported before (6, 12), the positions of “specificity element” helices are different in these enzymes, and only the helix of DAPDC interacts with the substrate (Tyr302-DAP amine) at this side.
Greater difference occurs at the DAP-carboxyl side (Fig. 3d). The only constant structure element at this side is the floor tyrosine beneath the ligand (Tyr358/DAPDC, Tyr389/ODC, and Tyr353/ADC). The ceiling of the pocket, however, exists only in DAPDC and ADC (Ile148/DAPDC and Val143/ADC). Moreover, the charge property of this side varies greatly among the three enzymes. In detail, at the center, Arg262/DAPDC is extending its side chain toward DAP to make a salt bridge with the l-center carboxyl, whereas Arg277/ODC and Arg255/ADC are retracting their side chains from the substrates probably to avoid electrostatic repulsion. In the specificity element helices, the basic residue in DAPDC (Arg298) is substituted by asparagine (Asn292/ADC) and/or acidic residues (Glu296/ADC and Asp332/ODC).
Considering the extreme diversity of the reactions catalyzed by PLP enzymes and the similar working mechanism shared by closely related PLP enzymes, the structural difference we described above might offer an opportunity to design selective DAPDC inhibitors for development of antibiotics.
Catalytic Role of the Active Site Loop—The active site loop of HpDAPDC (Asn137 to Asn155) is in a down conformation that completely shields the ligand from the bulk solvent. There is no crystal contact at this site, indicating the natural occurrence of this conformation. Structure-based sequence alignment (Fig. 4) and superposition (Fig. 5, 1st panel) both indicate that the active site loop is the most divergent element of DAPDC across species. As a consequence, the active pockets in various DAPDC structures are solvent-exposed to different extents (Fig. 5).
FIGURE 5.
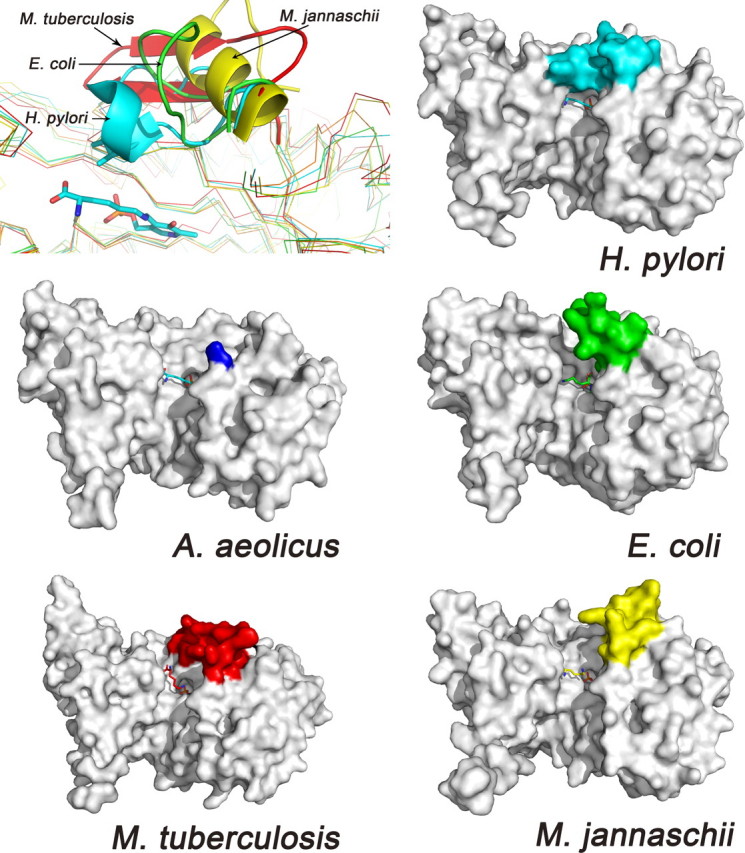
The active site loops of various DAPDCs. In the first panel, the five monomer structures are overlaid, and the active site loops are drawn as ribbon. In the next five panels, the structures are shown as surface, and the active site loops are colored differently. The ligand in AaDAPDC is taken from HpDAPDC to indicate the active site.
We speculate that HpDAPDC adopts an induced-fit catalytic mechanism, in which its active site loop cycles through down and up conformations to stabilize catalytic intermediates and release reaction products, respectively. This hypothesis is based on the following evidence. First, a highly similar loop of AaDAPDC (58% sequence identity, Fig. 4, boxed region) is flexible (crystallographically unobservable) in the absence of ligands. Second, superposition (Fig. 3d) and sequence alignment (Fig. 4) between HpDAPDC and cvADC reveal two well aligned active site loops both in down conformations, and the movement of the cvADC loop is believed to be kinetically linked with product release (12). Third, the protease-sensitive loop (residues 158-168) of human ODC has recently been shown to undergo large conformational change and cover the active site upon the binding of an inhibitor (38). Fourth, mutation of the active site loop in TbODC (K169A) resulted in a large decrease of the catalytic efficiency (39). In addition, we also carried out some biochemical experiments to verify our hypothesis.
The attempt to delete this loop failed because the truncated protein cannot express as soluble form, indicating the contribution of this loop to the overall stability of HpDAPDC. Thus, we mutated Ile148, the only ligand-contacting residue of this loop, to six different types of amino acid (Leu, Phe, Ala, Lys, Asp, and Gly) and found that all mutations caused great loss of the enzymatic activity. We also determined the crystal structure of I148L mutant. Detailed descriptions are as follows.
Enzymatic Assay—The kinetic constants of HpDAPDC (wild type and mutants) and EcDAPDC were determined by the reported coupled enzymatic assay approach (18). As shown in Table 3, the Km value of EcDAPDC is about 1.1 mm, comparable with the reported value of 1.7 mm measured by a different method (40), indicating the reliability of our assay system. The kcat value of HpDAPDC is 939 min-1, similar to the 1214 min-1 of EcDAPDC. However, the Km value of HpDAPDC (0.39 mm) is 2-fold smaller than that of EcDAPDC, probably because the substrate binding pocket of HpDAPDC is more compact.
TABLE 3.
Kinetic constants of various DAPDCs
| Km | kcat | kcat/Km | Relative activitya | |
|---|---|---|---|---|
| mm | min−1 | mm−1·min−1 | ||
| Wild type | 0.39 ± 0.02 | 939 ± 30 | 2441 ± 81 | 100 |
| I148L | 0.03 ± 0.01 | 23 ± 5 | 682 ± 125 | 28 |
| I148F | 0.11 ± 0.01 | 41 ± 5 | 378 ± 63 | 15 |
| I148A | 0.35 ± 0.04 | 20 ± 1 | 57 ± 5 | 2 |
| I148K | 0.36 ± 0.01 | 17 ± 1 | 47 ± 5 | 2 |
| I148D | 0.72 ± 0.03 | 4 ± 1 | 5 ± 1 | 0.2 |
| I148G | 0.36 ± 0.04 | 4 ± 1 | 12 ± 2 | 0.5 |
| EcDAPDC | 1.07 ± 0.13 | 1214 ± 211 | 1133 ± 135 | 46 |
The relative activities are calculated based on kcat/Km values.
The six mutations of Ile148, including the conservative substitution I148L, decreased the catalytic efficiency (kcat/Km) of HpDAPDC by 5-500-fold (Table 3). Based on the extent of activity loss, the six mutants could be classified into three groups. The first group, I148L and I148F, lost about 80% of the activity. Intriguingly, although the kcat values of these two mutants were impaired (40- and 23-fold, respectively), their Km values were enhanced (10- and 4-fold), indicating that the enzyme-substrate (ES) complex formed during reaction is more stable for the mutants. This point is supported by the structural analysis of I148L shown in the next section. The second group, I148A and I148K, suffered about 50-fold activity decrease, which is mainly caused by the reduced kcat values. The third group, I148D and I148G, retains only 1/500 and 1/200 of the original activity, respectively. The kcat values of these two mutants both decreased by more than 200-fold, whereas their Km values were close to that of the wild type. The 1-fold increase of the Km value for I148D might be caused by the electrostatic repulsion between Asp148 and the l-center carboxyl of the substrate. This is consistent with the observation that the side chain of Ile148 in wild-type structure lies closer to the l-center carboxyl group than to the l-center amine group, which also explains why the Km value of I148K was not affected. To further understand the deactivating effect of these mutations, we determined the crystal structure of I148L mutant.
Crystal Structure Analysis of I148L Mutant—The overall structure of I148L is basically the same as that of the wild type (Table 2). The only obvious change is the side-chain substitution of residue 148. However, combining the previous study about the catalytic mechanism of the closely related enzyme TbODC (41), we still find some explanations for the following questions.
Why Did the Mutations Cause Relatively Small Change of the Km Value?—As has been discussed, HpDAPDC is likely to adopt an induced-fit catalytic mechanism, which means the active site loop “closes” only after the proper anchoring of the substrate. In other words, this loop should have limited contribution to substrate binding. Thus, the mutations on this loop caused relatively small changes of the Km value.
Why Did the Mutations Greatly Decrease the kcat Value?—The active site loop is likely to be relevant with two kcat-determining processes. One is the stabilization of catalytic intermediates; the other is the release of the reaction product. Notably, the product release has been considered to be the rate-limiting step in the reaction catalyzed by TbODC (41). Mutations on the active site loop might interfere with both processes and result in decreased kcat values.
The mutations I148L, I148F, and I148K might have reduced the efficiency of product release. As shown in Fig. 6a, in the wild-type structure, the specificity element helix α14 is closely opposite to the small 310 helix α9 of the active site loop, and Tyr302 from α14 is hydrophobically packing against Ile148 from α9 with a buried surface area of 62.7 Å2. Thus α14 acts like a “buckle” to fasten the active site loop in its down conformation, and the affinity between Tyr302 and residue 148 is likely to determine the efficiency of loop opening and product release. In the structure of I148L mutant (Fig. 6b), however, the buried surface area between Leu148 and Tyr302 becomes larger (85.8 Å2), which might hinder the opening of the loop and product release. This also implies that the ES complex is probably more stable in I148L, resulting in enhanced Km value. Similar explanations could be applied to I148F and I148K.
FIGURE 6.

The active sites of wild-type (a) and I148L mutant (b) HpDAPDC. Tyr302 from the buckle helix α14 and residue 148 from the active site loop are shown as sticks with transparent spheres.
In contrast, the mutations I148A, I148G, and I148D might have compromised the ability of the enzyme to stabilize catalytic intermediates. First, no direct interaction (I148A and I148G), or even repulsion (I148D), could be made between the active site loop and the catalytic intermediates. Second, the affinity between the mutated residue (alanine, glycine, or asparagine) and Tyr302 might be weakened, destabilizing the down conformation of the active site loop. Thus, an isoleucine is probably the most appropriate residue at this position to delicately balance the close-to-stabilize and open-to-release processes of the active site loop.
As reflected by the mutagenesis study, altering the assumed dynamic behavior of this active site loop could have profound impact on the catalytic efficiency of HpDAPDC. Thus, restricting the movement of this loop could probably be an alternative strategy for designing specific HpDAPDC inhibitors.
Conclusion—The crystal structure of HpDAPDC complexed with the cofactor PLP and the reaction product l-lysine reveals the conformation of the catalytic intermediate (external aldimine), which provides insight into the substrate binding and catalytic mechanism of DAPDC. The aliphatic chain, the l-stereocenter carboxyl, and amine groups of the bound l-lysine are all properly anchored in the active pocket of HpDAPDC. The d-stereocenter carboxyl of the substrate DAP is very likely to locate at the si face of PLP. The recognition and cleavage of this carboxyl group might involve some highly conserved residues. The charge property of the active pocket of DAPDC is distinct from other group IV PLP-dependent decarboxylases, which provides structural clues for designing specific DAPDC inhibitors. The active site loop of HpDAPDC (Asn137 to Asn155) is in a down conformation and completely shields the ligand from solvent. Mutagenesis study and structural analysis suggest that HpDAPDC might adopt an induced-fit catalytic mechanism, in which this loop cycles through down and up conformations to stabilize catalytic intermediates and release the reaction product, respectively.
Acknowledgments
We are very grateful to Professor J. K. Bhattacharjee from Miami University (Miami, OH) and Professor Paul F. Cook from the University of Oklahoma for kindly providing us the plasmids encoding saccharopine dehydrogenase.
The atomic coordinates and structure factors (codes 2QGH, 3C5Q) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The nucleotide sequence(s) reported in this paper has been submitted to the GenBank™/EBI Data Bank with accession number(s) EU056336.
This work was supported by the State Key Program of Basic Research of China Grants 2004CB58905, 2007CB914304, and 2006AA09Z447, the National Natural Science Foundation of China Grants 30525024, 90713046, and 20721003, Shanghai Basic Research Project Grants 06JC14080 and 03DZ19228, and Foundation of Chinese Academy of Sciences Grant KSCX1-YW-R-18. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: DAP, meso-diaminopimelate; DAPDC, meso-diaminopimelate decarboxylase; PLP, pyridoxal 5′-phosphate; ODC, ornithine decarboxylase; ADC, arginine decarboxylase; ALR, alanine racemase; HpDAPDC, DAPDC from H. pylori; EcDAPDC, DAPDC from E. coli; MjDAPDC, DAPDC from M. jannaschii; MtDAPDC, DAPDC from M. tuberculosis; AaDAPDC, DAPDC from A. aeolicus; TbODC, ODC from T. brucei; cvADC, ADC from P. bursaria chlorella virus; BsALR, ALR from B. stearothermophilus; r.m.s.d., root mean square deviation; PBD, Protein Data Bank; MES, 4-morpholineethanesulfonic acid; Bicine, N,N-bis(2-hydroxyethyl)glycine.
References
- 1.Misono, H., Ogasawara, M., and Nagasaki, S. (1986) Agric. Biol. Chem. 50 1329-1330 [Google Scholar]
- 2.Sundharadas, G., and Gilvarg, C. (1967) J. Biol. Chem. 242 3983-3984 [PubMed] [Google Scholar]
- 3.De Lencastre, H., Wu, S. W., Pinho, M. G., Ludovice, A. M., Filipe, S., Gardete, S., Sobral, R., Gill, S., Chung, M., and Tomasz, A. (1999) Microb. Drug Resist. 5 163-175 [DOI] [PubMed] [Google Scholar]
- 4.Ray, S. S., Bonanno, J. B., Rajashankar, K. R., Pinho, M. G., He, G., De Lencastre, H., Tomasz, A., and Burley, S. K. (2002) Structure (Lond.) 10 1499-1508 [DOI] [PubMed] [Google Scholar]
- 5.Sandmeier, E., Hale, T. I., and Christen, P. (1994) Eur. J. Biochem. 221 997-1002 [DOI] [PubMed] [Google Scholar]
- 6.Lee, J., Michael, A. J., Martynowski, D., Goldsmith, E. J., and Phillips, M. A. (2007) J. Biol. Chem. 282 27115-27125 [DOI] [PubMed] [Google Scholar]
- 7.Eliot, A. C., and Kirsch, J. F. (2004) Annu. Rev. Biochem. 73 383-415 [DOI] [PubMed] [Google Scholar]
- 8.Christen, P., and Mehta, P. K. (2001) Chem. Rec. 1 436-447 [DOI] [PubMed] [Google Scholar]
- 9.Shaw, J. P., Petsko, G. A., and Ringe, D. (1997) Biochemistry 36 1329-1342 [DOI] [PubMed] [Google Scholar]
- 10.Kern, A. D., Oliveira, M. A., Coffino, P., and Hackert, M. L. (1999) Structure (Lond.) 7 567-581 [DOI] [PubMed] [Google Scholar]
- 11.Grishin, N. V., Osterman, A. L., Brooks, H. B., Phillips, M. A., and Goldsmith, E. J. (1999) Biochemistry 38 15174-15184 [DOI] [PubMed] [Google Scholar]
- 12.Shah, R., Akella, R., Goldsmith, E. J., and Phillips, M. A. (2007) Biochemistry 46 2831-2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gokulan, K., Rupp, B., Pavelka, M. S., Jr., Jacobs, W. R., Jr., and Sacchettini, J. C. (2003) J. Biol. Chem. 278 18588-18596 [DOI] [PubMed] [Google Scholar]
- 14.Momany, C., Levdikov, V., Blagova, L., and Crews, K. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 549-552 [DOI] [PubMed] [Google Scholar]
- 15.Schilling, C. H., Covert, M. W., Famili, I., Church, G. M., Edwards, J. S., and Palsson, B. O. (2002) J. Bacteriol. 184 4582-4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu, W., Luo, C., Han, C., Peng, S., Yang, Y., Yue, J., Shen, X., and Jiang, H. (2005) Biochem. Biophys. Res. Commun. 333 1078-1086 [DOI] [PubMed] [Google Scholar]
- 17.Liu, W., Han, C., Hu, L., Chen, K., Shen, X., and Jiang, H. (2006) FEBS Lett. 580 697-702 [DOI] [PubMed] [Google Scholar]
- 18.Laber, B., and Amrhein, N. (1989) Anal. Biochem. 181 297-301 [DOI] [PubMed] [Google Scholar]
- 19.Leslie, A. G. W. (1992) Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography 26
- 20.Evans, P. R. (1993) Proceedings of CCP4 Study Weekend, pp. 114-122, CLRC Daresbury Laboratory, Warrington, UK
- 21.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 22.Brunger, A. T. (2007) Nat. Protoc. 2 2728-2733 [DOI] [PubMed] [Google Scholar]
- 23.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 24.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 25.Roman, A., Laskowski, M. W. M., Moss, D. S., and Thornton, J. M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 26.Kabsch, W. (1976) Acta Crystallogr. Sect A 32 922-923 [Google Scholar]
- 27.Collaborative Computational Project, No. 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 28.DeLano, W. L. (2002) PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA
- 29.Jackson, L. K., Baldwin, J., Akella, R., Goldsmith, E. J., and Phillips, M. A. (2004) Biochemistry 43 12990-12999 [DOI] [PubMed] [Google Scholar]
- 30.Jackson, L. K., Brooks, H. B., Osterman, A. L., Goldsmith, E. J., and Phillips, M. A. (2000) Biochemistry 39 11247-11257 [DOI] [PubMed] [Google Scholar]
- 31.Jackson, L. K., Goldsmith, E. J., and Phillips, M. A. (2003) J. Biol. Chem. 278 22037-22043 [DOI] [PubMed] [Google Scholar]
- 32.Stamper, G. F., Morollo, A. A., and Ringe, D. (1998) Biochemistry 37 10438-10445 [DOI] [PubMed] [Google Scholar]
- 33.Morollo, A. A., Petsko, G. A., and Ringe, D. (1999) Biochemistry 38 3293-3301 [DOI] [PubMed] [Google Scholar]
- 34.Dunathan, H. C. (1966) Proc. Natl. Acad. Sci. U. S. A. 55 712-716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson, L. K., Brooks, H. B., Myers, D. P., and Phillips, M. A. (2003) Biochemistry 42 2933-2940 [DOI] [PubMed] [Google Scholar]
- 36.Kidron, H., Repo, S., Johnson, M. S., and Salminen, T. A. (2007) Mol. Biol. Evol. 24 79-89 [DOI] [PubMed] [Google Scholar]
- 37.Osterman, A. L., Brooks, H. B., Jackson, L., Abbott, J. J., and Phillips, M. A. (1999) Biochemistry 38 11814-11826 [DOI] [PubMed] [Google Scholar]
- 38.Dufe, V. T., Ingner, D., Heby, O., Khomutov, A. R., Persson, L., and Al-Karadaghi, S. (2007) Biochem. J. 405 261-268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Myers, D. P., Jackson, L. K., Ipe, V. G., Murphy, G. E., and Phillips, M. A. (2001) Biochemistry 40 13230-13236 [DOI] [PubMed] [Google Scholar]
- 40.White, P. J. (1971) Methods Enzymol. 17 140-145 [Google Scholar]
- 41.Brooks, H. B., and Phillips, M. A. (1997) Biochemistry 36 15147-15155 [DOI] [PubMed] [Google Scholar]
- 42.Baker, N. A., Sept, D., Joseph, S., Holst, M. J., and McCammon, J. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10037-10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gille, C., and Frommel, C. (2001) Bioinformatics (Oxf.) 17 377-378 [DOI] [PubMed] [Google Scholar]
- 44.Shindyalov, I. N., and Bourne, P. E. (1998) Protein Eng. 11 739-747 [DOI] [PubMed] [Google Scholar]