

Abstract
A new route to the chromene ring system has been developed which involves the reaction of an α,β-unsaturated Fischer carbene complex of chromium with a propargyl ether bearing an alkenyl group on the propargylic carbon. This transformation involves a cascade of reactions that begins with a benzannulation reaction and is followed by the formation of an o-quinone methide and finally, results in the emergence of a chromene upon an electrocyclization. This reaction was extended to the provide access to by employing an aryl carbene complex. This constitutes the first synthesis of chromenes in which both rings of the chromene system are generated in a single step and is highlighted in the synthesis of lapachenole and Vitamin E.
1. INTRODUCTION
As a consequence of its appearance in compounds with a broad spectrum of biological activity, the chromene ring system (1 in Scheme 1) has been identified as one of the privileged scaffolds for drug discovery1 and thus for combinatorial libraries.2 Nearly all of the methods for the synthesis of chromenes involve the closure of the pyran ring on a substrate containing a pre-formed phenol unit of the type 2. Recent representative methods include Claisen rearrangement3 or electrophile induced cyclization of aryl propargyl ethers,4 palladium-catalyzed5 and selenium-mediated2a cyclization of 2-butenylphenols, ring closing metathesis,6 cyclization of salicylaldehydes with enamines,7 Wittig olefination,8 Petasis reaction of salicylaldehydes9 and the reaction of salicylaldehydes with vinyltrifluoroborates,10 palladium catalyzed oxidative cyclization of aryl-3-butenyl ethers,11 ene12 and Baylis-Hillman13 reactions of salicylaldehydes, ylide induced annulation,14 enolization of vinylquinones,15 cyclization onto 3,4-epoxy alcohols,16 the reactions of phenols with α, β-unsaturated aldehydes,17 the oxa-Michael-aldol/Henry reaction of salicylaldehydes18 and the dehydrative cyclizations of 2-(1-hydroxy-2-propenyl)phenols.19 Of all the methods of the myriad, to the best of our knowledge, there is not a single one in which both rings of the chromene core are constructed at the same time. We report here an efficient method for the preparation of chromenes in which both rings of the chromene unit are generated from the reaction of the carbene complex 3 and the alkoxyenyne 4 in a benzannulation/o-quinone methide formation/electro-cyclization cascade.
Scheme 1.

The anticipated events in the transformation of carbene complex 3 and alkoxyenyne 4 into a chromene are the benzannulation to give the phenol complex 5,20 the elimination of an alcohol to generate the o-quinone methide complex 6,21,22 and finally, an electrocyclization to give the chromene chromium tricarbonyl complex 723 (Scheme 2). The chromene oxygen in 7 has as its origin one of the carbon monoxide ligands of the carbene complex and the overall integration of the pieces is indicated diagrammatically in the assembly 8.
Scheme 2.

In previous studies we had shown that the reaction of alkenyl carbene complexes would react with propargyl ethers of the type 10 with a tethered alkene unit to generate hexahydrodibenzopyrans of the type 11.21 Significant yields of 11 were only observed if the reaction was performed in the presence of Hunig’s base that presumably aided in the elimination of the propargyl oxygen unit from the benzannulated product 12 to generate the o-quinone methide complex 13. The intramolecular Diels-Alder reaction that concludes the cascade must have occurred via the intermediate 13 with an E-alkene such that the trans-stereochemistry of 11 is established. This in turn requires that during o-quinone methide formation the alkyl group moves away from the phenol unit in 12 to establish the E-stereochemistry in the o-quinone methide. This was a source of concern in the original planning of the chromene synthesis in Scheme 2 since the ultimate electrocyclic ring-closure would require the Z-configuration of the alkene in the o-quinone methide unit.
2. Chromenes from Alkenyl Carbene Complexes
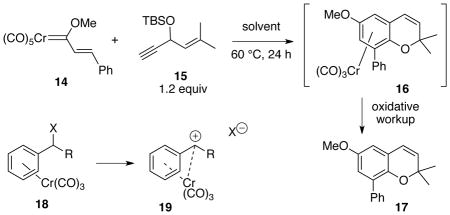
In our initial foray into exploring the cascade process in Scheme 2, we chose to examine the reaction of the trans-styrenyl carbene complex 14 and the siloxyenyne 15. The first reaction was carried out in toluene with 5 equiv of Hünig’s base and the reaction mixture was subjected to oxidation with ferric chloride DMF complex to remove the metal from the product and simplify purification. This reaction gave a 35% yield of chromene 17 but the yield could be improved to 62% yield if the base was excluded (Table 1, entries 1 vs 2). This was a surprise since we had previously found that o-quinone methide formation was facilitated by the presence of a base (Scheme 3).21 The more facile elimination in the present case may be related to the fact that a chromium tricarbonyl unit in an arene complex can stabilize a benzylic cation (19 in Table 1).24 Substitution reactions on the chromium tricarbonyl complex of benzyl chloride are 105 times faster than on benzyl chloride itself.24a The benzylic cation 19 (R = alkenyl) derived from alkoxyenyne 15 would be both benzylic and allylic rather than just benzylic (R = alkyl) as in previous reactions where base was employed to effect o-quinone methide formation which was presumably initiated via base induced deprotonation in phenol complexes of the type 12 (Scheme 3).21 The fact that base is not needed in the present case could be explained if o-quinone methide formation could be initiated by a chromium induced loss of an alkoxide in the complex 5 (Scheme 2).
Table 1.
Optimization of chromene formation from chromium carbene complex 14.a
 | |||
|---|---|---|---|
| entry | solvent | oxidative workup | % yield 17b |
| 1c,d | toluene | FeCl3•DMF | 35 |
| 2c | toluene | FeCl3•DMF | 62 |
| 3 | benzene | FeCl3•DMF | 74 |
| 4 | hexane | FeCl3•DMF | 70 |
| 5 | THF | FeCl3•DMF | 65 |
| 6 | MeCN | none | 95 |
| 7 | CH2Cl2 | FeCl3•DMF | 76 |
| 8 | CH2Cl2 | CAN | nde |
| 9 | CH2Cl2 | none | ndf |
Unless otherwise specified, all reactions were run at 0.03 M in 14 with 1.2 equiv 15 at 60 °C for 24 h and worked-up with 7.5 equiv of the indicated oxidant (if employed)
Isolated yield after chromatography on silica gel. All yields are the average of two runs except for entry 7. nd = not determined.
Reaction performed at 80 °C for 24 h.
Reaction performed with 5 equiv of (i-Pr)2EtN.
TLC indicated the absence of 16 and 17 and the presence of compounds more polar than either.
A mixture of 16 and 17 from which 16 could not be separated to purity due its slow and continuous decomposition to 17.
Scheme 3.

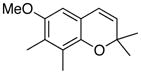
A survey of different solvents for this reaction shown in Table 1 found that acetonitrile was the optimum for this particular reaction and had the additional advantage that an oxidative workup was not needed since the solvent was capable of completely displacing the metal from the product under the reaction conditions. Ceric ammonium nitrate was too strong an oxidant since neither the chromene complex 16 nor the chromene 17 could be detected in the crude reaction mixture. In the absence of an oxidative workup, a mixture of 16 and 17 were obtained from which 16 could not be purified since it slowly oxidizes to 17 in air. The chromium tricarbonyl arene complex 21 derived from the trans-2-butenyl carbene complex 20 was much more stable to loss of the metal and could be isolated in 61% yield after purification by chromatography on silica gel (Scheme 4). The corresponding chromene 22 could be obtained free of the metal in 88% yield if an oxidative workup with FeCl3•DMF complex was employed.
Scheme 4.

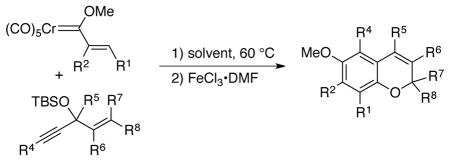
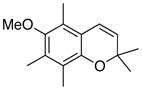
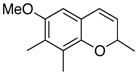
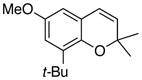
The formation of chromenes from α, β-unsaturated carbene complexes and alkoxyenynes is efficient for a number of substituent patterns and with substituents of varying sizes (Table 2). It should be noted that alkoxyenynes with an internal alkyne function (R4 ≠ H) gave a single regioisomer of the chromene, specifically, that in which the R4 substituent is ortho to the methoxy group in complex 5 in Scheme 2.20 Most of the cases examined involved chromenes in which the sp3 ethereal carbon of the chromene bears two substituents but we were particularly pleased to note that chromenes with either one (26) or no substituents (24) could also be generated. The concern was that alkoxyenynes with either one or both of the substituents R7 and R8 as hydrogen would fail since the cation 19 would be less stable. While this was not the case, the fact that the yield of 24 is lower than 17 and that the yield of 26 is lower than 22 suggests the substitution pattern in cation 19 may play some role in this reaction. While Hünig’s base was detrimental (Table 1), it was found that the yield of 28 could be slightly improved if the reaction was performed in the presence of 10 equivalents of aniline but the generality of this effect was not examined.
Table 2.
Chromene Synthesis via Carbene Complexes and Alkoxyenynes.a
 | ||
|---|---|---|
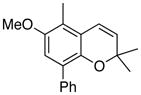 23 69% (CH2Cl2) 66% (CH3CN) |
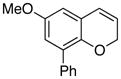 24 47% (CH3CN) |
 22 88% (CH2Cl2) 65% (CH3CN) |
 25 84% (CH2Cl2) 87% (CH3CN) |
 26 70% (CH2Cl2) |
 27 78% (CH2Cl2) 78% (CH3CN) |
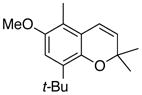 28 74% (CH2Cl2) 86% (CH2Cl2) b 65% (CH3CN) c,d |
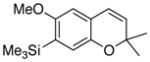 29 56% (CH2Cl2) 23% (CH3CN) |
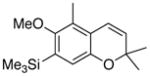 30 84% (CH2Cl2) 35% (CH3CN) |
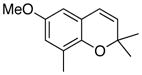 31 41% (CH2Cl2) 83% (CH3CN) c |
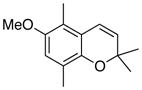 32 61% (CH2Cl2) 48% (CH3CN) c |
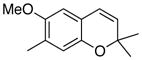 33 45% (CH2Cl2) 26% (CH3CN) 55% (hexane) |
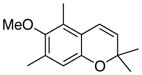 34 68% (CH2Cl2) 34% (CH3CN) 74% (hexane) |
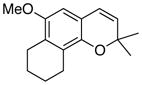 35 72% (CH2Cl2) 65% (CH3CN) c |
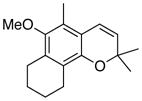 36 80% (CH2Cl2) 68% (CH3CN) c |
Unless otherwise specified, all reactions were run at 0.03 M in carbene complex in the indicated solvent with 1.2 equiv of the enyne at 60 °C for 24 h. If oxidation was used it was carried out with 7.5 equiv of FeCl3•DMF complex. All yields are isolated yields after chromatography on silica gel.
Reaction performed with 10 equiv of aniline.
Oxidative workup not used.
Isolation after treatment of crude reaction mixture with trifluoromethane sulfonic acid.
In many of the reactions, the solvents CH2Cl2 and CH3CN were found to give similar yields of the chromenes. However, for carbene complexes that only had a substituent in the α-position (R2), CH2Cl2 was superior to CH3CN as solvent (29, 30, 33, 34). In the case of the chromenes 33 and 34 which are obtained from the carbene complex with R2 = methyl, the best solvent was found to be hexane. While the source of these differences is not fully appreciated, α, β-unsaturated carbene complexes that do not have a substituent at the β-carbon tend to be less stable as a result of a tendency to undergo polymerization.25 While the reactions in CH3CN do not need an oxidative workup to remove the chromium tricarbonyl group from the chromene, these reactions produce (CO)5Cr(CH3CN) which slowly air oxidizes and which can co-elute with the chromene product. Therefore, many of the reactions in CH3CN also utilized the FeCl3•DMF complex in an oxidative workup to prevent (CO)5Cr(CH3CN) from complicating product isolation.
A side-product was isolated from the reaction of the trans-t-butyl alkenyl carbene complex 37 with the enyne 38. The phenol 39 is the major product for the reaction in acetonitrile formed in a 3:2 ratio over the chromene 28. Clearly, this product is the result of the failure of the o-quinone methide to form. Nonetheless, phenol 39 can be quantitative converted to the chromene 28 by treatement with triflic acid. The yield given for chromene 28 in acetonitrile that is indicated in Table 2 involved treatment of the crude reaction mixture with triflic acid before purification. A possible explanation for the partial failure of o-quinone methide formation in these reactions is that the H-bonding indicated in structure 40 prevents the proper anti-orientation of the benzylic oxygen with respect to the chromium for the assisted elimination to generate a benzylic cation of the type 19 (Table 1). The role of t-butyl group in this process may be related in some fashion to the orientation of the chromium tricarbonyl group relative to the arene carbons.26 As a test, this reaction was repeated in the presence of increasing amounts of iso-propanol which was added to dissrupt the H-bonding and, indeed, at 100 equiv no detectable amount of 39 was observed. Acetonitrile would not be expected to disrupt this hydrogen bonding since the pKa of protonated acetonitrile has been reported to be −10.27 Instead, the role of the acetonitrile in favoring the phenol product 39 is suspected of being related to its ability to displace the chromium tricarbonyl from the benzannulated product before it has the chance to completely ionize the benzylic oxygen.
3. Naphthopyrans from Aryl Carbene Complexes
The benzannulation/o-quinone methide formation/electrocyclization cascade of an aryl carbene complex with a propargyl enyne has the potential for providing access to 2H-benzo[h]chromenes (naphthopyrans) 43 in a single step (Sheme 6). The 2H-benzo[h]chromene core 45 is quite common and occurs in a large number of natural and unnatural products.28 One of the simplest members is the natural product lapachenole 46 which has been isolated from different sources including Avicennia rumphiana.29 This compound has been used as a fluorescent photoaffinity label30 and has been shown to have cancer chemopreventitive activity.29 It occurs in Tabebuia heptaphylla which is the source of the Paraguayan traditional medicine “Tayï Pytá” used in the treatment of wounds, cancer and inflammations.31 Both 2H-benzo[h]chromenes 4530 and 3H-benzo[f]chromenes 4432 are of interest for their photo-chromic properties which are associated with photo-induced electrocyclic ring opening to o-quinone methides. We have previously reported an approach to 3H-benzo[f]chromenes via the simple benzannulation reaction of a chromene carbene complex and an alkyne.33 This approach required the preparation of the chromene carbene complex 49 which was accomplished in 6 steps from o-methoxybenzaldehyde. The proposed route to 2H-benzo[h]chromenes would potentially be much more efficient since aryl carbene complex of the type 41 can be prepared in one step from the corresponding aryl bromide or iodide in good to excellent yields.
The key reaction for the synthesis of lapchenole via the benzannulation/o-quinone methide formation/electrocyclization cascade is that of carbene complex 53 and enyne 15 (Scheme 7). This reaction only produced the natural product in 37% yield but it was found that the yield could be improved slightly to 48% if the reaction was performed in the presence of 10 equivalents of aniline. Despite the moderate yield, it represents a very short synthesis of lapachenole: 2 steps from bromobenzene, or 2 to 3 steps from the commercially available prenal.34 No significant by-products were observed to form along with the desired product lapachenole. Collection of other fractions from the silica gel column yielded a complexed mixture of compounds none of which were predominate or separable. This is suggestive of incorporation of multiple units of the enyne and this has been observed in other reactions to produce phenols, trisubstituted benzenes or oligiomers.35 Previous experience suggests that if this was the case, improved yields could be achieved by contolling concentration. However, in the present case, there was not a great response in the yield to changes in the concentration. The yield was 37% at 0.035 M and, while this fell as expected when the concentration was increased (26% at 0.1 M), it also fell when the concentration was reduced (31% at 0.005 M). If multiple insertions of alkyne 15 containing a terminal alkyne function is responsible for the moderate yields of lapachenole, then increased yields would be expected for similar reactions with internal alkynes. Indeed, the synthesis of 5-methyllapachenole 54 was possible with a much higher yield (85%) than lapachenole.
Scheme 7.

An alternative approach to lapchenole 46 that has the potential to be more efficient is the reaction of the carbene complex 53 with the enyne 55 bearing an internal alkyne as the trimethylsilylated analog of enyne 15 (Scheme 8). In analogy with enyne 38 bearing an internal alkyne, the product from the reaction of enyne 55 would be expected to be the naphthopyran 56 from which the trimethylsilyl group could be removed by protonolysis to give lapachenole 46. However, it was found that the reaction of the silylated enyne 55 gave the indene product 57 rather than the expected naphthopyran 56. The indene 57 was isolated 65% yield as a 1.14:1.0 mixture of diastereomers and was the only product that was observed that was mobile on TLC. This type of five-membered ring cyclized product is perhaps the most common of the many side-products that have been observed in the benzannulation reaction.20 There is a tendency to see increased amounts of five-membered ring products with increased steric bulk of the two acetylene substituents. Mechanistically, this reaction should occur by initial insertion of the alkyne function of 55 into the metal-carbene bond in carbene complex 53 to give the vinyl carbene complexed intermediate 58. The subsequent events normally would be migratory insertion of a CO ligand to give the chromium complexed vinyl ketene 59 and then electrocyclic ring-closure to give the phenol chromium tricarbonyl complex 60. Apparently, in the case of the vinyl carbene complexed intermediate 58, there is a preference for direct cyclization to 57 rather than CO insertion to give 59. The reasons for this are not clear but one might expect that it may be related to the increase in bond angles for the sp2 carbons of 58 as cyclization occurs to give a 5- membered ring and the associated decrease in strain energy as the large substituents move further apart.
Scheme 8.

4. Synthesis of Vitamin E via the Benzannulation/o-Quinone Methide Formation/Electrocyclization Cascade
As an illustration of the utility of the benzannulation induced cascade in the synthesis of chromenes, we undertook the synthesis of Vitamin E outlined in Scheme 9.36,37 The most commercially important form of Vitamin E is (all-rac)-α-tocopherol 65 which is a mixture of eight stereoisomers.37 The synthesis begins with a Swern oxidation of commercially available (all-rac)-phytol 61 followed by reaction of the a,b-unsaturated aldehyde with propynyl Grignard and then protection of the alcohol as a TBS ether to give the key enyne 62. The chromene 63 is generated in 85% yield directly from the reaction of carbene complex 20 and the internal alkyne 62. Reduction of the double bond is quantitative and cleavage of the methyl ether with BF3•SMe2 complex and aluminum chloride gives Vitamin E in 73% overall yield from the carbene complex 20.
Scheme 9.

5. CONCLUSIONS
The formation of chromenes from the reaction of a chromium carbene complex with a 3-siloxypent-4-en-1-yne has been shown to proceed via an initial benzannulation reaction that produces a chromium tricarbonyl complexed phenol. Spontaneous loss of a silanol generates an o-quinone methide that undergoes a six-electron electrocyclic ring-closure to the chromene. The reaction is general giving good to high yields of a variety of 4-alkoxychromenes with mono- and di-substituted alkenyl carbene complexes and mono-, di- and tri-substituted 3-silyloxypent-4-en-1-ynes. The primary product of the reaction is a chromium tricarbonyl complexed chromene that can be isolated, but is typically stripped of the metal either by ligand exchange with acetonitrile as solvent or by workup with an oxidizing agent. The reaction can also be extended to aryl carbene complexes for the synthesis of 2-H-benzo[h]chromenes and this process was employed in the synthesis of lapachenole and 5-methyllapachenole. The benzannulation/o-quinone methide formation/electrocyclization cascade was featured in a synthesis of Vitamin E in which both rings of Vitamin E were generated in a single step.
Supplementary Material
Scheme 5.

Scheme 6.

Acknowledgments
This work was supported by NSF Grant CHE-0750319 and the National Institute of General Medical Sciences (GM094478).
Footnotes
Supporting Information. Synthetic procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Welsch ME, Snyder SA, Stockwell BR. Curr Opin Chem Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Nicolaou KC, Pffefferkorn JA, Roecker AJ, Cao GQ, Barluenga S, Mitchell HJ. J Am Chem Soc. 2000;122:9939. [Google Scholar]; b) An H, Eum SJ, Koh M, Lee SK, Park SB. J Org Chem. 2008;73:1752. doi: 10.1021/jo702196f. [DOI] [PubMed] [Google Scholar]; c) Oh S, Jang HJ, Ko SK, Ko Y, Park SB. J Comb Chem. 2010;12:548. doi: 10.1021/cc100044w. [DOI] [PubMed] [Google Scholar]; d) Kapeller DC, Bräse S. Synlett. 2011:161. [Google Scholar]
- 3.a) Hlubeck J, Ritchie E, Taylor WC. Tetrahedron Lett. 1969:1369. [Google Scholar]; b) Bigi F, Carloni S, Maggi R, Muchetti C, Sartori G. J Org Chem. 1997;62:7024. [Google Scholar]
- 4.a) Pastine SJ, Youn SW, Sames D. Org Lett. 2003;5:1055. doi: 10.1021/ol034177k. [DOI] [PubMed] [Google Scholar]; b) Worlikar SA, Kesharwani T, Yao T, Larock RC. J Org Chem. 2007;72:1347. doi: 10.1021/jo062234s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Savitha G, Felix K, Perumal PT. Synlett. 2009:2079. [Google Scholar]
- 5.a) Iyer M, Trivedi GR. Synth Commun. 1990;20:1347. [Google Scholar]; b) Labrosse JR, Lhoste P, Sinou D. Synth Commun. 2002;32:3667. [Google Scholar]
- 6.a) Chang S, Grubbs RH. J Org Chem. 1998;63:864. doi: 10.1021/jo9712198. [DOI] [PubMed] [Google Scholar]; b) Harrity PPA, La DS, Cefalo DR, Visser MS, Hoveyda AH. J Am Chem Soc. 1998;120:2343. [Google Scholar]; c) van Otterlo WAL, Ngidi EL, Kuzvidza S, Morgans GL, Moleele SS, de Koning CD. Tetrahedron. 2005;61:9996. [Google Scholar]
- 7.Varman RS, Dahiya R. J Org Chem. 1998;63:8038. [Google Scholar]
- 8.Hanamoto T, Shindo K, Matsuoka M, Kiguchi Y, Kondo M. J Chem Soc Perkin. 2000;1:103. [Google Scholar]
- 9.a) Wang Q, Finn MG. Org Lett. 2000;2:4063. doi: 10.1021/ol006710r. [DOI] [PubMed] [Google Scholar]; b) Petasis NA, Butkevich AN. J Organometal Chem. 2009;694:1747. doi: 10.1016/j.jorganchem.2008.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu F, Evans T, Das BC. Tetrahedron Lett. 2008;49:1578. doi: 10.1016/j.tetlet.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Youn SW, Eom JI. Org Lett. 2005;7:3355. doi: 10.1021/ol051264z. [DOI] [PubMed] [Google Scholar]
- 12.Prado S, Janin YL, Bost PE. J Heterocyclic Chem. 2006:1605. [Google Scholar]
- 13.a) Kaye PT, Nocanda XW. J Chem Soc Perkin. 2000;1:1331. [Google Scholar]; b) Ravichandran S. Synth Commun. 2001;31:1233. [Google Scholar]; C) Kaye PT, Nocanda XW. J Chem Soc Perkin. 2002;1:1318. [Google Scholar]
- 14.Ye LW, Sun XL, Zhu CY, Tang Y. Org Lett. 2006;8:3853. doi: 10.1021/ol0615174. [DOI] [PubMed] [Google Scholar]
- 15.Parker KA, Mindt TL. Org Lett. 2001;3:3875. doi: 10.1021/ol0167199. [DOI] [PubMed] [Google Scholar]
- 16.Goujon JY, Zammattio F, Chretien JM, Beaudet I. Tetrahedron. 2004;60:4037. [Google Scholar]
- 17.a) Chauder BA, Kalinin AV, Snieckus V. Synthesis. 2001:140. [Google Scholar]; b) Lee YR, Choi JH, Yon SH. Tetrahedron Lett. 2005:7539. [Google Scholar]; c) Dintzner MR, Lyons TW, Akroush MH, Wucka P, Pzepka AT. Synlett. 2005:785. [Google Scholar]; d) Adler MJ, Baldwin SW. Tetrahedron Lett. 2009;50:5075. [Google Scholar]; e) Gembus V, Sala-Jung N, Uguen D. Bull Chem Soc Jpn. 2009;82:843. [Google Scholar]; f) Aponick A, Biannic B, Jong MR. Chem Commun. 2010:6849. doi: 10.1039/c0cc01961e. [DOI] [PubMed] [Google Scholar]
- 18.a) Govender T, Hojabri L, Moghaddam FM, Arvidsson PI. Tetrahedron: Asymmetry. 2006;17:1763. [Google Scholar]; b) Li H, Wang L, E-Nunu T, Zu L, Jiang W, Wei S, Wang W. Chem Commun. 2007:507. doi: 10.1039/b611502k. [DOI] [PubMed] [Google Scholar]; c) Sunden H, Ibrahem I, Zhao GL, Eriksson L, Cordova A. Chem Eur J. 2007;13:574. doi: 10.1002/chem.200600572. [DOI] [PubMed] [Google Scholar]; d) Xu DQ, Wang YF, Luo SP, Zhang S, Zhong AG, Chen H, Xu ZY. Adv Synth Catal. 2008;350:2610. [Google Scholar]; e) Luo SP, Li ZB, Wang LP, Guo Y, Xia AB, Xu DQ. Org Biomol Chem. 2009;7:4539. doi: 10.1039/b910835a. [DOI] [PubMed] [Google Scholar]; f) Das BC, Mohapatra S, Campbell PD, Nayak S, Mahalingam SM, Evans T. Tetrahedron Lett. 2010;51:2567. doi: 10.1016/j.tetlet.2010.02.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Aponick A, Biannic B, Jong MR. Chem Commun. 2010:6849. doi: 10.1039/c0cc01961e. [DOI] [PubMed] [Google Scholar]; b) Rueping M, Uriia U, Lin MY, Atodiresei I. J Am Chem Soc. 2011;133:3732. doi: 10.1021/ja110213t. [DOI] [PubMed] [Google Scholar]
- 20.a) Waters ML, Wulff WD. Organic Reactions. 2008;70:121–623. [Google Scholar]; b) Dötz KH, Stendel J., Jr Chem Rev. 2009;109:3227. doi: 10.1021/cr900034e. [DOI] [PubMed] [Google Scholar]
- 21.Korthals KA, Wulff WD. J Am Chem Soc. 2008;130:2898. doi: 10.1021/ja077579m. [DOI] [PubMed] [Google Scholar]
- 22.For a review of non-metal complexed o-quinone methides, see: van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367.
- 23.For leading references to electrocyclizations of non-metal complexed o-quinone methides, see: Bishop LM, Winkler M, Houk KN, Bergman RG, Trauner D. Chem Eur J. 2008;14:5405. doi: 10.1002/chem.200800662.
- 24.a) Davies SG, Donohoe TJ. Synlett. 1993:323. [Google Scholar]; b) Uemura M, Kobayashi T, Isobe K, Minami T, Hayashi Y. J Org Chem. 1986;51:2859. [Google Scholar]
- 25.Macomber DW, Hung MH, Liang M, Verma AG, Madukar P. Macromolecules. 1988;21:1187. [Google Scholar]
- 26.Chamberlin S, Majumdar N, Wulff WD, Muntean JV, Ostrander RL, Rheingold AL. Inorganica Chemica Acta. 2010;364:205. [Google Scholar]
- 27.Pearson RG. J Am Chem Soc. 1986;108:6109. [Google Scholar]
- 28.Ellis GP, editor. Chromenes, chromanones, and chromones. Vol. 31. John Wiley & Sons, Inc; 1977. [Google Scholar]
- 29.Itoigawa M, Ito C, Tan HTW, Okuda M, Tokuda H, Nishino H, Furukawa H. Cancer Lett. 2001;174:135. doi: 10.1016/s0304-3835(01)00707-8. [DOI] [PubMed] [Google Scholar]
- 30.Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD. Biochemistry. 2005;44:1833. doi: 10.1021/bi048228c. [DOI] [PubMed] [Google Scholar]
- 31.Schmeda-Hirschmann G, Papastergiou F. Z Naturforsch. 2003;58c:495–501. doi: 10.1515/znc-2003-7-809. [DOI] [PubMed] [Google Scholar]
- 32.For citations to the literature, see reference 31.
- 33.Rawat M, Prutyanov V, Wulff WD. J Am Chem Soc. 2006;128:11044–11053. doi: 10.1021/ja0568852. [DOI] [PubMed] [Google Scholar]
- 34.For a synthesis and leading references to the synthesis of lapachenole, see: Lee YR, Kim YM. Helv Chim Acta. 2007;90:2401.
- 35.For examples and leading references, see: Wulff WD, Bax BM, Brandvold TA, Chan KS, Gilbert AM, Hsung RP. Organometallics. 1994;13:102.
- 36.For a two-stage synthesis of Vitamin E with Fischer carbene complexes, see; Dötz KH, Kuhn W. Angew Chem Int Ed Engl. 1983;22:732.
- 37.For a comprehensive review of the syntheses of Vitamin E, see: Vitamins and Hormones–Advances in Research and Applications. Vol. 76. 2007. pp. 155–202.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.