

Abstract
Metal neurotoxicity is a global health concern. This paper summarizes the evidence for metal interactions with synaptic transmission and synaptic plasticity. Presynaptically metal ions modulate neurotransmitter release through their interaction with synaptic vesicles, ion channels, and the metabolism of neurotransmitters (NT). Many metals (e.g., Pb 2+, Cd 2+, and Hg +) also interact with intracellular signaling pathways. Postsynaptically, processes associated with the binding of NT to their receptors, activation of channels, and degradation of NT are altered by metals. Zn 2+, Pb 2+, Cu 2+, Cd 2+, Ni 2+, Co 2+, Li 3+, Hg +, and methylmercury modulate NMDA, AMPA/kainate, and/or GABA receptors activity. Al 3+, Pb 2+, Cd 2+, and As 2 O 3 also impair synaptic plasticity by targeting molecules such as CaM, PKC, and NOS as well as the transcription machinery involved in the maintenance of synaptic plasticity. The multiple effects of metals might occur simultaneously and are based on the specific metal species, metal concentrations, and the types of neurons involved.
1. Introduction
Metals and their compounds are distributed in ecosystems as a result of natural processes as well as anthropogenic activities. Metals are used in their elementary form as well as in compounds for various human needs. Therefore, a number of these metals enter our environment as a consequence of their widespread use in preservatives, biocides, and paints [1]. They are taken up by organisms through inhalation or by ingestion of food and water contaminated with these metals. For living systems, metals can be divided in those which are essential for life, such as cobalt (Co), copper (Cu), zinc (Zn), manganese (Mn), and iron (Fe); which are potentially toxic only at higher concentration, and those which have no known biological function, which can be toxic at all concentrations such as cadmium (Cd), chromium (Cr), mercury (Hg), and lead (Pb) [13] (for all abbreviations used in the review please refer to the abbreviations section; to facilitate reading, the names of the specific metals discussed are given in italics).
Since the uptake mechanisms of the body are not able to distinguish between “physiologically required” and harmful metals, the toxic metals absorbed consequently might interact with the functions of the central nervous system (CNS), liver, kidneys, and hematopoietic system, thus presenting a significant health hazard. In this review, we will examine the effects of these metals in the CNS, specifically at the synapse.
The human brain has about 1011 neurons, which interconnect and “communicate” with each other through synapses. It is estimated that each neuron has approximately 7000 synapses. At the presynaptic side of the synapse the incoming electrical signal, in from of action potentials, is transformed to a chemical signal in the form of neurotransmitter release. Synaptic transmission depends on the timely opening of membrane channels, the precise functioning of intracellular signaling pathways, and metabolic pathways involved in the synthesis and the release of neurotransmitters. Postsynaptically the binding of neurotransmitters changes the membrane potential, resulting in a hyper- or depolarization of the neuron and in the generation of an action potential when the threshold potential is reached. These are crucial process and the basis of all higher cognitive functions including learning and memory.
Therefore, we highlight the mechanisms by which metals and their compounds interfere with the processes of synaptic transmission and synaptic plasticity. This review covers the effects of metals on signal transmission from the presynaptic to the postsynaptic membrane, as well as the effects on synaptic plasticity with an emphasis on learning and memory, since subtle alterations in synaptic transmission due to the interaction of metals may have profound toxic effects in the CNS [14].
Some metals, which have already been shown to alter synaptic transmission, are discussed in this review. The metals are listed in an alphabetical order below with a short description of their neurotoxic effects, to show their relevance to this study (for more details regarding the neurotoxicity of these metals see [15]).
Aluminum was found present in high concentrations in brains of patients with Alzheimer's disease, Parkinson's disease, and dialysis encephalopathy and could contribute to neurogenerative disorders [21]. In animals the administration of aluminum salts results in neurofibrillary degeneration, a condition similar to the encephalopathy in Alzheimer's disease [3].
Arsenic, one of the oldest known poisons, due to its cholera-like symptoms, became a favorite poisoning agent and earned the title the “Poison of Kings” [15]. An acute ingestion of arsenic affects many systems of the body including gastrointestinal, cardiovascular, respiratory, and the nervous system. Even today, chronic low-dose exposure to arsenic is very common in countries like Bangladesh, India, Taiwan, and other parts of South East Asia due to contamination of groundwater by arsenic. It is a major cause of infant mortality in Bangladesh [8]. Chronic manifestations of arsenic poisoning are pigmentation changes, gastrointestinal problems, anemia, liver disease, black foot disease, and Mees' lines on the nails. Central neuropathy due to Arsenic poisoning usually manifests as impairment of learning, short-term memory and concentration. However, peripheral neuropathy is more frequently observed and this might last for several years. It manifests as a rapid and severe ascending weakness and sometimes these patients require mechanical ventilation [8, 15].
Cadmium and manganese also have neurotoxic effects, where cadmium damages cells of the cerebellar cortices of young rats as well as rabbits and chronic manganese poisoning causes extrapyramidal symptoms much like those of Wilson's disease and Parkinsonism [9]. Moreover, increased total cadmium levels in human hair were associated with mental retardation and impairment in visual motor abilities [4]. Similar toxicities also occur in humans.
Lead, whose mechanisms of neurotoxicity have been extensively studied, was discovered more than 5000 years ago and was used in the ancient world for lead water piping, as utensils, to sweeten food and wine, and as a constituent of eye paints [2]. It was discovered that acute exposure to Pb could cause lead colic and mental disturbances and even chronic exposure to low concentrations of lead in children caused several cognitive and behavioral disturbances. Since Pb crosses the placenta, prenatal exposure to lead can have especially severe consequences [4, 5, 15].
Exposure to dietary methylmercury leads to Minamata disease, which manifested in patients as paresthesias followed by irreversible impairment of vision, hearing, speech, gait, and ultimately leads to death. In addition, cognitive impairment ensued with prenatal exposure to methylmercury [4].
Organo-tins are industrially produced in large quantities for applications as PVC stabilizers, glass coverings, silicone, wood preserver additives, and antifouling paints. Moreover, considerable amounts of organo-tins are released in the environment causing large concern about their impact on human health. Due to their lipophilicity organo-tins are taken up by humans and distributed in different tissues. In mammalian organs such as brain, liver, and kidneys, organo-tins are biotransformed and this process may increase their toxicity [7]. Specifically alkyl-tins have been shown to cause neurotoxicity [15].
Even though metals are well known for their various toxicities, they are also used as therapeutic agents. Lithium salts have been used in the treatment and prophylaxis of bipolar affective disorder [10, 16]. Arsenic in the form of arsenic trioxide is used for the treatment of leishmaniasis, leukemia, and trypanosomiasis [8, 15]. The specific toxicities of some metals are actually being used to man's benefit, especially for the treatment of cancers. Cisplatin (cis-diammine-dichloro-platin = CDDP) is used as an anticancer drug and testicular cancer, endometrial cancer, prostatic tumors, bladder carcinoma, and small cell bronchial carcinoma [17] are successfully treated with this drug.
With the wide description of harmful effects of metals as well as their irreplaceability in modern life and medicine, it becomes essential to demarcate the level at which metals become toxic. This includes concentrations of metals as well as their targets of actions. Recognizing the targets sites at which metals interact can serve as a stepping-stone for the development of therapeutic agents to counteract metal toxicity as well as the side effects of anticancer drugs such as arsenic and cisplatin compounds.
This paper aims to review the literature available of the mechanisms of actions of metals at targets presynaptically, postsynaptically, and on long-term potentiation (LTP) and summarizes the findings in a logical and easily comprehensible manner. In the first part the toxic effects of organic and inorganic metals on the presynaptic part will be described (Section 2), followed by a review of their postsynaptic actions (Section 3), and the review finally looks at the impairment of synaptic plasticity (Section 4) before concluding remarks are made (Section 5).
2. Presynaptic Targets of Toxic Metals
Presynaptically, the action potential, which is an electrical signal, is transduced to a chemical signal in the form of neurotransmitter release. Generally, the action potential induces a membrane depolarization, which opens voltage gated calcium channels allowing the influx of Ca2+. Ca2+ activates calmodulin (CaM) and therefore CaM kinases (CamK) are activated, which leads to the phosphorylation of synaptic vesicle associated proteins and the conversion of the reserve pool of synaptic vesicles to a readily releasable pool of vesicles. Ca2+ also binds synaptotagmin, a calcium sensor protein in the vesicle membrane and triggers neurotransmitter vesicle fusion and the release of neurotransmitter [18] (Figure 1).
Figure 1.
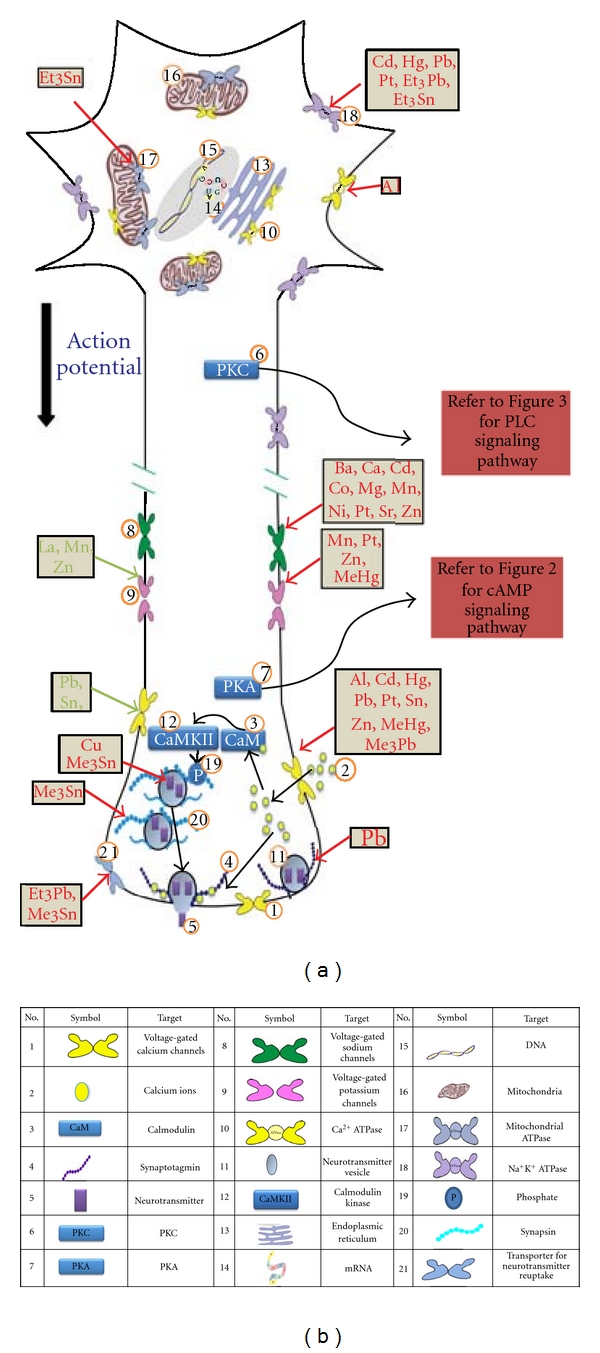
Presynaptic targets of neurotoxic metals. Events at the synapse from the arrival of the action potential which results in the membrane depolarization-induced opening of voltage-activated calcium channels and the entry of calcium which activates CaM, which activates CaM kinases and causes the phosphorylation of synaptic vesicle-associated proteins and an increase in readily releasable neurotransmitter vesicles. Calcium also binds synaptotagmin and causes exocytosis of neurotransmitter from the vesicles. Shown in boxes are the metals and the targets at which they act in the synaptic transduction pathway. A table at the end indicates the symbols and what they indicate. Green indicates activation or upregulation while red indicates inhibition or downregulation. Please refer to the section of Abbreviations and metals.
Metals interact with specific targets in these pathways and the same metals might even interact with various targets simultaneously. For instance, aluminum blocks voltage gated calcium channels, decreases the biological activity of CaM, and also inhibits Ca2+ ATPase [19, 20]. In addition, if a metal interacts with an upstream target of a pathway, it may influence all the processes succeeding it. For example, cadmium reduces voltage activated calcium channel currents, therefore, it can influence the intracellular calcium concentration and consequently the activation of CaM and calcium-dependent intracellular signaling pathways [6, 11]. Notably, cadmium caused a decrease in release of excitatory neurotransmitters glutamate and aspartate while it caused an increase in the release of inhibitory neurotransmitters GABA and glycine [12].
The upcoming sections (Sections 2.1 to 2.6) highlight the literature relating to the toxic effects of metals on presynaptic targets including voltage-activated ion channels (Section 2.1), signaling cascades (Section 2.2), transporters (Section 2.3), synaptic vesicle associated proteins (Section 2.4), neurotransmitters (Section 2.5), and neurofilaments and microtubules (Section 2.6). For ease of access, wherever possible metals are described in alphabetical order in each Section.
2.1. Voltage-Activated Channels
2.1.1. Voltage-Activated Calcium Channels
Voltage activated calcium channels open by a depolarization. They are subdivided into high- and low-voltage activated channels. The high-voltage activated channels, which have to be depolarized to more positive voltages than −30 mV for activation, include the L-type, P/Q-type, N-type, and R-type, where the L-type has a “long-lasting” current. The other types are divided on the basis of their inactivation and their susceptibility to various peptide toxins. There are also low-voltage activated channels which are mainly composed of the T-type channels which have a small, fast inactivating and therefore transient current [22].
Aluminum (Al3+) blocked N- and L-type voltage activated calcium channels in cultured rat dorsal root ganglions, with a threshold concentration of 20 μM and a Hill's coefficient of 3 (Table 1). It also required an open channel for its actions thereby indicating that the possible site of action of Al3+ was inside the channel. The current-voltage relation was shifted to depolarizing voltages in the presence of Al3+ [19]. Aluminum also blocked voltage-activated calcium channels in vivo in rats when given 10 mg per kg body weight per day intraperitoneally for 4 weeks. Inhibition was nearly 85% in the corpus striatum, 58% in the cerebral cortex, and 46% in the hippocampus [20].
Table 1.
Effects of metals on voltage-activated ion channels (↑—activation/upregulation, ↓—inhibition/downregulation).
| Voltage-gated channels | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Target | Calcium channels | Sodium channels | Potassium channels | |||||||||
| L | N | T | R | All(ii) | Tetrodotoxin sensitive | Tetrodotoxin resistant | All(i) | |||||
| Al | Effect | ↓ | ↓ | ↓ | ||||||||
| Conc | 20 μM | 20 μM | 50 μM | |||||||||
| Ref | [1] | [1] | [2] | |||||||||
|
| ||||||||||||
| Cd | Effect | ↓ | ↓ | ↓ | ↓ | |||||||
| Conc | 20 μM | 2.2, 125 μM | 5 mM | 0.2 mM | ||||||||
| Ref | [3] | [4], [5] | [6] | [6] | ||||||||
|
| ||||||||||||
| Co | Effect | ↓ | ||||||||||
| Conc | 500 μM | |||||||||||
| Ref | [7] | |||||||||||
|
| ||||||||||||
| Hg | Effect | ↓ | ↓ | ↓ | ↓ | |||||||
| Conc | 2.2 μM | 0.5–2 μM | 0.7 μM | 1.1 μM | ||||||||
| Ref | [8] | [9] | [8] | [10] | ||||||||
|
| ||||||||||||
| La | Effect | ↑ | ||||||||||
| Conc | 10 μM | |||||||||||
| Ref | [11] | |||||||||||
|
| ||||||||||||
| Mn | Effect | ↓ | ↑ | |||||||||
| Conc | 10 mM | 1 mM | ||||||||||
| Ref | [12] | [12] | ||||||||||
|
| ||||||||||||
| Pb | Effect | ↓ | ↓ | ↓ | ↓ | |||||||
| Conc | 30 nM(i), 0.7, 0.64, 0.1 μM | 80 nM(i), 0.64, 0.1 μM | 1.3 μM, 6 μM, 6 μM | 1, 1, 0.6, (1–30) μM | ||||||||
| Ref | [13], [14], [15], [1] | [13, 15] [1], [5], [1] | [14], [15], [4] | [16], [15], [4], [17] | ||||||||
|
| ||||||||||||
| Pt | Effect | ↓ | ↓ | ↓ | ||||||||
| Conc | 23.9 μM | 10 μM | 10 μM, 100 μM | |||||||||
| Ref | [18] | [18] | [18], [18] | |||||||||
|
| ||||||||||||
| Sn | Effect | ↓ | ↑ | |||||||||
| Conc | 50 μM | 50 μM | ||||||||||
| Ref | [19] | [20] | ||||||||||
|
| ||||||||||||
| Zn | Effect | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ | ||||
| Conc | 5,69 μM | 5,69 μM | 20 μM | 2 mM, 69 μM | 2 mM | 50 μM | 30 μM | |||||
| Ref | [1, 21] | [1, 21] | [21] | [10, 16] | [6] | [6] | [22] | |||||
(i)Extent of block and EC50 differ based on concentration of charge carrier used.
(ii)Paper does not describe which sub-type is affected.
Cadmium (Cd2+) effectively reduced voltage-activated calcium channel currents, which were high threshold and fast inactivating types in cultured chick dorsal root ganglion cells, at concentrations of 20 μM. This block was released at hyperpolarizing voltages, which may be due to shifts in gating and permeability of the channels. When the membrane potential was hyperpolarized, the channels conducted transiently, as Cd2+ exited the channels, but closed again thereafter. The channels can close with Cd2+ in the channel pore, therefore implying that Cd2+ does not affect the closing mechanisms of the channels [6]. Similar results were obtained in squid giant fiber neurons. In addition, a kinetic model was created and the binding site for Cd2+ was determined to be near the outer end of the pore, and the entry of Cd2+ into the pore was voltage independent while its exit was voltage dependent [11].
Lead (Pb2+) is a potent blocker of voltage-activated calcium channels in invertebrate Aplysia neurons as well as in mammalian neurons [19, 23, 25, 27, 28, 30]. There is no change in the voltage dependence of activation or inactivation of the channels in mammalian neurons, which suggests an external binding site for Pb2+ [25, 27, 28]. In mammals Pb2+ blocked N-, L- and T-type voltage activated calcium channels [19, 27, 28, 30]. The block of L- and T-type channels was concentration dependent and reversible in N1E-115 mouse neuroblastoma cells [30]. The concentration for 50% inhibition (IC50) of L-type channels was 30 nM, and for N-type channels it was 80 nM free Pb2+ where 10 mM Ba2+ was used as the charge carrier in cultured E-18 rat hippocampal neurons [28]. Some contradictory data were obtained in a study of mouse N1E-115 neuroblastoma cells, where in five of the fifteen cells studied, 2.3 μM Pb2+ enhanced L-type calcium channel currents and also enhanced the inactivation of L-type channels at holding potentials of −60 to −40 mV [30]. A study on human neuroblastoma cells SH-SY5Y determined that lead acetate at concentrations of 1 to 30 μM blocked voltage-activated calcium channels, both N- and L-types in a concentration-dependent and reversible way. More importantly, the concentrations used in the study were inclusive of the blood level concentrations at which children present with neuropsychological disorders (between 1.5–2.5 μM) [29].
Mercury (Hg2+) blocked voltage-activated calcium channels with an IC50 of 1.1 μM in vitro in rat pup dorsal root ganglion cells, and it required a partially open channel for its block [24]. Mercury (Hg2+) blocked neuronal N- and R-type calcium channels transiently expressed in human embryonic kidney 293 cells with an IC50 of 2.2 and 0.7 μM. This effect was partially reversible in N-type but not in R-type channels [26]. Mercury also blocked T-type calcium channel currents in the concentration range of 0.5–2 μM in cultured rat dorsal root ganglion cells. In addition the current-voltage relation was shifted to positive voltages implying that mercury may have an effect on channel gating [40].
Platinum in the form of cis-diammine-dichloro-platin (CDDP) reduced voltage-activated calcium channel currents in dorsal root ganglion cells of rats in vitro. CDDP reduced peak calcium current with an IC50 of 23.9 ± 4.5 μM and sustained current with an IC50 of 38.8 ± 6.1 μM in small neurons with a diameter of ≤20 μm. Surprisingly, in large neurons with a cross-sectional diameter of ≥25 μm, the peak calcium current was only reduced by 14.1 ± 2.3% even with a concentration of 100 μM CDDP. It is unlikely that the voltage-activated calcium channel currents were blocked directly since the small and large cells were unequally affected and the Hill's coefficient was not 1. CDDP probably decreases voltage-activated calcium channel currents by acting through an intracellular pathway more prominent in small neurons, possibly through IP3 receptor activation as described later [17].
Tin (Sn2+) used as stannous chloride (SnCl2) decreased voltage-activated calcium channel currents in vitro in rat dorsal root ganglion cells in a concentration-dependent manner with a threshold of 1 μM. These effects were found to be irreversible [41]. However, contradictory results were obtained in a study of motor nerve terminals of frog, where nerve muscle preparations were exposed to 50 μM SnCl2, which caused an increased inward Ca2+ current [34].
Zinc (Zn2+) blocked voltage-activated calcium channels in cultured rat dorsal root ganglion cells [19, 24, 31]. The IC50 for this effect on N- and L-type channels was 69 μM Zn2+ while the Hill's coefficient was 1. T-type currents were more sensitive, and the block was partly reversible in 50% of the neurons [31]. Zinc did not require an open channel for this blocking effect [24]. The current voltage relationship shifted to more depolarizing voltages in the presence of Zn2+, implying that the mechanism of action of Zn2+ may involve the screening of charges in the vicinity of the channels [19].
Methylmercury (MeHg) caused an increase in calcium influx and therefore [Ca2+]i through nifedipine and ω-conotoxin sensitive mechanisms, that is, through either, L-, N-, or Q-type calcium channels [36]. However, methylmercury caused an irreversible time and concentration dependent block of calcium channel currents at concentrations between 0.25 and 1 μM in vitro in rat cerebellar granule neurons. The block did not require depolarization, indicating that it did not require an open channel. Increasing the frequency of stimulation of cells increased the magnitude of block at 0.25 μM and 0.5 μM but not at 1 μM, which may suggest the presence of other counteracting effects. None of the calcium channel antagonists used—ω-conotoxin GVIA, ω-conotoxin MVIIC, ω-agatoxin IVA, calcicludine, and nimodipine, were able to decrease the MeHg-induced block of calcium channel currents [38]. MeHg blocked N-, R-, and L-type voltage-activated calcium channels [26, 39]. MeHg blocked human neuronal N- and R- type calcium channel currents transiently expressed in human embryonic kidney 293 cells with an IC50 of 1.3 μM and 1.1 μM respectively (Table 4). This block was determined to be irreversible [26].
Table 4.
Effects of organic metals on presynaptic targets (↑—activation/upregulation, ↓—inhibition/downregulation).
| Target | MeHg | Me3Pb | Et3Pb | Me3Sn | Et3Sn | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | Conc | Ref | Effect | Conc | Ref | Effect | Conc | Ref | Effect | Conc | Ref | Effect | Conc | Ref | |||
| Voltage-gated channels | Calcium channels | L | |||||||||||||||
| N | ↓ | 1.3 μM | [8] | ||||||||||||||
| T | |||||||||||||||||
| R | ↓ | 1.1 μM | [8] | ||||||||||||||
| All(i) | ↓ | 0.25–1 μM | [44] | ↓ | 0.5–50 μM | [45] | |||||||||||
| Potassium channels | ↓ | 2.2 μM | [46] | ||||||||||||||
|
| |||||||||||||||||
| ATPases | ↓ | 260 μM | [47] | ||||||||||||||
| Na+K+ATPase | ↓ | 5–20 μM | [48] | ↓ | 5–20 μM | [48] | ↓ | ||||||||||
|
| |||||||||||||||||
| Transporters | Na+-dependent GABA transporter | ↓ | 10 μM | [49] | ↓ | 10 μM | [49] | ↓ | |||||||||
|
| |||||||||||||||||
| Pathways | IP3 | ↑ | 3 μM | [50] | |||||||||||||
| Intracellular Calcium | ↑ | (0.5–1) μM | [51] | ||||||||||||||
| Synapsin I | ↓ | [52] | |||||||||||||||
| p38 | ↓ | [52] | |||||||||||||||
|
| |||||||||||||||||
| Neurotransmitters | GABA | ↑(iii) | 10 μM | [49] | ↑(ii) | 75 μM | [53] | ↓ | |||||||||
| Dopamine | ↓ | [54] | |||||||||||||||
| Norepinephrine | ↑(ii) | 43 μM | [53] | ↓ | |||||||||||||
| Serotonin | ↑(ii) | 24 μM | [53] | ↓ | |||||||||||||
(i)Paper does not describe which subtype is affected
(ii)Decreases uptake of neurotransmitter into synaptosomes, thereby probably increasing the amount in cleft
(iii)Release of neurotransmitter from vesicles is being measured.
Trimethyl lead blocked voltage-activated calcium channels with a threshold concentration of 0.5 μM in vitro in rat dorsal root ganglion cells. This block was irreversible and concentration dependent but not voltage dependent. It required an open channel and the IC50 was between 1–5 μM [33].
2.1.2. Voltage-Activated Sodium Channels
There are 9 subtypes of voltage-activated sodium channels Nav 1.1–Nav 1.9 distinguished by their sensitivity to tetrodotoxin and their rate of inactivation. Nav 1.8 and Nav 1.9 have relatively slower inactivation [35]. Nav 1.1, Nav 1.2, Nav 1.3, and Nav 1.7 are expressed in neurons and are highly sensitive to tetrodotoxin. Nav 1.5, Nav 1.8, and Nav 1.9 are relatively tetrodotoxin resistant and are found in heart and dorsal root ganglion neurons. Nav 1.4 and Nav 1.6 are mostly expressed in skeletal muscle and the CNS, respectively [37].
Zinc (Zn2+) and cadmium (Cd2+) reduced both tetrodotoxin-sensitive and tetrodotoxin-insensitive voltage-activated sodium channel currents in voltage clamp experiments in neuronal, cardiac, and skeletal muscle cells [42]. Tetrodotoxin-resistant channels were more sensitive to Zn2+ and Cd2+ with IC50 of the block being 50 μM and 0.2 mM, respectively; tetrodotoxin-sensitive channels were less resistant with IC50 of the block being 2 mM and 5 mM for Zn2+ and Cd2+, respectively [42] (compare effects in Table 1). It was suggested that the site of action of Zn2+ contains cysteine sulfhydryl groups in or near the saxitoxin binding site since Zn2+ was able to relieve the saxitoxin-induced block of the channel in a competitive manner, and the blocking action of zinc was inhibited by sulfhydryl-specific alkylating reagents. These experiments were done in voltage-activated sodium channels taken from the hearts of dogs or calves [32].
SnCl2 had an effect on voltage-activated sodium channel currents of the mollusk Lymnaea stagnalis in vitro where it shifted the current voltage curve to the left. SnCl2 increased voltage-activated sodium channel currents at a concentration of 10 μM, but caused a depression in current at concentrations above 25 μM. Organic tin in the form of (CH3)3SnCl decreased significantly the Na current only at high concentrations above 100 μM. Additionally the current voltage curve was shifted to the left. These effects were time dependent and irreversible [43].
Cobalt, manganese, nickel, calcium, magnesium, strontium, and barium in divalent cation form blocked both tetrodotoxin-sensitive and tetrodotoxin-insensitive channels in channels incorporated into planar bilayers in the presence of batrachotoxin. The block was voltage dependent and the sequence of affinity to block was Co2+≅ Ni2+>Mn2+> Ca2+> Mg2+> Sr2+> Ba2+. The suggested mechanisms of block included a specific divalent cation binding site and surface charge screening [44]. Also 10 μM of the anticancer drug CDDP reduced voltage-activated sodium channel currents by 9.2% ± 7.2% in rat dorsal root ganglions in vitro [17].
2.1.3. Voltage-Activated Potassium Channels
The family of voltage-activated potassium channel includes Kv 1–6, Kv 8, and Kv 9, where the principal subunit of the channels contains 6 transmembrane domains. All these channels are expressed in brain tissue [45]. Whole cell patch-clamp measurements of transient voltage-dependent potassium currents in rat suprachiasmatic nucleus neurons showed that Zn2+ potentiated current when activated from a holding potential of −60 mV (approximately the resting membrane potential). This potentiation was voltage dependent and arose from a shift of the inactivation current to more positive voltages. Zn2+ (30 μM) shifted the half-inactivation voltage by 20 mV from −80 mV to −60 mV [46]. Kuo and Chen showed that at hyperpolarized voltages Zn2+ inhibited voltage-dependent transient K+ currents which can be accounted for by the selective binding of Zn2+ to closed K+-channels with a dissociation constant of approximately 3 μM, which kept the channels closed and slowed the activation of the current [47].
Whole cell patch clamp studies in central neurons of Drosophila third instar larvae showed that millimolar Ca2+ and Mg2+ concentrations and micromolar concentrations of Zn2+ increased the peak inactivation current and shifted the steady-state inactivation curve of voltage gated potassium channels to more positive voltages, but had no effect on the voltage dependence of activation. A micromolar concentration Cd2+ had the same effect; however, millimolar concentrations of Cd2+ had an effect on both steady state inactivation and activation curves, where the midpoint of the activation curve was shifted more positively. The potency of effect on the inactivation current in terms of amount of shift of steady state inactivation curves was Zn2+ (2 mM) >Cd2+ (2 mM) >Ca2+ (20 mM) >Mg2+ (20 mM). The mechanism of action was most likely through specific binding to the channels at extracellular sites [48].
10 μM cisplatin in the form of CDDP reduced voltage-gated potassium channel currents by 20.9 ± 4.8% in small dorsal root ganglion neurons while 100 μM CDDP reduced the peak current by 12.8 ± 3.4% [17]. Micromolar concentrations of lanthanum (La3+) enhanced outward voltage-gated potassium channel currents evoked by depolarization steps from −50 mV in rat cerebellar granule neurons. 10 μM La3+ shifted the steady state inactivation curve by approximately 40 mV in the depolarizing direction and increased the slope factor slightly [49].
Mayer and Sugiyama showed that fast activating transient potassium channel currents were reduced by 10 mM manganese (Mn2+) in cultured rat sensory neurons. This reduction was due to a depolarizing shift of the activation curve and a slight reduction in maximum conductance. At the same concentration, steady state inactivation curves were also shifted to depolarizing voltages. The positive shift of steady state inactivation and activation curves were obtained for other metals as well, where the potency of shift was Cd2+ >Mn2+ = Co2+ >Ca2+ > Mg2+. Lower concentrations of Mn2+ (1 mM), however, increased the amplitude of fast inactivating transient potassium channel currents at prepulse potentials from −50 to −70 mV, which was due to a shift of the inactivation curve with no significant shift in the activation curve. These effects may have been due to binding to a specific site within the channel or to phospholipids in close proximity of the gating apparatus [50].
Organic metals also affect voltage-gated potassium channels. Methylmercury (MeHg) blocked voltage-gated potassium channels irreversibly, with an IC50 of 2.2 ± 0.3 μM in a concentration-dependent manner. The Hill's coefficient for this block was ∼1 [51].
2.2. Signaling Cascades
2.2.1. The cAMP System
G-protein-coupled receptors (GPCR) are coupled to Gs or Gi/o, where Gs acts as a stimulator of adenylate cyclase and the Gα subunit of Gi/o acts as an inhibitor of PKA (Figure 1). PKA phosphorylates Ca2+-channels, thereby enhancing the influx of Ca2+ and this increases the release of neurotransmitters [52]. Also PKA phosphorylates SNAP-25 and this leads to a larger pool of readily releasable vesicles [52]. The cAMP-system appears to enhance the release of neurotransmitter in response to a stimulus (Figure 2).
Figure 2.
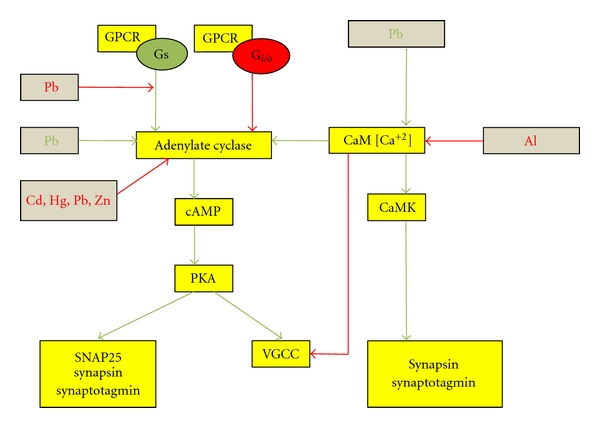
Effects of metals on the PLC signaling pathway at presynaptic terminal (green: activation/increase, red: inhibition/decrease). The PLC system consists of GPCRs coupled to Gq, which activate PLC, which activates DAG and IP3. IP3 increases intracellular calcium ([Ca2+]i) and activates DOC2 and synaptotagmin which leads to increased evoked release and readily releasable pool size. DAG activates PKC, which activates voltage-gated calcium channels. PKC phosphorylates Munc-18, which negatively regulates vesicle fusion and syntaxin. PKC activation leads to the increased spontaneous and evoked neurotransmitter release. The effects of metals on this pathway are shown in this figure where a green color indicates an activation/upregulation and a red color indicates an inhibition/downregulation.
In vitro and in vivo exposure to lead acetate decreased cAMP-dependent synaptic vesicle protein phosphorylation in rat brain which is most likely a contributing mechanism of lead toxicity [53].
Gs —
Rodrigues and colleagues determined the effect of lead acetate on rat cerebral cortex membranes using 5′ Guanylylimidodiphosphate (Gpp(NH)p). Gpp(NH)p is a nucleotide phosphorylase-resistant GTP analogue, which is known to stimulate adenylate cyclase by saturating Gs. On preincubation of membranes with lead acetate, the stimulatory effect of Gpp(NH)p on the adenylate cyclase activity was inhibited [10].
Adenylate Cyclase —
The same group [10] also determined the effects of lead acetate on adenylate cyclase activity in the cerebral cortex membranes and found that lead caused a concentration-dependent inhibition of adenylate cyclase activity with an IC50 of 2.5 ± 0.1 μM (Table 2) [10].
In another series of experiments, Ewers and Erbe [54] determined the effects of lead, cadmium, and mercury on adenylate cyclase of the cerebrum, cerebellum, and the brain stem, in vitro and in vivo. Adenylate cyclase activity was determined in terms of the number of moles of cAMP formed. Concentrations between 0.1 and 30 μM lead nitrate, cadmium nitrate, or mercury nitrate inhibited adenylate cyclase activity in vitro in homogenates of the cerebrum, brain stem, and the cerebellum. In vivo studies were performed on rats, which received lead acetate dissolved in sterile demineralized water, cAMP formation was determined 1 hour, 4 hours, and 24 hours after treatment. In the cerebellum, and brainstem, adenylate cyclase activity increased after one hour by about 25% but was unaffected in the cerebrum. After four hours, adenylate cyclase declined by 29%, 33%, and 21% in the cerebrum, cerebellum, and brainstem respectfully. By 24 hours adenylate cyclase activity had returned to normal in the cerebrum and brainstem but not in the cerebellum [54]. These differences in the effects of lead acetate on adenylate cyclase in different parts of the brain may be an indicator of the varied effects of lead on different isoforms of adenylate cyclase.
Zinc (Zn2+) inhibited adenylate cyclase with an IC50 of 1-2 μM and a Hill's coefficient of 1.33, which was not competitive with Mg2+ or Mg2+ATPase [82]. Both the CI and the CII domains of adenylate cyclase bind Zn2+ with high affinity which is correlated with Zn2+ inhibition of enzyme activity [83].
Table 2.
Effects of metals on presynaptic signaling pathways (↑—activation/upregulation, ↓—inhibition/downregulation).
| Target | Pathways | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PKC | Adenylate cyclase | Phosphodiesterase | CaM | IP3 | Intracellular calcium | ||||
| Al | Effect | ↓ | ↑ | ↓ | ↑ | ||||
| Conc | 0–100 μM | 0–1000 microM | 0–1000 μM | ||||||
| Ref | [2] | [23] | [2] | [2] | |||||
|
| |||||||||
| As | Effect | ↑ | |||||||
| Conc | 1 μM | ||||||||
| Ref | [24] | ||||||||
|
| |||||||||
| Cd | Effect | ↓ | ↓ | ↑ | |||||
| Conc | 0.4, 0.9, 1.4 (i) μM | 0.1–1 mM | |||||||
| Ref | [25], [25], [25] | [25] | [26] | ||||||
|
| |||||||||
| Hg | Effect | ↓ | ↓ | ||||||
| Conc | 0.8, 0.5, 0.9 (i) μM | ||||||||
| Ref | [25], [25], [25] | [25] | |||||||
|
| |||||||||
| Ni | Effect | ↓ | |||||||
| Conc | 30 μM | ||||||||
| Ref | [27] | ||||||||
|
| |||||||||
| Pb | Effect | NC | ↓ | ↓ | |||||
| Conc | 1500–10000 ppm | 2.5, 8.6,1.9, 8.0 (i) μM | |||||||
| Ref | [25] | [28], [25], [25], [25] | [25] | ||||||
|
| |||||||||
| Pt | Effect | ↑ | ↑ | ||||||
| Conc | 1 nM–10 μM | 1 nM–10 μM | |||||||
| Ref | [29] | [29] | |||||||
|
| |||||||||
| Zn | Effect | ↓ | |||||||
| Conc | 1-2 μM | ||||||||
| Ref | [30] | ||||||||
(i)The three different concentrations indicate actions in different areas of the brain—cerebrum, cerebellum, and brainstem.
2.2.2. The PLC System
The PLC system consists of GPCRs coupled to Gq, which activate DAG and IP3 through PLC. IP3 causes an increase of intracellular calcium ([Ca2+]i) and the activation of DOC2 and synaptotagmin which leads to increased evoked release and readily releasable pool size. DAG through PKC causes an activation of voltage-gated calcium channels. PKC phosphorylates Munc 18, which negatively regulates syntaxin and synaptic vesicle fusion [52, 84]. PKC activation eventually leads to an increase of spontaneous and evoked neurotransmitter release and more readily releasable pool of vesicles (Figure 3) [52].
Figure 3.
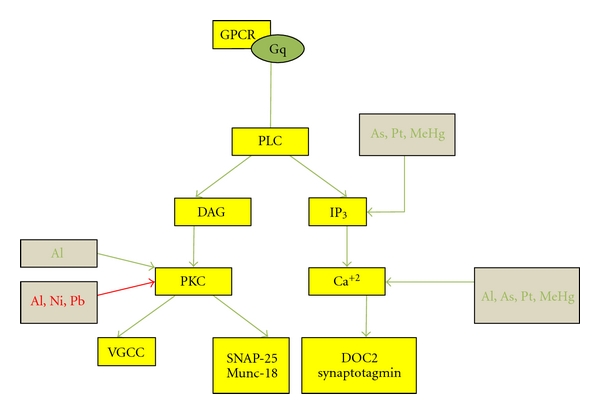
Effects of metals on the PLC signaling pathway at presynaptic terminal (green: activation/increase, red: inhibition/decrease). The PLC system consists of GPCRs coupled to Gq, which activate PLC, which activates DAG and IP3. IP3 increases intracellular calcium ([Ca2+]i) and activates DOC2 and synaptotagmin which leads to increased evoked release and readily releasable pool size. DAG activates PKC, which activates voltage-gated calcium channels. PKC phosphorylates Munc 18, which negatively regulates vesicle fusion and syntaxin. PKC activation leads to the increased spontaneous and evoked neurotransmitter release. The effects of metals on this pathway are shown in this figure where a green color indicates an activation/upregulation and a red color indicates an inhibition/downregulation.
PKC —
Metals that inhibit PKC include lead, aluminum, and nickel. Pb2+ inhibits PKC enzymes through interactions with its catalytic domains [85]. The effect of aluminum on PKC is debated: Julka and Gill demonstrated that aluminum lactate given to male albino rats for four weeks, caused an inhibition of PKC at all concentrations used (up to 100 μM). This was shown both in vivo and in vitro. The largest inhibition was observed in the cerebral cortex (47.73%) followed by the hippocampus (45.95%) and the corpus striatum (38.74%) [20]. However, contrasting findings were determined by Johnson and coworkers who showed that aluminum sulfate, when given orally for a period of 4 months to male Sprague-Dawley rats, showed an increase in PKC specific activity by 60% and total activity by 70% in the soluble fraction of cerebral cortex homogenates [69]. The different effects of aluminum could be attributed to the mode of intake reflecting differences in concentration of aluminum absorbed and its distribution to the brain or the duration of exposure.
Microarray analysis in neuronal PC12 cells indicated that exposure to Nickel (Ni2+) caused a decline in the transcription of two isoforms of PKC- prkcc, prkz, and two regulatory binding proteins prkcbp1 and prkcdbp, and also caused temporary upregulation and downregulation of prkcq at 24 hours and 72 hours, respectively [86]. These effects are important in terms of the events at the synapse because PKC activates voltage-activated calcium channels, and increases the secretion of neurotransmitter through effects on proteins involved in neurotransmitter exocytosis- Munc-18, and SNAP25 (a SNARE protein) [87].
IP3 —
Increase of [Ca2+]i in human cervix adenocarcinoma cells by cisplatin (0.001–10 μM) was dependent on extracellular Ca2+ and was blocked by an IP3 receptor blocker. The types 1–3 IP3 receptors were at the cellular membrane of these cells, which suggests a possible mechanism of cisplatin-induced calcium entry through IP3 receptor activation. This was supported by the observation that the same results were not obtained in human osteosarcoma cells, which in addition did not show the presence of types 1–3 IP3 receptors at cell membrane [88]. Arsenic trioxide (As2O3) similarly caused an increase in intracellular calcium which was dependent on calcium release from the intracellular calcium stores through the activation of IP3 receptors [88]. MeHg also causes an increase in intracellular calcium, which may be due to release from intracellular stores through inositol phosphate. MeHg doubled intracellular inositol phosphate levels at concentrations above 3 μM in vitro in rat cerebellar granule neurons [89].
2.2.3. Intracellular Calcium ([Ca2+]i)
Aluminum (Al3+) caused an increase of [Ca2+]i in rat synaptosomes, which could be a consequence of the inhibition of the Ca2+-ATPase [20]. 1 μM arsenic trioxide (As2O3) caused an irreversible increase in [Ca2+]i in human neuroblastoma cells (SY-5Y) and in human embryonic kidney 293 cells. This rise of [Ca2+]i was independent of extracellular calcium, but dependent on intracellular calcium stores. Blocking of IP3 receptor and ryanodine receptors with their specific blockers reduced the increase in [Ca2+]i indicating their involvement in this process [88]. Cisplatin also increased [Ca2+]i in a concentration-dependent manner in human cervix adenocarcinoma cells but not in human osteosarcoma cells. It is unlikely that the increase in [Ca2+]i is induced by entry of extracellular calcium, but more likely through activation of IP3 receptor as described above [88].
In addition, [Ca2+]i could indirectly be affected by several mechanisms. For example, [cis-(NH3)2Pt(H2O)2]2+, a form of platinum, caused an uncoupling of oxidative phosphorylation one minute after exposure in a concentration-dependent manner, which resulted in a release of Ca2+ from the mitochondria. Cisplatin did not produce the same effect even at a concentration of 500 μM [90]. However, another study by Gemba et al. showed that mitochondrial uptake of Ca2+ in rat kidney cortical mitochondria was decreased 24 hours after exposure to 500 μM cisplatin [91].
Methyl-mercury (MeHg) 0.5–1 μM caused an increase in [Ca2+]i in vitro in rat cerebellar granule neurons, which decreased cell viability (Table 4). This increase in cell death was prevented 3.5 hours after exposure by using two calcium channel blockers (ω-conotoxin and nifedipine) and a calcium chelator (1,2-bis(2-aminophenoxy) ethane-N,N,N9,N9-tetracetic acid tetrakis (acetoxymethyl) ester (BAPTA)). The effect of the calcium channel blockers could indicate that they inhibit the MeHg interaction with the target site or block of the entry of MeHg in addition to the effects on [Ca2+]i. BAPTA may have reduced calcium-induced cell death at 3.5 hours after exposure but was unable to prevent methylmercury-induced cell death at 24.5 hours. That may indicate that calcium-independent pathways of cell death are involved [92]. The increase of [Ca2+]i by methylmercury is caused by release from intracellular stores and through an influx of Ca2+ into the cell [89].
In HeLa cells, trimethyl-tin caused spikes in [Ca2+]i as well as sustained increases. The spikes were of variable size and duration and required 0.25 μM trimethyl tin. The sustained increase in intracellular calcium was partially reversible and dependent on the concentration of trimethyl tin used, where a 5 μM concentration caused an 8% increase in [Ca2+]i. These effects were independent of external calcium concentrations; however, the increase in [Ca2+]i was reduced when the internal calcium stores were compromised [7].
Overall, any of the metals affecting any channel or active transport mechanism that involves calcium, at the cellular membrane or the internal stores (as described above) could potentially change [Ca2+]i.
2.2.4. Calmodulin (CaM)
Calmodulin is a calcium binding protein. Ca2+/calmodulin activates CaMK, which phosphorylates synapsin I and opens voltage-activated calcium channels by phosphorylation. Thereby Ca2+-influx is increasing which is crucial for releasing the neurotransmitter from vesicles [52, 93].
Aluminum (Al3+) decreased the biological activity of CaM both in vitro and in vivo where inhibition in vivo is largest in the hippocampus (36.56%), followed by the cerebral cortex (31.76%) and the corpus striatum (22.49%) [20]. Lead, however, had an opposite effect as lead acetate enhanced CaM activity both in vitro and in vivo resulting in an increase in CaM-dependent synaptic vesicle protein phosphorylation including the phosphorylation of proteins such as synapsin I. This was proposed as a mechanism for increased spontaneous release of neurotransmitter and depletion of neurotransmitters norepinephrine and acetylcholine following exposure to lead [53].
2.3. Transporters
2.3.1. Ca2+-ATPase
Ca2+-ATPase activity in male albino rat synaptic plasma membranes was reversibly inhibited by Al3+ (up to100 μM). This inhibition was concentration dependent with an IC50 of 10 μM and resulted in an increase of [Ca2+]i [20] (Table 3).
Table 3.
Effects of metals on transporters, neurotransmitters, and neuropeptides (↑—activation/upregulation, ↓—inhibition/downregulation).
| Target | Transporters | Neurotransmitters | Neuropeptides | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ca2+ into mitochondria | Ca2+ ATPase | Dopamine transporter | Glutamate | Aspartate | GABA | Glycine | Dopamine | Acetylcholine | Substance P, neuropeptide K, and neurokinin | |||
| Al | Effect | ↓ | ||||||||||
| Conc | 0–100 μM | |||||||||||
| Ref | [2] | |||||||||||
|
| ||||||||||||
| Cd | Effect | ↓( v ) | ↓( v ) | ↑(v) | ↑(v) | ↓ | ||||||
| Conc | 10–30 μM | 10–30 μM | 10–30 μM | 10–30 μM | 5(iii) μM | |||||||
| Ref | [31] | [31] | [31] | [31] | [32] | |||||||
|
| ||||||||||||
| Cu | Effect | ↓ | ||||||||||
| Conc | 10(i) μg m−1 | |||||||||||
| Ref | [33] | |||||||||||
|
| ||||||||||||
| Hg | Effect | NC | h | |||||||||
| Conc | 400 μM | 6 mg/kg, 400 μM | ||||||||||
| Ref | [34] | [35], [34] | ||||||||||
|
| ||||||||||||
| Mn | Effect | ↓ | ↓ | ↓ | ↓ | |||||||
| Conc | 20–200 nm(iv) | 20–200 nm | 20–200 nm | 10(ii) μg mL−1 | ||||||||
| Ref | [36] | [36] | [36] | [33] | ||||||||
|
| ||||||||||||
| Ni | Effect | ↑ | ||||||||||
| Conc | ||||||||||||
| Ref | [37] | |||||||||||
|
| ||||||||||||
| Pb | Effect | ↑ | ↑ | ↑ | ||||||||
| Conc | 50 μmol L−1 | 100 μmol L−1 | ||||||||||
| Ref | [38] | [39], [38] | [39] | |||||||||
|
| ||||||||||||
| Pt | Effect | ↓ | NC | |||||||||
| Conc | 0.5 mM | 500 μM | ||||||||||
| Ref | [40] | [41] | ||||||||||
|
| ||||||||||||
| Sn | Effect | ↑ | ||||||||||
| Conc | 10–100 μM | |||||||||||
| Ref | [42] | |||||||||||
(i)Upregulates expression of monoamine oxidase, decreases production, and increases depletion
(ii)Downregulates expression of tyrosine hydroxylase gene
(iii)Reduces expression of precursor gene
(iv)Another study shows that uptake of glutamate into astrocytes is reduced through the decreased expression of glutamate aspartate transporter; this may result in increase in glutamate levels in the synapse [43]
(v)Shows effect on neurotransmitter release.
2.3.2. Na+/K+-ATPase
Cisplatin caused a concentration and time-dependent decrease in Na+-K+ATPase activity in liver and kidney cells [90]. Lead also affected Na+-K+ATPase activity, and one study showed lowered RBC membrane Na+/K+-ATPase activity below 60% in 77% of patients with Pb-Rbc ≥ 40 μg/100 mL while only 40% had the same decrease in activity who had a Pb-Rbc ≤ 40 μg/100 mL [94]. Mercury compounds, cadmium, triethyltin, and trimethyl-tin also inhibit Na+-K+ATPase activity [95–97]. Triethyl lead altered the microviscosity of the plasma membrane of ascites tumor cell and also completely inhibited Na+-K+-ATPase at concentrations 5–20 μM possibly through direct interaction with its catalytic subunit [98].
2.3.3. Mitochondrial ATPase
Mitochondrial ATPase was inhibited in adult rat brain homogenates with an IC50 of 260 μM by triethyltin [99]. Trimethyl tin has also been shown to affect mitochondrial ATPases in vitro [97].
2.4. Synaptic Vesicle Associated Proteins
2.4.1. Synaptotagmin I
Synaptotagmin I is a membrane protein, which is hypothesized to be a Ca2+-sensor in Ca2+-dependent neurotransmitter exocytosis. It has a short intravesicular N-terminus and the cytoplasmic part is composed mostly of two C2 domains, C2A and C2B. The C2A domain is known to bind two Ca2+-ions and the binding affinity shows a correlation with the Ca2+ dependence of exocytosis [100]. Synaptotagmin I binds phospholipids and syntaxin in a Ca2+-dependent manner. The binding to syntaxin is associated with exocytosis. The C2B domain of synaptotagmin I also has Ca2+ binding sites and is involved in the Ca2+ dependent self-association of synaptotagmin I into multimers [100].
Synaptotagmin I was recently described as a target site for lead. Bouton and coworkers suggested a competitive interaction between Pb2+ and Ca2+ for the Ca2+ binding sites in the C2A domain of synaptotagmin I. At nanomolar concentrations Pb2+ induced the binding of synaptotagmin I to phospholipids with an EC50 of 8 nM. This made it a thousand times more potent than Ca2+ at increasing phospholipid binding to synaptotagmin I. Binding of Pb2+ also increased the stability of the secondary structure of synaptotagmin I. A concentration of 2 μM free Pb2+ protected a 32 kDa fragment of synaptotagmin I from proteolytic degradation. It required 11 μM free Ca2+ to protect the same size of synaptotagmin I. The same authors showed that Pb2+, unlike Ca2+, did not induce the interaction of synaptotagmin I and syntaxin. Overall, the interaction of Pb2+ was competitive with Ca2+ and nanomolar concentrations of Pb2+ could inhibit the ability of micromolar concentrations of Ca2+ to induce the interaction of synaptotagmin I and syntaxin [101].
Four binding sites of Cu2+ in the cytoplasmic C2A domains of synaptotagmin I are discussed, three of which are common to Ca2+, and one of which is unique to Cu2+. It was suggested that Cu2+ has a competitive interaction with Ca2+, but Cu2+ has a greater affinity for the binding sites common to these metals. Also it was determined that Cu2+ caused a conformational change in the protein, which may make it less susceptible to trypsin cleavage [102]. Kathir and colleagues looked at the interactions between Cu2+ and the C2B domain of p40 synaptotagmin I, which is formed by an alternative translation of the synaptotagmin I gene at the Met103 of the p65 synaptotagmin I. They determined that these interactions stabilized synaptotagmin I bound to phosphatidyl serine vesicles [103].
2.4.2. Synapsin I and p38
Synaptic vesicle associated proteins, synapsin I and p38, in rat CNS decreased on acute exposure of rat to trimethyl tin. This decrease was both concentration and time dependent; however, 12 weeks after the exposure, the levels returned to normal. The decline was not a result of loss of tissue which also occurs with trimethyl tin exposure but was significantly greater that the reduction in tissue [104].
2.5. Neurotransmitters
Release of neurotransmitters is modulated by multiple mechanisms. How metals interfere with some of these pathways has been described above. The following paragraph focuses how metals modulate neurotransmitter levels, their release, and uptake in the presynaptic button.
2.5.1. Effect on Neurotransmitter Metabolism
Treatment of PC-12 dopaminergic neuronal cells with 10 μg/mL copper nanoparticles (Ø 90 nm) caused a decrease in dopamine and its metabolites 3.4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA). This indicates that the decrease in dopamine may be attributed to a decrease in production and an increase in the breakdown of dopamine [105].
In the same cell line, 10 μg/mL manganese nanoparticles (Ø 40 nm) caused a suppression of the tyrosine hydroxylase gene expression, which is involved in the synthesis of dopamine [105]. Aluminum has been shown to decrease striatal dopamine content and inhibit the enzyme dopamine-β-hydroxylase, which converts dopamine to norepinephrine [106].
Among organic metals, trimethyl tin hydroxide treatment of rats on alternate days from days 2–29 of life was shown to decrease the amount of dopamine in the striatum without affecting dopamine metabolites homovanillic acid and dihydroxyphenylacetic acid [107].
2.5.2. Effect on Neurotransmitter Release
Stannous chloride increased the amplitude of end-plate potentials in frog neuromuscular junction. A concentration of 10–100 μM SnCl2 increased the quantum of end plate potentials (EPP). However, the miniature end plate potential (MEPP) was not affected. Hattori and Maehashi (1988) suggested that this was due to an increase in the evoked neurotransmitter release while there was no effect on spontaneous release of neurotransmitter. Also, SnCl2 did not increase MEPP amplitude or acetylcholine (Ach) potential, indicating that the sensitivity to ACh was not altered [108].
Lead (Pb2+) in concentrations of at least 100 nM was found to increase the spontaneous release of glutamate and GABA from the presynaptic terminal of rat hippocampal neurons. This effect was found to be concentration dependent and partially reversible and the suggested mechanism of action was through an intracellular signaling pathway [109]. Similarly, it is likely that other metals also affect neurotransmitter release through their interaction with the voltage-gated ion channels, intracellular signaling pathways, and synaptic vesicle associated proteins.
One study looked at the effects of cadmium on synaptic transmission by perfusing the amygdala of rats with 10–30 μM CdCl2. There was an inhibitory effect on the release of excitatory neurotransmitters glutamate and aspartate while the release of inhibitory neurotransmitters glycine and GABA was stimulated [12]. Aluminum, as described in previous chapters, affects [Ca2+]i and, therefore, as expected, inhibits the release and uptake of GABA from synaptosomes by inhibiting Ca2+/calmodulin-dependent calcineurin. It also inhibited pyruvate-supported calcium-evoked acetylcholine release in synaptosomes while in differentiated SN56 cells it decreased acetylcholine release on short-term exposure and increased release on long-term exposure [106].
2.5.3. Effect on Neurotransmitter Reuptake
Trimethyl tin, in vitro, inhibited the uptake of neurotransmitters GABA, norepinephrine and serotonin, with an IC50 of 75, 43, and 24 μM in a concentration-dependent manner in mouse forebrain synaptosomes. In vivo, at 2 and 14 hours after trimethyl tin exposure, uptake of GABA, and serotonin was decreased whereas there was no significant decline in norepinephrine. These changes in uptake of neurotransmitters could explain their altered levels in the synaptic cleft [110]. However, unlike trimethyl tin, triethyl tin sulfate had no effect on the levels of dopamine, GABA or acetylcholine in rat brain on exposure for 6 days a week from days 2 to 29 of life in mice [107].
Triethyl lead caused a concentration-dependent inhibition of Na+-dependent high-affinity GABA uptake with an IC50 of 10 μM in rat brain synaptosomes. These results were not dependent on Na+ and GABA concentration-indicating that competition with Na+ and GABA were not the mechanism of action. Triethyl lead also caused a time- and chloride-dependent decrease in ATP [111]. Skilleter showed that trialkyl lead at 1 μM causes a decrease in pyruvate uptake by mitochondria in a KCl medium which could possible explain the decline in ATP [112]. However, since the inhibition of GABA uptake occurs before a significant decline in ATP, Seidman and Verity suggested that the inhibition could be due to a defect in GABA binding to uptake site [111].
2.6. Neurofilaments and Microtubules
In vivo exposure of Wistar rats to arsenic caused a dose-dependent decrease in neurofilament M and L proteins in the sciatic nerve [113]. These components are required for the formation of a heteropolymer in the cytoskeleton. Since the mRNA expression of these proteins was unaffected, it is possible that the decrease was a consequence of proteolysis. Caplain, which is a calcium-activated cytoplasmic protease, has been implicated in this phenomenon due to the increase in cytoplasmic calcium caused by trivalent arsenic [8].
Triethyl lead also affects microtubules [114–116] and neurofilaments [117]. 50 μM triethyl lead caused an inhibition of assembly and a disassembly of microtubules in vitro in porcine brain [114]. Triethyl lead also caused a reversible perinuclear coil formation of neurofilaments in vivo in mouse neuroblastoma cells, which was not associated with a significant change in the microtubules. In vitro, triethyl lead caused bulging and constriction of isolated neurofilaments from porcine spinal cord, and an unraveling of fibers in preformed filaments [117].
2.7. Summary of Presynaptic Effects of Metals
To summarize, presynaptically, voltage-gated sodium, potassium and calcium ion channels are affected by metals such as Al3+, Cd2+Pb2+, Hg2+, cisplatin, Sn2+, Zn2+, Co2+, Ni2+, Mn2+, Ca2+, Mg2+, Sr2+, Ba2+, and La3+. Mechanisms of effect included binding to a specific target, charge screening, shift of current-voltage curves, and competitive inhibition with the physiological ion or a combination of mechanisms [6, 11, 17, 19, 20, 23–32, 34, 41, 42, 44, 46–50]. Metals also interact with intercellular signaling pathways to modulate synaptic transmission. Lead modulated Gs, adenylate cyclase, PKC, and CaM [10, 53, 54, 85]. Adenylate cyclase activity was also modulated by cadmium, mercury, and zinc [54, 82, 83] while PKC was inhibited by lead, aluminum, and nickel, [20, 69, 85, 86], and IP3 was inhibited by cisplatin and arsenic [88]. Intracellular calcium was affected through interference with several targets including voltage-gated calcium channels, Ca2+ ATPases, and intracellular pathways. Al3+, As2O3, and cisplatin modulated intracellular calcium [20, 88, 90, 91], and aluminum, and lead affected Calmodulin activity [20, 53]. Ca2+-ATPase activity was inhibited by aluminum and Na+/K+-ATPase activity was modulated by lead, cisplatin, mercury, and cadmium [20, 90, 94–96]. Synaptotagmin I, a synaptic vesicle associated protein, was a target site for interaction with Pb2+ and Cu2+ [101–103]. Neurotransmitter release was possibly affected through interaction with many of the targets above as well as through interaction with synthesis and degradation of neurotransmitters and enzymes in the metabolic pathway, which resulted in modulation of neurotransmitter release by metals such as copper, manganese, and tin [101–103]. The mRNA expression of neurofilaments was affected by arsenic [113]. Often contradictory results were obtained regarding the effects of metals, which may indicate that metals had different effects on targets depending on the state of the metal, its concentration, the medium, the area of the brain, and whether the experiment was in vivo or in vitro.
3. Postsynaptic Targets
The activation of ligand-gated receptor channels is vital for controlling nerve cell inhibition or excitation and, therefore, fashioning the response of individual neurons, neuronal networks, and, ultimately, the entire brain. Consequently the resulting currents through the associated channels will either depolarize or hyperpolarize the postsynaptic terminal under different physiological conditions. The major excitatory neurotransmitter in the brain is L-glutamate. There are three classes of ionotropic glutamate receptors named according to their potent excitatory amino acids: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate, and N-methyl-D-aspartate (NMDA). The AMPA and kainite-activated channels are designated non-NMDA receptor-channels and will be further discussed in Section 3.2 while metal actions at the NMDA/receptor channel complex are analyzed in the upcoming Section 3.1.
The most abundant inhibitory neurotransmitter in the brain is γ-aminobutyric acid (GABA), which acts on its own class of ligand-gated channels. However, these receptors are subject to modulation by other compounds and ions, including metals [118] (Figure 4). As mentioned in the introduction, biological systems utilize metals because of their catalytic versatility, but the high affinity of these metals to specific binding sites could possibly severely impair synaptic transmission and, therefore, cause a malfunction of neuronal networks which might result in changes in perception, learning and memory, and finally change behavior, even at very low and environmentally relevant concentrations.
Figure 4.
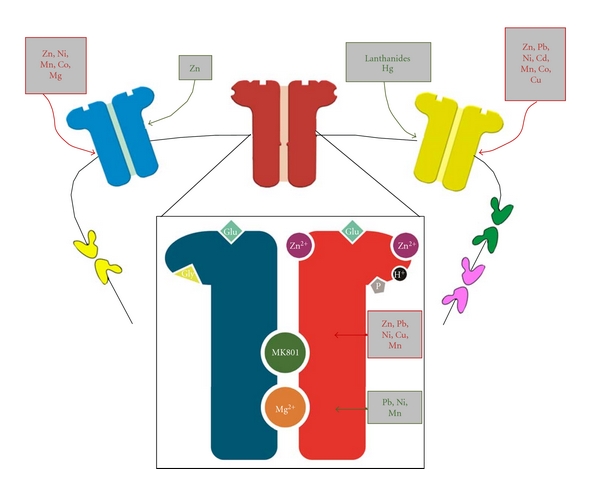
Postsynaptic ligand-gated ion channels as targets of neurotoxic metals. The main postsynaptic channels are the AMPA/kainate and NMDA receptors whereas the main inhibitory receptors are the GABAARs. Each receptor represents a target for multiple metals. The NMDAR has many modulatory sites identified as it is more extensively studied. NMDAR is composed of a heteromer made of NR1 and NR2, each having multiple subtypes. In the diagram blue arm represents NR1 while the red arm represents NR2, the main modulatory subunit. Most metals have been shown to have effects on NR2 subunit (for values regarding the specific subtypes refer to Section 3.1 and Table 5).
3.1. The NMDA-Receptor/Channel-Complex
The N-methyl-D-aspartate receptor (NMDAR) is a subtype of glutamate ionotropic receptors. The most widely distributed and studied NMDARs are tetrameric assemblies composed of two NR1 subunits and two of the four different NR2 types (named A, B, C, and D), of which NR2A and NR2B are most common [119, 120]. The physiological and pharmacological properties of these receptors are dependent on the NR2 subunit, although different NR1 splice variants may also influence channel performance [121, 122]. NMDAR subunits have a characteristic modular architecture consisting of two extracellular domains, the regulatory amino terminal domain (ATD) and the agonist-binding domain (ABD), and three membrane-spanning segments (M1, M3, and M4) and a reentrant hairpin-like pore loop, M2 [123].
The associated NMDAR channel requires simultaneous binding of two agonists, glutamate (Glu) and glycine (Gly), for opening (for review, [124]). Gly has its binding site in the ABD region of NR1 whereas NR2 ABD binds Glu [120]. The receptor-channel complex has unique properties such as a high Ca2+ permeability. Also, the functional activation of NMDAR channels is linked to a voltage-dependent magnesium- (Mg-) mediated block [125, 126]. Extracellular Mg2+ inhibits NMDA responses at membrane potentials close to the resting membrane potential [127]. Studies of the site of action of Mg2+ reveal that the N and N+1 site on NR2 subunit are important for the Mg2+ block [128]. When the membrane potential is sufficiently depolarized, Mg2+ leaves its binding site and even potentiates NMDA responses in low glycine concentrations. This potentiation is shown to be due to increased NMDAR affinity to glycine, in all neurons [129]. However, there was also glycine-independent potentiating effect of Mg2+, which appeared to be largely voltage-independent and subunit specific, being seen only with NR2B-containing receptors. This potentiation has an EC50 of ∼2 mM [130].
All of these effects reveal the complicated modulation by Mg2+ on NMDAR currents. Some data suggest that Mg2+ and spermine may completely or partially share a common binding site; similar observations are obtained using spermine [130, 131]. Three different steps in the action of these two substances could be distinguished: (1) increase in glycine affinity, seen in all neurons; (2) voltage-dependent block, also seen in all neurons; and (3) glycine-independent potentiation that was subunit specific [132, 133].
NMDARs contain a number of distinct recognition sites for other endogenous and exogenous ligands, which modulate their functions, such as divalent metal cations, as explored in the later sections (Figure 4).
Zinc is the second most prevalent trace element in the body. Most of the zinc ions (Zn2+) are trapped within proteins, but some of it is loosely bound (chelatable zinc) [134]. In the mammalian brain, chelatable zinc is distributed mainly in the forebrain and localized almost exclusively within synaptic vesicles of a subset of glutamatergic axon terminals [135]. Since it is accumulated in synaptic vesicles, it has been assumed that zinc is released, with glutamate, during neuronal activity. Many studies have showed evidence of quantal corelease of zinc and glutamate (for review, [134]).
NMDARs are the best characterized synaptic zinc targets. At low micromolar concentrations, Zn2+ selectively inhibited NMDAR-mediated responses. The major effect was through voltage-independent, noncompetitive inhibition seen as a decrease in the opening probability of the channel [136–138]. However, at concentrations higher than 20 μM, Zn2+ could also produce voltage-dependent inhibition, probably by binding inside the pore at the Mg2+ blocking site [139]. It had been proposed that zinc is an endogenous ligand controlling NMDARs functions [140].
An important consideration in NMDAR function and pharmacology is that the Zn2+ binding to NR2A and NR2B subunits is associated with discrete subunit selectivity [141]. NMDARs containing the NR2A subunit had a very high sensitivity to extracellular Zn2+ (IC50 ∼15 nM) [133, 142]; however, this inhibition never exceeded 60–80% [142]. The mechanisms of this inhibition occurred in different steps [143]: in the first step Zn2+ bound in the interlobe cleft of the NR2A-NTD promoting its closure, which would exert tension on the linkers connecting NTDs to ABDs. This effect would secondarily cause a disruption of the ABD dimer interface. In turn, this disruption relieved the strain on the transmembrane segments, and with proton binding, it allows the closure of the channel gate [133]. This mechanism of enhancement of proton inhibition was supported by subsequent work [144].
Zinc has a much lower affinity to the NR2B subunit, compared to NR2A, with voltage-independent inhibition (IC50 ∼1 μM) [122, 145]. It was suggested that the mechanism of inhibition might be similar to the mechanism described for NR2A receptors. However, zinc inhibition of NR2B receptors appeared to not be dependent on pH [122], suggesting that this inhibition might occur through a different mechanism [133]. Studies showed that Zn2+ bound with high affinity to a site in NR2A ATD region [146] and with a lower affinity to a site in the same region of the NR2B [145]. The affinities of NR2C and NR2D to Zn2+ described to be even higher (IC50> 10 μM) [134].
Lead (Pb2+) is an exogenous heavy metal, which has been a public health concern due to its widespread contamination and its multiple toxic effects. Effects of acute exposure to Pb2+ in the micromolar range were originally described in cultured and acutely dissociated neurons as a reversible inhibition of the NMDAR current [19, 147–150]. These studies outlined several features of the effects of Pb2+ (for details, see review [151]). First, the inhibition was specific for NMDA channels, which were significantly more sensitive to Pb2+ inhibition than other glutamate channels. Secondly, the channel block was independent of voltage [148–150], and therefore the interaction site was likely to be located away from the electric field, or outside the conducting pore. Thirdly, the effect was noncompetitive since increasing the glutamate or glycine concentration could not overcome the block of the current [148, 152]. Biochemical studies suggested that the inhibitory effects of Pb2+ on NMDA receptors were age- and brain-region specific [152–154]. One important observation in Pb2+ neurotoxicity was that the hippocampus appears to be more sensitive than other brain regions [153, 155, 156].
The effect of Pb2+ on glutamate and NMDA-evoked currents depended on the subunit composition of the receptor-channel complex. Concentration-dependent Pb2+ inhibited the currents activated by either Glu or NMDA in oocytes expressing NR1-2A or NR1-2B (Table 1, [157]). Yamada and colleagues [158], however, showed that higher concentrations were needed than mentioned before, although, there were methodological differences between the two studies, which could account for the different results (for details, [157]).
Also, Pb2+ at low concentrations (<1 μM) acted as a positive modulator of agonist action on NR1-2AB and NR1-2AC receptors whereas at higher concentration lead inhibited NR1-2AB and NR1-2AC receptors, but with less potency compared to NR1-2A or NR1-2B [157, 159].
There is incongruity whether lead acts via the zinc binding site or through a different site. A set of experiments demonstrated that in the presence of increasing amounts of Pb2+, there was a concentration-dependent downward shift of the Zn2+ inhibition curve; also, the values of IC50 for Zn2+ inhibition decreased as a function of increasing Pb2+ concentrations. The effects of Zn2+ on Pb2+ curve and IC50 were analogous [160]. These findings suggested that the two metals act via independent binding sites, which is in line with the observation that increasing concentrations of Pb2+ did not affect the Zn2+ IC50 [154]. However, these results were in contrast with other findings which report that the two cations compete for the same binding site [161, 162].
Nickel (Ni2+) is a trace element, which is essential for many biological organisms, but could also induce toxicity. The effects of Ni2+ on NMDA channel activity were described as a voltage-dependent and “Mg2+-like” inhibition [127]. Later work showed a potentiation of homomeric NR1a channels [121] and an inhibition of NR1-2A channels [13, 146]. In more recent studies, it is suggested that, besides the voltage-dependent Mg2+-like inhibition, Ni2+ causes a potentiation of NR2B-containing channels and a voltage-independent inhibition (Zn2+-like inhibition) in those neurons containing NR2A [163].
Nickel also caused a reduction of single channel current amplitude at negative voltages while the dependence on membrane voltage was slightly steeper for NR2A than NR2B [13]. Several analogies with Mg2+-like inhibition indicated that it might interact with either the N or N+1 site in the pore-forming region of the NR2 subunit [13]. Support for the above theory came from an experiment showing that a single mutation in the NR2B site at the N+1 site would completely abolish the voltage-dependent block Ni2+ [123]. The N+1 residue had been shown to be a critical binding site for Mg2+ block in NR2A subunit [128]. Moreover, at positive potentials the effects of Ni2+ were highly subunit dependent. NR2A-containing channels were blocked in a voltage-independent manner whereas NR2B containing channels were facilitated [13] (see Abbreviation section). However, at higher concentrations (IC50 = 442 μM), a voltage-independent inhibition was also present in NR1-2B channels [123]. The voltage-independent inhibition site of Zn2+ was investigated as a potential site for Ni2+ inhibition, but this did not seem to be the case. Besides the difference in blocking affinity, Zn2+ inhibition was pH dependent [122, 164] while Ni2+ inhibition was not [163]. Also, Zn2+ inhibition was never more than 60–80% (as described in section 3.2.1), while Ni2+ inhibition approached 100% at positive potentials [123]. Other results also showed that mutations that affect the inhibition of Zn2+ did not modify Ni2+ sensitivity [146].
The NR2B-selective potentiation was suggested to share the site of action with spermine, as Ni2+ partially obscured the effect of spermine when they were applied concurrently [123].
Although the actions of zinc, lead, and nickel on NMDARs were intensively investigated, there are also some reports that other metals have an effect on these receptors and channel activity.
Copper (Cu2+) is an endogenous metal in the human brain [165], and it is an established fact that copper represents an integral part of neurotransmission [166]. It is released from synaptic vesicles following neuronal depolarization [167]. The concentration of copper in the synaptic cleft could reach up to 100 μM [168]; later studies estimated the concentration of copper released into the synaptic cleft to be in the range of ~15 μM [169, 170]. However, the topographic distribution showed marked variations between different brain areas [171]; the highest concentration of copper has been found to be in the hypothalamus [172].
Cu2+ acts on NMDA receptors and reduces the current—induced by 50 μM NMDA—in a concentration-dependent manner with an IC50 of 15.9 μM. This block was completely and quickly reversible, even in the absence of antioxidant dithiotreitol, suggesting that the inhibition was not an oxidizing effect [173]. Further studies showed that Cu2+ inhibition was characterized by voltage-independent, but use-dependent mechanism of action, as the degree of inhibition was dramatically decreased in the absence of agonists [174].
Another trace metal required for normal brain function is manganese (Mn2+). In the human brain, Mn2+ is most concentrated in the globus pallidus, caudate, and putamen, but also found in other areas [175]. Mn2+ produces a strong voltage-dependent block in response to NMDA [176]. It was, also, a competitive antagonist of MK-801 binding to the NMDAR-channel. Its inhibitory effects were activity-dependent since Mn2+ was a more potent inhibitor in the presence of NMDA coagonists (Glu and Gly) than in their absence [177]. Taking these studies together, they indicate that Mn2+ is an NMDAR channel blocker. Interestingly, the inhibitory constant for Mn2+, in the absence of Glu and Gly, was significantly different in neuronal membranes from the cerebellum relative to other brain regions; however, in the presence of the agonists, Mn2+ was equally potent in inhibiting NMDARs in different brain regions [177].
3.1.1. The Glycine-Binding Site of the NMDA-Receptor/Channel Complex
The NMDAR glycine-binding site was susceptible to modulation by divalent cations, especially when the glycine site was not saturated. Low, extracellular concentrations of Mg2+ potentiated NMDAR currents. The potentiation was the result of an increase in the affinity of NMDAR for glycine [129, 130, 178]. The mean glycine EC50 value was 100–133 nM in control conditions and was reduced to 60–62 nM in the presence of 10 μM Mg2+ [178]. This increase in affinity was also demonstrated by decreasing the inhibitory potency of NMDAR glycine-site antagonists upon the addition of potentiating concentrations of Mg2+[129, 178, 179]. Ca2+ had the same effect as Mg2+ [129, 178, 179].
Concentrations of Pb2+ and Zn2+ higher than 10 μM inhibited NMDAR potentiation by Ca2+ and Mg2+ [178]. These findings were supported by other studies, which showed that increasing concentrations of Ca2+ diminished the inhibition of NMDAR currents by Zn2+ [136], or Pb2+ [180]. It is suggested that these divalent cations act on the same site, and Ca2+ and Mg2+ have opposite effects on glycine binding compared to Pb2+ and Zn2+ [178].
Even the presence or absence of glycine modulated the effects of some of the cations: for example, Cu2+ and Mn2+ are both potent NMDAR channel inhibitors (as shown above), but in the presence of glycine and glutamate, Cu2+ was more potent than Mn2+, and in the absence of glycine, Mn2+ was slightly more potent [177].
3.2. AMPA and Kainate Receptors
The two classes of ionotropic glutamate receptor-channels, which are designated non-NMDA channels, are AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) and kainate receptors, named after their most potent excitatory amino acids. The AMPAR channel is also activated by kainate (for review, [181]). Also, AMPARs mediate the fast excitatory synaptic transmission in the CNS [182].
AMPA/kainate receptor-gated channels are permeable to Na+ and K+ and more or less impermeable to Ca2+; however, there is a subpopulation of central neurons, which contain AMPA/kainate receptors with enhanced Ca2+-permeability [183, 184]. This Ca2+ conductance triggered by the AMPA/kainate receptors seemed to be dependent on the absence of the GluR2 subunit [119, 182, 185].
Extracellular calcium ions produced rapid and reversible voltage-independent inhibition of AMPARs, with both Ca2+ permeable and Ca2+ impermeable AMPAR being equally sensitive [186]. The Ca2+ effects were agonist dependent, more prominent in the case of AMPA compared to Glu or kainate. These data suggested that Ca2+ enhances desensitization, as two well-known antidesensitization agents prevented Ca2+ inhibition through Ca2+ binding to a modulatory site in the AMPAR [186].
Effects of zinc on AMPA/kainate receptors have also been explored. Zn2+ appeared to have a dual effect on AMPAR: at micromolar concentrations, it enhances AMPA receptor responses whereas at millimolar concentrations, it inhibits them [136, 187]. These effects of Zn2+ appear to be subunit specific as well. Experiments using cloned AMPAR expressed in oocytes demonstrate that, in normal calcium-containing solution, zinc could potentiate current from homomeric GluR3 receptors over a narrow range of 4–7.5 μM Zn2+ while homomeric GluR1 receptors could not be potentiated, but are inhibited by 10 μM Zn2+ [188]. Additionally, in calcium-free solution, the inhibition caused by Zn2+ on GluR1 shifted to ≥1 mM and potentiation was attainable reaching a maximum of ∼200% at 50 μM Zn2+. Also, GluR3 showed maximum potentiation not significantly different from GluR1 potentiation. The presence of GluR2 subunit in heteromeric expression of GluR2/GluR3 prevented the potentiation by Zn2+, but also allowed inhibition (with 500 μM Zn2+, current was 39% of control). The presence of GluR2 rendered the effects of Zn2+ independent of Ca2+ levels (for details, [189]).
The effects of other divalent metals effects were explored on these channels but less extensively compared to NMDARs. Various metals caused inhibition of Ca2+ impermeable AMPAR with the following rank order of inhibition: Ni2+> Zn2+> Co2+> Ca2+> Mn2+> Mg2+ (for values, refer to Table 5) [190]. The proposed mechanism of action is that complexes of divalent cations and AMPAR agonists compete with the free agonists rather than the cations themselves. This mechanism fits the data in which a competitive type of inhibition is observed; in addition, an increase in agonist concentration reduce the inhibitory effects of divalent metals less than that of DNQX (the classical competitive AMPAR antagonist) [190].
Table 5.
Effects of metals on postsynaptic ligand-gated ion channels (↑—activation/upregulation, ↓—inhibition/downregulation).
| Target | NMDA | AMPA/kainate | GABA-A | |
|---|---|---|---|---|
| Lead | Effect | (1) ↓ open channel probability | (1) ↓ (35%) | |
| (2) 60% ↓ in current (reversible) | ||||
| (3) >80% block | ||||
| (4) ↓ | ||||
| (5) ↓ | ||||
| (6) ↓ receptor binding | ||||
| (7) ↓ | ||||
| Conc | (1) 1–10 | (1) 1m M | ||
| (2) 50 | ||||
| (3) 100 | ||||
| (4) IC50 = 1.52–8.19 | ||||
| (5) IC50 = 8.78 (in 0 Zn); IC50 = 1.26 (10 Zn) at high site, 94 at low site | ||||
| (6) IC50 = 300 (adult); 60 (neonatal) | ||||
| (7) IC50 (free) = 0.55 | ||||
| Ref | (1) [16] | (1) [10] | ||
| (2-3) [14] | ||||
| (4) [20] | ||||
| (5) [9] | ||||
| (6) [12] | ||||
| (7) [5] | ||||
|
| ||||
| Zinc | Effect | (1) ↓ open | (1) ↑ | (1) ↓ |
| channel probability | ||||
| (2) Channel block | (2) ↓ | ↓ current in voltage independent, noncompetitive manner | ||
| (3) NR2A block | (3) ↑ | |||
| (4) NR2B block | (4) ↓ | |||
| (5) ↓ | (5) ↑ (16% to kainate, 15% to glu peak and steady state) | |||
| (6) ↓ receptor binding (76%) | (6) ↓ | |||
| (7) ↓ | (7) ↑ AMPA response | |||
| (8) ↓ | (8) ↑ desensitized Kainate responses | |||
| (9) ↓ AMPA and kainate responses | ||||
| Conc | (1) 1–10 μM | (1) 50 | (1) 100 (dose dependent) | |
| (2) >20 | (2) 1 mM | (2) IC50 = 19 | ||
| (3) nM | (3) <300 | |||
| (4) μM | (4) >500 | |||
| (5) High affinity: IC50 = 0.77; low affinity: IC50 = 153 | (5) 10 | |||
| (6) 1 mM | (6) IC50 = 700 | |||
| (7) IC50 (free) = 1.3 | (7) EC50 = 30 | |||
| (8) IC50 = 42.9 | (8) EC50 = 13 | |||
| (9) IC50 = 1.2-1.3 mM | ||||
| (1-2) [11] | (1-2) [17] | (1) [13] | ||
| Ref | (3) [17, 21] | (3-4) [7, 25] | (2) [10] | |
| (4) [17] | (5-6) [15] | |||
| (5) [9] | (7-9) [22] | |||
| (6) [12] | ||||
| (7) [5] | ||||
| (8) [22] | ||||
|
| ||||
| Magnesium | Effect | (1) ↑ NMDA-R affinity to glycine in all receptors | (1) ↓ (27%) | |
| (2) ↓ elementary current at +ve potentials(+20 to +80) | ||||
| (3) ↑ glycine and voltage-independent and subunit specific | ||||
| (4) external channel block, voltage dependent | ||||
| Conc | (1) 10 mM | (1) 20 mM | ||
| (2) 10 mM | ||||
| (3) 2 mM | ||||
| (4) IC50 (−100 mV) = 2–15 | ||||
| Ref | (1–3) [6] | (1) [15] | ||
| (4) [2] | ||||
|
| ||||
| Manganese | Effect | (1) ↓ (activity dependent, channel blocker) | (1) ↓ (46%) | (1) Little or no effect |
| Conc | (1) Ki = 35.9 (presence of glu and gly); Ki = 157 (no glu nor gly) | (1) 25 mM | (1) 1 mM | |
| Ref | (1) [8] | (1) [15] | (1) [10] | |
|
| ||||
| Copper | Effect | (1) ↓ | (1) ↓ | (1) ↓ (voltage independent) |
| (2) ↓ receptor binding (54%) | (2) ↓ kainite-induced current | |||
| (3) ↓ (channel block) | (3) ↓ efficacy of kainate | |||
| (4) ↓ | ||||
| (5) ↓ voltage independent, noncompetitive | ||||
| Conc | (1) ND | (1) | ||
| (2) 1 mM | (2) IC50 = 4.3 | |||
| (3) Ki = 195 (no coagonists); | (3) 30 | |||
| two sites (9.4, 248) with glu and gly | (1) IC50 = 5 | |||
| (4) IC50 = 15 | ||||
| (5) IC50 (free) = 0.27 | ||||
| Ref | (1) [17, 23] | (1) [17] | (1) [10] | |
| (2) [12] | (2-3) [30] | |||
| (3) [8] | ||||
| (4) [30] | ||||
| (5) [28] | ||||
|
| ||||
| Cobalt | Effect | (1) ↓ | (1) ↓ | (1) ↓ |
| (2) ↓ | (2) ↓ (29%) | |||
| Conc | (1) 2 mM | (1) 2 mM | (1) 2 mM | |
| (2) IC50 = 6.1 mM | (2) 1 mM | |||
| Ref | (1) [3] | (1) [3] | (1) [3] | |
| (2) [15] | (2) [10] | |||
|
| ||||
| Nickel | Effect | (1) NR2A: ↓, NR2B: ↑ | (1) ↓ (kainite-induced current) | (1) ↓(20%) |
| (2) NR2A ↓ (100% at +ve potentials) | (2) ↓ (glu-induced current) | |||
| (3) NR2B ↓ | ||||
| (4) NR2B ↑ (voltage independent) | ||||
| Conc | (1) 30 | |||
| (2) IC50 = 36 at −60 mV and 81 at +40 mV | ||||
| (3) IC50 138 at −60 mV and 442 at +40 mV | ||||
| (4) 3 | ||||
| (1) IC50 = 420 | (1) 1 mM | |||
| (2) IC50 = 2.6 mM | ||||
| Ref | (1) [16] | (1-2) [15] | (1) [10] | |
| (2–4) [21] | ||||
|
| ||||
| Mercuric chloride | Effect | (1) ↑ 130% | ||
| (2) ↑ (270%) | ||||
| Conc | (1) 1 | |||
| (2) 100 | ||||
| Ref | (1) [1] | |||
| (2) [19] | ||||
|
| ||||
| Methyl mercury | Effect | (1) ↓ receptor binding | (1) ↓ amplitude to 82.4% | |
| Conc | (1) IC50 = 0.95 (neonatal); 70 (adult) | (1) 100 | ||
| Ref | (1) [12] | (1) [55] | ||
|
| ||||
| Cadmium | Effect | (1) ↓ receptor binding (58%) | (1) ↑ (kainate to 108% and QA to 115%) | (1) ↓ (18%) |
| (2) ↓ (39% of control) | (2) ↓ (kainate to 79% and QA to 60%) | |||
| (3) ↓ (4% of control) | ||||
| Conc | (1) 1 mM | (1) 50 | (1) 1 mM | |
| (2) 50 | (2) 1 mM | |||
| (3) 1 mM | ||||
| (1) [12] | (1-2) [18] | (1) [10] | ||
| Ref | (2-3) [18] | |||
|
| ||||
| Lanthanide | Effect | (1) ↓ NMDA response in a voltage-independent manner | (1) ↑ | (1) ↑ (300% max) and ↑ as the potential more −ve |
| (2) ↓ | ||||
| Conc | (1) IC50 = 2 | (1) 1–100 | (1) EC50 = 231 | |
| (2) >100 | ||||
| Ref | (1) [27] | (1-2) [27] | (1) [10] | |
|
| ||||
| Trimethyl-tin (TMT) | Effect | (1) ↓ (35%) reversible | (1) ↓ (20%) irreversible | (1) ↓ (30%) irreversible |
| Conc | (1) 100 | (1) 100 | (1) 100 | |
| Ref | (1) [4] | (1) [4] | (1) [4] | |
3.3. GABA Receptor
γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the mammalian central nervous system. The synaptic transmission mediated by GABAA receptor-channel complex leads to a hyperpolarization of the cell membrane due to the fast activation of postsynaptic chloride channels upon the exposure to GABA [191]. The GABAAR is comprised of pentameric combination of α1–6, β1–4, γ1–3, δ 1, and/or ε1 subunit subtypes that form an intrinsic chloride ion channel, and each subunit comprises four domains. GABAARs have some recognized allosteric binding sites such as barbiturates, benzodiazepines, and picrotoxin [192, 193]. The properties of the allosteric binding sites were influenced by the subunit subtype composition of GABAAR (for review [194]).
3.3.1. Inhibitory Effects of Metal Ions
The GABAAR response to GABA-evoked currents was modulated by a number of divalent metal cations. Zinc, cadmium, nickel, manganese, cobalt, lead, and copper inhibited the response to GABA. The inhibition by divalent metals has consistently been shown to be reversible with no or little voltage dependence [14].
Zn2+ had the potential to directly interact with the GABAAR to influence inhibitory postsynaptic currents (IPSC) amplitudes and kinetics [118]. Zn2+ suppressed the GABA-induced chloride current with a K d of 19 μM in a noncompetitive, voltage-independent manner, and without interference with any of the allosteric sites on the GABA-R [195]. Using cultured hippocampal neurons, studies showed that Zn2+ reduced the amplitude, slowed the rise time, and accelerated the decay of mIPSCs. Evidence indicated that inhibition of mIPSCs by Zn2+ was attributed to an allosteric modulatory site located on the extracellular domain of GABAA receptors [196, 197]. In accordance with the previous hypothesis, single-channel studies have also shown that Zn2+ reduced the opening frequency with no evidence of flickering [198–200].
From another perspective, the effects of Zn2+ were subtype-specific. The zinc-sensitivity of the channels seemed to be dependent on the absence of γ subunits, as its presence in any combination with other subunits led to the formation of GABAA receptors almost insensitive to Zn2+ [201, 202]. Furthermore, the presence of a δ subunit enhanced zinc sensitivity [202, 203]. The exchange of a particular subunit with other members of the same subunit family (α1 versus α3, β1 versus β2, and γ1 versus γ2) did not alter the large difference in Zn2+ sensitivity between GABAAR containing or lacking γ subunit [202]. Later studies showed that recombinant GABAAR, which contain α4, α5, and α6 subunits, were more sensitive to zinc than those that contain α1 subunits [204–207]. Given that the majority of synaptic GABAAR are of the α-, β-, γ-isoform [208], together with the above studies, indicate that a likely target of Zn2+ modulation is an extrasynaptic α-, β- or δ-receptor [118].
In rats dorsal root ganglion (DRG) neurons, Cu2+ at concentration of 15 μM, suppressed the peak amplitude of the GABA-induced current to approximately 50%; the blocking was exerted and reversed quickly, and it was independent of membrane potential [195].
The similar blocking profiles of Cu2+ and Zn2+ led to the question whether they shared a common binding site. Competition experiments showed that Zn2+ suppression of GABA-induced current was decreased with increasing concentrations of Cu2+, suggesting that Zn2+ and Cu2+ act on the same allosteric site to inhibit GABAAR [55].
In a later study, the copper-induced block of GABAAR in Purkinje cells developed slowly, was poorly reversible, and decreased with increasing GABA concentrations. The block occurred at low concentrations indicating a high affinity with an IC50 ~35 nM [209]. The copper block of GABAAR in Purkinje cells seemed to have a higher affinity compared to the block in DRG [195] and olfactory bulb neurons [210]. Another difference between these tissues was that Cu2+ in DRG cells interacts in a noncompetitive manner while in Purkinje, Cu2+ decreased the potency of GABA without affecting the maximal response. A possible explanation for this discrepancy might be different subunit composition of the GABAAR [210].
The effects of multiple divalent metals (cadmium, nickel, manganese, zinc, and barium) were shown in a study of GABA responses of embryonic chick spinal cord neurons. The results were suggestive of an allosteric mechanism of inhibition of GABAAR currents. Through combination experiments they showed that the ions acted at a common site but possessed different intrinsic efficacies with the following rank: Zn > Cd> Ni> Mn. Ba was thought to bind to the site but lacked efficacy as an inhibitor of the GABA response [14]. The rank of efficacy was supported by other experiments [195].
3.3.2. Excitatory Effects of Metal Ions
Lanthanides comprise a series of 15 metals starting with lanthanum (La) and ending with lutetium. In sub-millimolar concentrations, lanthanum ions modulate GABA-induced currents [55, 191, 211].
La3+ increased the affinity of GABA for the receptor in a concentration-dependent manner with an EC50 = 231 μM and maximum enhancement to about 300% of control with 1 mM. This potentiation was completely and quickly reversed, but was more enhanced as the potential became more negative (1.6% per 10 mV) [195]. Also, this effect was independent of the presence or absence of barbiturates, benzodiazepines, picrotoxin, or Zn2+/Cu2+, indicating that it was bound to a site different from all of the binding sites of the above substances [55]. La3+ did not activate transmembrane currents, it only potentiated GABA-induced currents; also La3+ did not affect the amplitude of the maximum response induced by GABA. These data together suggested that La3+ increased the affinity of GABAAR to its agonist [212]. Other lanthanides exhibited enhancing actions, and the efficacy increased with increasing the atomic number, such that Lu3+ (1 mM) increased the current to 1230% of control [55]. However, previously it was reported that lanthanides generate inward currents on their own in the absence of GABA [55]. This controversy might be due to use of different tissues; as La3+ did not activate transmembrane currents in CA1 hippocampal pyramidal neurons whereas La3+ generated inward currents in DRG neurons.
Additionally, recombinant GABAAR studies suggested that changing the α-subunit subtype from α1 to α6 alters the effects of lanthanum from potentiation to inhibition at comparable concentrations. These studies also suggested that the maximal inhibition of GABAAR current by La3+ in α6-containing receptors is greater in the presence of δ subunit (83%) than in the presence of γ subunit (32%) [213].
Another cation that might affect GABAAR is mercury (Hg). In its inorganic form, GABAAR channel complex was strongly stimulated by low concentrations of Hg [214]. Mercuric chloride (100 μM) increased the GABA-induced current to 270% of control, and increased it to 115% of control with 0.1 μM [55] indicating its high potency.
3.4. Summary of Postsynaptic Effects
The main postsynaptic targets are the ligand-gated receptors including, but not limited to, NMDA, AMPA/kainite, and GABA receptors. Of those, NMDAR channels are the most widely studied receptors due to their association with disease status. Relatively fewer studies have been done on other targets, which could lead to underestimation of their roles in metal toxicity.
Two main mechanisms established for metals effects on NMDAR: Mg2+-like inhibition, which is voltage dependent, or Zn2+-like inhibition, which is voltage independent. Lead and copper were found to inhibit NMDAR in Zn2+-like pattern. However, copper-mediated inhibition of NMDAR was use dependent which was also true for manganese-mediated inhibition [160–162]. Nickel on the other hand, showed an Mg2+-like inhibition at negative potentials. However, it had different effects on NR2B and NR2A containing channels at positive potentials. It caused a potentiation of NR2B-containing channels and a Zn2+-like inhibition in those containing NR2A. However, at high concentrations, NR2B-containing receptors also showed Zn2+-like inhibition at positive potentials [163]. Another major target on the NMDAR was glycine-binding site with multiple metals affecting it. Ca2+ and Mg2+ potentiated NMDAR currents whereas Pb2+ and Zn2+ inhibited NMDAR currents. All of these four divalent metals might act on the same binding site with different effects [178]. Also, the presence or absence of glycine affected the potency of Cu2+ and Mn2+, such that in the presence of Gly, Cu2+ was more potent, whereas in the absence of Gly, Mn2+ was slightly more potent [177].
The main inhibitory receptors in the brain, GABAA, are also modulated by a variety of metals. Certain metals suppressed the GABA-induced chloride current while others augmented it. Multiple divalent metals had inhibitory effects on GABA-induced current. Cu2+ and Zn2+ suppressed GABAAR in an equipotent manner, and there was some evidence that they act on the same site. Other divalent metals also showed inhibitory effects through an allosteric mechanism of inhibition, and they demonstrated a common site of action but with different intrinsic efficacies with the rank Zn2+> Cd2+> Ni2+>Mn2+ [14].
Interestingly, Lanthanides exhibited enhancing effects of GABA-induced currents. The effect was completely and quickly reversible. The efficacy increased with increasing atomic number [55]. However, recombinant GABAAR studies suggested that changing the α-subunit subtype could alter the effects of lanthanum from potentiation to inhibition in a comparable concentration range.
Mercury was another metal, which had inhibitory and excitatory effects. In its inorganic form, GABAAR channel complex was strongly stimulated by low concentrations of Hg. However, methyl mercury was a potent inhibitor of GABA-induced current, and this effect was irreversible [55]. This showed how organic metals might behave differently compared to inorganic cations; however, there is less information in the literature about the effects of organic metals. One of the most toxic organic metal is trimethyl-tin (TMT). At a concentration of 100 μM, around 20–30% of AMPAR and GABAAR currents were inhibited whereas 35% of NMDAR ion currents were blocked [215].
4. Disruption of Synaptic Plasticity
Multiple reports have demonstrated that human exposure to environmentally concentrations of certain metals can result in cognitive deficits.
Arsenic consumption, mainly through contaminated water, has been found to be associated with impairment of long-term memory and a reduction in the verbal IQ of children [216, 217]. Lead has been studied extensively for its role in disruption of synaptic plasticity in an attempt to explain the cognitive deficits observed in children with elevated blood lead levels. The CDC currently considers blood Pb2+ level of 10 μg/dL to be the threshold for impairment of cognitive function in children [218], although recent studies have observed that cognitive impairment can occur even at blood lead levels <10 μg/dL [219]. There have been reports that aluminum also affects synaptic plasticity, which has been implicated in the pathogenesis of Alzheimer's disease, although this topic is highly debated. It has been argued that these detrimental effects on learning, memory, and cognition, which are associated with exposure to metals, may be linked to the disruption of processes that are involved in synaptic plasticity. The formation of long-term potentiation (LTP) is impaired in mice that have inborn low learning capacity indicating the crucial role for synaptic plasticity as the basis of learning and memory. Impairment of LTP has been observed with exposure to lead [220]. Moreover, studies have shown that aluminum also impaired hippocampal long-term potentiation (LTP) and long-term depression (LTD) in rats both in vivo and in vitro [221, 222]. Also, multiple metals have been shown to have different concentrations in patients with Parkinson's disease compared to healthy individuals, and the levels of aluminum have been identified as a potential diagnostic marker [223].
To understand how metals and their compounds affect learning and memory, their effects on different stages of LTP and LTD were compared to identify specific sites of interaction for particular metals as well as targets common to more than one metal.
Recently, it has been shown that LTP consists of different succeeding forms: early-phase LTP (E-LTP), which lasts only a few hours, and late-phase LTP (L-LTP), which lasts for several days [224–226]. Several of the molecules required to produce these different forms of LTP have been identified and are targets for metal toxicity [224, 225, 227] (refer to Figure 5 and Table 6).
Figure 5.
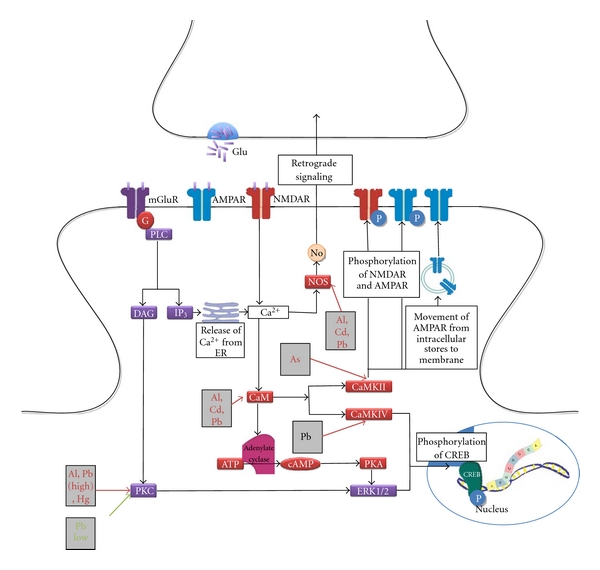
Proteins involved in the formation of long-term potentiation (LTP) and toxic effects of metals. LTP consists of different forms: early-phase LTP (E-LTP), which lasts only a few hours, and late-phase LTP (L-LTP), which lasts for several days. E-LTP includes short-term potentiation (STP), which is dependent on NMDA receptor activation and Ca2+/calmodulin and LTP-1 that involves protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase- (CaMK-) dependent phosphorylation. While STP can be formed by activation of NMDA and calmodulin dependent enzymes, LTP-1 requires activation of PKC via DAG that is produced after the activation of mGluRs. PKC and CaMKII then phosphorylate AMPA and NMDA receptors. L-LTP consists of the later phases of LTP, which are LTP-2 and LTP-3. LTP-2 requires synthesis of new proteins and receptors whereas LTP3 requires gene transcription. Activation of adenylate cyclase and cAMP-dependent activation of PKA are required for the formation of the later phases of LTP. LTP-3 depends on the activation of extracellular signal- related kinase 1/2 (ERK1/2) and CaM kinase IV, which in turn phosphorylate CREB and lead to new protein synthesis. Other factors such as p38 mitogen-activated protein kinase (p38 MAPK) leads to the formation of long-term depression (LTD). Several of the molecules required to produce these different forms of LTP have been identified and are targets for metal toxicity, which have been shown (red arrows indicate inhibition whereas green arrows indicate activation by metals. Black arrows indicate activation that occurs during normal formation of LTP).
Table 6.
| Target | Al | As | Cd | Hg | Pb | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Effect | Concentration | Ref | Effect | Concentration | Ref | Effect | Concentration | Ref | Effect | Concentration | Ref | Effect | Concentration | Ref | ||
| Pumps | Ca2+ ATPase | ↓in vitro | 10 mg/kg/day Al3+ | [1] | ||||||||||||
|
| ||||||||||||||||
| Protein Synthesis | NMDAR NR1 | ↕ dev. | 750 ppm PbAc | [13] | ||||||||||||
| ↑ dev. | 750 ppm PbAc | [14] | ||||||||||||||
| NMDAR NR2A | ↓ dev. | 750 ppm PbAc | [13] | |||||||||||||
| ↓ dev. | 750 ppm PbAc | [14] | ||||||||||||||
| NMDAR NR2B | ↕ dev. | 750ppm PbAc | [13] | |||||||||||||
| ↕ dev. | 750 ppm PbAc | [14] | ||||||||||||||
|
| ||||||||||||||||
| Enzymes | CaM | ↓in vitro | 10 mg/kg/day Al3+ | [1] | ↓in vivo | 6 mg/kg/day CdCl2 | [15] | ↑ in vitro | 30 μM Pb2+ | [21] | ||||||
| ↓in vitro | IC50 —15 μM AlCl3 | [3] | ↓in vitro | IC50 —0.47 mM CdCl2 | [8] | |||||||||||
| ↓in vitro | AlCl3 —240 μM | [9] | ↓in vitro | 10 nM CdCl2 | [4] | |||||||||||
| PKC | ↓in vitro | 10 mg/kg/day Al3+ | [1] | ↓in vitro | IC50 —1.5 μM Hg; IC50 — 0.2 2μM CH3Hg | [2] | ↓ dev. | 0.1% PbAc | [5] | |||||||
| ↓in vivo | 0.3% AlSO4 for 4 months | [18] | ↓in vitro | IC50 —0.08 μM HgCl2; IC50 —1.32 μM CH3Hg; IC50 —0.90 μM C6H5Hg+ | [7] | ↓in vitro | IC50 —2.12 μM PbAc | [10] | ||||||||
| ↓in vivo | 10 mg MeHg/kg | [16] | ↓in vivo | 1500 ppm PbAc | [17] | |||||||||||
| ↑ in vitro | 10−13 to 4 × 10−4 M PbAc | [19] | ||||||||||||||
| ↓in vitro | > 4 × 10−4 M PbAc | |||||||||||||||
| NOS | ↓in vivo | 2.5% AlSO4 for 3–5 weeks | [20] | ↓in vivo | 37 ppm NaAsO2 | [6] | ↓in vitro | 100 μM CdCl2 | [11] | ↓in vivo | 125 ppm PbAc | [12] | ||||
| ↓dev. | 3% AlSO4 | [27] | ↓ dev. | 0.2 % PbAc | [22] | |||||||||||
| ↓in vitro | IC50—0.36 Pb2+ | [11] | ||||||||||||||
| CaMK II | ↓dev. | 1500 ppm PbAc | [25] | |||||||||||||
| CaMK IV | ↓in vivo | 4 ppm As2O3 for 60 days | [23] | |||||||||||||
|
| ||||||||||||||||
| Transcription factors | P38 MAPK | ↑ in vitro | 5–100 μM CdCl2 | [28] | ↑ in vitro | 5 μM Pb2+ | [30] | |||||||||
| ERK1/2 pho-sphorylation | ↑ in vitro | 100–200 μM CdCl2 | [28] | ↑dev. | 2 mg/kg/day PbAc | [30] | ||||||||||
| ↑ in vitro | 5 μM Pb2+ | [30] | ||||||||||||||
| CREB phospho-rylation | ↓ dev. | 1500 ppm PbAc | [29] | |||||||||||||
Julka and Gill [20], Nihei and Guilarte [56], Guilarte and McGlothan [57], Vig et al. [58], Sandhir and Gill. [59], Siegel and Haug [60], Cox and Harrison [61], Levi et al. [62], Ohtani-Kaneko et al. [63], Rajanna et al. [64], Xu et al. [65], Inoue et al. [66], Rajanna et al. [64], Saijoh et al. [67], Nihei et al. [68], Johnson et al. [69], Coppi et al. [70], Hermenegildo et al. [71], Zarazua et al. [72], Mittal et al. [73], García-Arenas et al. [74], Chetty et al. [75], Llansola et al. [76], Wang et al. [77], Toscano et al. [78], Rigon et al. [79], Cordova et al. [80], Toscano et al. [81].
Early-LTP —
it includes short-term potentiation (STP), which is dependent on NMDA receptor activation Ca2+/calmodulin; and LTP-1 that involves protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase- (CaMK-) dependent phosphorylation. While STP can be formed by activation of NMDA and calmodulin-dependent enzymes, LTP-1 requires activation of PKC via DAG that is produced after the activation of mGluRs. The activity of mGluR-PKC is important for both increasing activity as well as increasing number of AMPA receptors. PKC and CaMKII then phosphorylate AMPA and NMDA receptors.
Late-LTP —
there are two later phases of LTP named LTP-2 and LTP-3. LTP-2 requires synthesis of new proteins and receptors whereas LTP-3 requires gene transcription. Activation of adenylate cyclase and cAMP-dependent activation of PKA are required for the formation of the later phases of LTP. LTP-3 depends on the activation of extracellular signal-related kinase 1/2 (ERK1/2) and CaM kinase IV, which in turn phosphorylate CREB, and this leads to new protein synthesis. p38 mitogen-activated protein kinase (p38 MAPK) is involved in the formation of long-term depression (LTD), and c-JUN-N-terminal kinase (JNK) is thought to participate in LTD [228, 229].
4.1. Disruption of Long-Term Potentiation by Exposure to Metals in Adults
4.1.1. Calmodulin
Calmodulin (CaM) is a regulatory protein that is activated by [Ca2+]i. This protein is found in high concentrations in CNS neurons and is involved in the activation of several other proteins. Some of the CaM-regulated proteins that modulate synaptic plasticity include adenylyl cyclases (AC1 and AC8), protein kinases, calcineurin, calmodulin kinases (CAMK I, II, and IV), nitric oxide synthase, and Ca2+ conducting channels. CaM has four Ca2+ binding sites. Ca2+ binding to CaM leads to a conformational change that exposes a hydrophobic domain which enhances the binding of CaM to other target proteins [226]. It has been hypothesized that in absence of Ca2+, the concentration of free CaM is regulated by neurogranin that binds CaM and releases free CaM in response to PKC and Ca2+ [226, 230, 231]. This important molecule has been identified as a target of several neurotoxic metals such as aluminum, cadmium, and lead.
CaM, when incubated with increasing concentrations of aluminum (Al3+) (from 0–1000 μM), showed decreased activity. This decrease in activity, measured by the ability of CaM stimulate activator-deficient cAMP phosphodiesterase was concentration dependent, [20]. Yet, another study showed that an [Al3+]: [CaM] ratio of 3 : 1 resulted in 50% decrease in phosphodiesterase activity, and maximal inhibition was observed at a ratio of 4 : 1 [60].
Recently, using highly specific monoclonal antibodies that detect the different conformational states of CaM and monoclonal antibodies against Al-CaM complex, researchers found that on dissolving CaM with AlCl3 ·6H2O (in increasing concentrations from 0–480 μM), the antibody specific to Ca2+ calmodulin conformation (the active form) mAb CAM-1, did not recognize the Al-CaM complex (at Al concentrations of 240–300 μM) indicating that the CaM was in the inactive conformation. Moreover, the antibodies against the Al-calmodulin complex were found to bind to their antigen in the presence of Ca2+. This shows Al binds CaM, even in the presence of Ca2+, and CaM undergoes a conformational change into an inactive form. Equilibrium dialysis and atomic adsorption studies indicated that Ca2+ remained bound to CaM simultaneously with Al. When the Al-chelator citrate was added to the solution only partial restoration of CaM activity occurred, suggesting that some of the Al ions became inaccessible for chelation [62].
The effects of Cadmium (Cd2+) were observed in vivo, where adult male rats, received 6 mg Cd2+/kg body weight daily for four weeks. Brain CaM activity was determined by measuring the stimulation of phosphodiesterase activity. A significant decrease in the CaM activity was observed after Cd2+ treatment. CaM bound to Cd2+ was also detected in the brains of rats exposed to CdCl2. It was proposed that, since Cd2+ has an ionic radius similar to Ca2+, it might interact with the Ca2+-binding sites on the CaM [58, 61].
Lead was also found to interfere with CaM activity in vitro and in vivo. In vitro incubation of CaM with lead (Pb2+) increased the activity of calmodulin in terms of its ability to stimulate cAMP phosphodiesterase and a maximum increase was observed at 30 μM lead concentration whereas at higher concentrations the calmodulin activity was inhibited. CaM-dependent cAMP phosphodiesterase activity increased up to a concentration of 100 μM, following which there was a sharp decline in activity with higher concentrations of lead. The involvement of phenomenon of mimicry of calcium by lead as a mechanism of toxicity has been proposed. The affinity of lead to CaM is stronger than that of calcium and lead can displace calcium from calmodulin [59]. An in vitro study done on CaM purified from bovine brain showed that Pb2+ mimics a natural ligand and raises the maximal activation slightly above the activation by Ca2+ [232].
4.1.2. Protein Kinase C (PKC)
Protein Kinase C is a Ca2+ and phospholipid-dependent serine/threonine kinase that is a receptor for DAG and phorbol esters. There are two classes of PKC. The classical group of PKC consisting of four isozymes: PKC-α, PKC-βI, PKC-βII, and PKC-γ, are Ca2+-dependent and require Ca2+ as well as DAG or phorbol ester for their activation. The second group of PKC isoforms consists of five isozymes: PKC-δ, PKC-ε, PKC-η, PKC-θ, and PKC-μ. These do not require Ca2+ for their activation by DAG or phorbol ester. Various isozymes of PKC are involved in the formation of LTP. For instance, a null mutation in PKC-γ prevented the induction of LTP [233]. PKC is activated postsynaptically when metabotropic glutamate receptors (mGluR) are activated leading to the formation of DAG and release of intracellular Ca2+, which activates PKC. The mGluR- PKC pathway then increases the number and activity of AMPA receptors [224]. The PKC activity is affected by metal ions such as Al3+, Pb2+, Hg, and organic metals such as methylmercury (MeHg; refer to Figure 5).
Aluminum (Al3+; 0–100 μM) decreased in vitro PKC activity (determined by transfer of 32P from γ-32P-ATP to lysine rich histone in the presence of Ca2+ and phosphatidyl serine), and this effect was concentration dependent [20]. In vivo, rats fed aluminum (AlSO4) orally were found to have more PKC in the particulate fraction of the brain homogenate compared to the soluble fraction. Normally PKC is translocated from the cytosol to the membrane when it is activated. Application of Al3+ caused a 70% increase in the total activity of PKC resulting in a greater fraction of it being translocated to the membrane, and hence the presence of greater fraction of PKC in particulate fraction compared to the soluble fraction [69].
Lead acetate upon in vitro incubation with PKC from adult rat brains significantly inhibited PKC activity with an IC50 of 2.12 μM [64]. However, it was found that while very low concentrations of Pb2+ (10−13 to 4 × 10−4 M) increased PKC activity, higher Pb2+ concentrations (>4×10−4 M) caused an inhibition of PKC activity. When recombinant human PKC iso-enzymes were examined, low concentrations of Pb2+ had very little activating effect on PKC-γ but inhibited it at higher concentrations (>4 × 10−4 M) [70]. In vivo, on exposure of adult rats to 1500 ppm lead acetate, there was a decrease in protein expression of PKCγ by 32% in the cytosol of hippocampal cells and 25% in the membrane fraction [68]. Another study comparing the effects of Pb2+ on the PKC in the brain in vivo and in vitro found a considerable increase in PKC activity in vitro, but failed to find a considerable change in PKC activity in vivo [234].
In vivo methylmercury chloride administration in rats in five doses of 10 mg/kg body weight leads to a decrease in the enzymatic activity of the cytosolic PKC extracted from the brain, although it did not induce any change in second messenger binding as measured by binding of [3H]PDBu [67].
4.1.3. Ca2+/Calmodulin Kinases
There are two types of Ca2+/calmodulin kinases (CaMK) involved in LTP: CaMKII and CaMKIV. The Ca2+-CaM complex generally activates these kinases. CaMKII is a serine/threonine protein kinase consisting of 12 subunits that are activated when activated calmodulin is associated with them [235]. Studies have shown that CaMKII blockers impede the ability to generate LTP. CaMKII can also be autophosphorylated at Thr286 and its activity becomes independent of Ca2+-CaM. This autophosphorylation occurs after the formation of LTP. It is suggested that after activation, CaMKII phosphorylates the AMPA receptor subunit as well as GluR1 and NMDA receptors and therefore enhances their conductance [236, 237].
CaMKIV is also activated similarly but the downstream targets are different for CaMKIV. Experiments have shown that upon activation, CaMKIV can phosphorylate CREB, which in turn mediates the transcriptional control of protein synthesis required for the long-term maintenance of LTP [238].
In vivo, rats exposed to 4 ppm arsenic trioxide (As2O3) for 60 days showed about a 4-fold decrease in expression of CaMKIV compared to a control group of rats as elucidated by microarray analyses. Western blot analyses reflected similar findings. Moreover, the decrease in expression of the β-subunit of CaMKIV was greater than the decrease in α-subunit expression [77].
4.1.4. Nitric Oxide Synthase
Nitric oxide synthase (NOS) is an enzyme that produces nitric oxide (NO) by oxidizing L-arginine using molecular oxygen and NADPH [239, 240]. There are different kinds of NOS expressed in several cell types. Endothelial cells express constitutive endothelial NOS (eNOS) that is activated by Ca2+. Macrophages express inducible NOS (iNOS), and its expression is inducible by cytokines. Neurons express constitutive Ca2+-activated neuronal NOS (nNOS). Following the activation of NMDA receptors and the influx of Ca2+, it is believed that nNOS produces NO, a retrograde signal, that diffuses into the presynaptic membrane to enhance presynaptic neurotransmitter release by the production of cGMP during the formation of LTP [241, 242].
In vivo, chronic exposure to aluminum (Al3+) resulted in the reduced formation of NO after activation of NMDA in the rat cerebellum, as a consequence of decreased calmodulin and NOS [71].
In vivo arsenic exposure to 37 ppm sodium arsenite for 10 days, reduced NMDA-induced NOS activity (as measured by sampling of extracellular fluid by means of microdialysis). The maximal NMDA-induced increase of NOS activity (estimated by measuring the changes in extracellular citrulline in the exposed groups) was only 170 ± 24% while under control conditions it reached 278 ± 27% (P< 0.001) [72].
In vitro incubation of NOS with 100 μM of cadmium (CdCl2) resulted in a significant reduction in brain NOS activity as measured by the conversion of radioactive arginine to citrulline. When incubated, the activity of NOS was decreased with an IC50 value of 0.22 mM [73].
In vitro incubation of NOS with lead. inhibited NOS activity with an IC50 of 0.36 mM [73]. In vivo, the cNOS activity in the hippocampus and cerebellum (measured by citrulline radioactivity following incubation with radioactive arginine) was decreased in rats that were exposed to 125, 250 and 500 ppm lead acetate for 14 days. This decrease was completely reversible by increasing the free Ca2+-concentration. The decrease in NOS activity correlated with blood lead levels [74].
4.1.5. Extracellular Signal-Regulated Kinases (ERK1/2)
The extracellular signal-regulated kinases (ERK1/2) are serine threonine kinases that are activated when extracellular signals lead to an increase in intracellular Ras-GTP (GTP-bound form of Ras). Ras-GTP produced by an increase in guanyl nucleotide exchange factors (GEF), a decrease in activity of GTPase-activating proteins (GAPs) or a combination of both then leads to the activation of the enzyme MAPK/ERK kinase (MEK). MEK than activates ERK1 and ERK2 by phosphorylating them. ERK1 and ERK2 (also known as p44 and p42 MAPK) target transcription factors, cytoskeletal proteins, regulatory enzymes, as well as other kinases. In the postsynaptic membrane, calcium influx through the NMDA receptors leads to production of Ras-GTP that can then trigger the cascade leading to phosphorylation of ERK1 and ERK2. CREB maybe one of the targets of the ERK1/2 pathway involved in LTP [229, 243]. CREB, a member of the basic leucine zipper (bZip) family, is a transcription factor that is responsible for initiating new protein synthesis for the maintenance of L-LTP. PKA, CAMK and MAPK can activate CREB by phosphorylation at serine-133. On phosphorylation other proteins such as CREB binding protein are recruited to form a complex, which initiates transcription of CRE containing genes [244–250] (refer to Figure 6).
Figure 6.
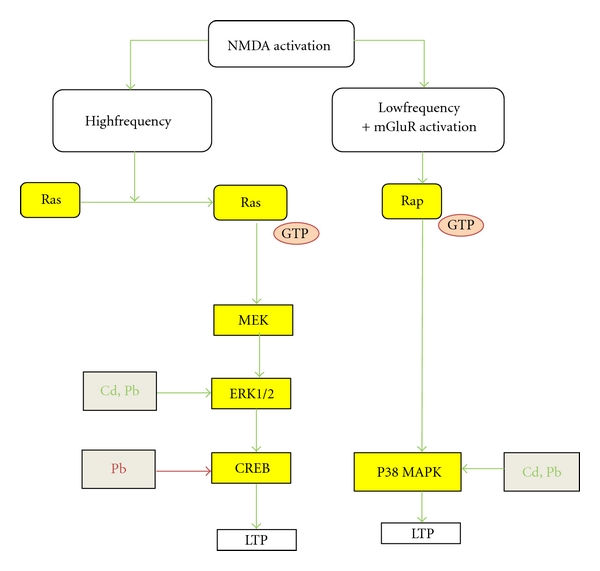
Molecules involved in the transcriptional control of LTP and LTD and effects of metals. LTP-3 depends on the activation of ERK1/2 and CaM kinase IV, which in turn phosphorylate CREB, and this leads to new protein synthesis. Other factors such as p38 mitogen-activated protein kinase (p38 MAPK) are involved in the formation of long-term depression (LTD) and c-JUN-N-terminal kinase (JNK) is thought to participate in LTD. A green color indicates an activation or an increase, and a red color indicates an inhibition or a decrease.
Exposure of hippocampal slices of rats to Cd2+ activates ERK1 and ERK 2 but only at very high concentrations (100–200 μM CdCl2) [79, 229].
On incubation of ERK1/2 with 5 μM Pb2+ in vitro for 3 hours, there was significant increase in ERK1 and ERK2 phosphorylation in hippocampal homogenates [80].
4.1.6. P38 Mitogen-Activated Protein Kinase (p38 MAPK)
Parallel to the ERK1/2 pathway, which is involved in long-term potentiation, another MAPK cascade, which involves p38 MAPK is involved in long-term depression (LTD). Inhibition of p38 MAPK was shown to inhibit a form of hippocampal LTD that involved the activation of mGluR. Inhibition of ERK1/2 by blocking MEK had no effect on this form of synaptic plasticity. The pathways upstream and downstream of p38 MAPK are yet to be elucidated [229, 251] (refer to Figure 6).
Hippocampal slices of postnatal day 14 rats were exposed to Cd2+ in concentrations between 5–100 μM for 3 hours. A western blot analysis showed that this increased the activity of p38 MAPK, which is involved in the inhibition of LTP [79, 229].
Incubation of ERK1/2 with 5 μM Pb2+ for 3 hours resulted in a significant increase in p38 MAPK phosphorylation in hippocampal homogenates [80].
4.2. Disruption of Long-Term Potentiation by Exposure to Metals during Development
Developmental exposure to “neurotoxic” metals differs from exposure in an adult in various ways. The developing brain is more vulnerable than the adult one. The basic circuitry of the brain is laid down during development and any disruption of receptors, neurotransmission, and neurogenesis can prevent the brain from maturing normally. Inappropriate activation of the unspecific receptors in the developing brain can interfere with the normal “tuning.” Moreover, the blood-brain barrier is not laid down till approximately six months of age in humans. This absence of blood brain barrier allows toxic agents to enter the brain freely and interfere with its development. Developmental exposure to metals also raises the issue of what Costa et al. labeled as “silent” neurotoxicity. This is when the deleterious effects of various neurotoxic insults do not manifest until several months or years post-partum. For instance, in Guam's disease, unknown neurotoxic agents cause damage to the CNS, which do not become apparent until decades later. Here we discuss developmental exposure of metals and their effects on the molecules involved in the formation of LTP, which are important for the development of memory and learning [252].
N-methyl-D-aspartate receptors (NMDAR) are Ca2+ channels, which play an essential role in several forms of synaptic plasticity (see Section 3.1). They have glutamate receptors present which are involved in excitatory synaptic transmission in various parts of the brain. Its unique properties, such as Mg2+ block and high permeability to Ca2+, give NMDAR the ability to contribute to the formation of long-term potentiation and long-term depression. Several subunits of NMDA receptors have been identified: NR1 that is ubiquitously expressed; NR2 subunit family that has four distinct types (A, B, C, and D) and two NR3 subunits. The expression of the various subunits is different in different stages of development. For instance, NR2B and NR2D expression is present during the neonatal period and NR2A and NR2C are present in the later stages of development [253]. Due to its many binding sites (especially those for divalent cations), which change in their affinity to their agonists during development, a variety of (toxic non-physiologic) metals might bind to these NMDAR with a high affinity and thereby impair their function.
Lead causes impairment of long-term potentiation in different regions of the hippocampus following chronic lead exposure [254, 255]. This has been associated with a disruption in the normal functioning of the NMDA receptors (NMDAR). NMDAR currents decrease after in vitro exposure to 5 μM lead in hippocampal cells [152]. This can be attributed to the observation that Pb2+ alters expression of the different subunits of NMDAR, which has been observed in the hippocampus and cerebral cortex. Additionally, a decrease in expression of NR2A subunit mRNA and proteins in the hippocampus have been seen [56, 57]. Also, the expression of NR1 subunit mRNA in the hippocampus and the cerebral cortex of rats increases [57], but this finding was not supported by another study by Nihei and Guilarte [56], which found no change in the expression of the NR1 subunit protein.
The effects of cadmium (Cd2+) on calmodulin expression were determined in an in vitro study done on embryonic rat (ED 15) cerebral cortex, where the cortical slices were incubated with 10 nM cadmium chloride for 24 hours. This experiment showed a reduced the amount of calmodulin expression following cadmium exposure [63].
Nitric oxide synthase (NOS) was affected by the developmental exposure of rats to aluminum (Al3+) and lead (Pb2+). Prenatal exposure of developing rats to aluminum sulfate (3%) decreased the content of neuronal NOS by 62 ± 12% in the cerebellum [76].
Perinatal exposure to Pb2+ decreased NOS activity, as well as NOS expression. Chetty et al., using western blot analysis of nNOS in developing rat brain after perinatal exposure to 0.2% lead acetate, found a significant decrease in nNOS protein levels at postnatal day (PND) 21 and 35 in cerebellum, and at PND 21 in hippocampus [75].
Developmental lead exposure also affected PKC-γ and CaMKII function. PKC-γ is activated by binding of Ca2+ or DAG and on activation, it translocates to the membrane. To determine the effects of Pb2+ on PKC-γ and other PKC-subtypes, pregnant rats were exposed to 0.1% lead acetate, dissolved in distilled deionized water (DDW) from gestation day 6 to postnatal day 21 (PND). With western blot analysis the expression on PKC-γ was determined. Pb2+ reduced PKC-γ mRNA expression significantly in hippocampus and frontal cortex at PND 1, 5, and 10, with greater effect on the membrane PKC-γ than on the cytosolic PKC-γ. Additionally there was a decrease in the activity of PKC-γ following exposure to lead. The PKC-γ activity was determined by measuring the amount γ-32P transferred to histone per min per mg protein. In the hippocampus and the frontal cortex, both total and calcium-dependent PKC activities were significantly inhibited [65].
Moreover, rats exposed to 1500 ppm Pb2+ during development demonstrated a reduction in the V max of CaMKII (examined by measuring the phosphorylation of a biotinylated substrate for CaMKII) and reduced expression of CaMKII β subunit in the hippocampus, but showed no changes in the sensitivity of calmodulin to CaMKII. In other words, the decrease in CaMKII activity was not due to impairment in its ability to bind CaM [78].
Various metals also inhibited the enzymes related to the transcription of new proteins involved in the formation of LTP. Two such targets are ERK1/2 and CREB.
Prenatal exposure to aluminum sulfate (3%) slightly increased the content of ERK [76]. Also, in vivo developmental exposure to 2 mg/kg of Pb2+ increased both ERK1 and ERK2 phosphorylation in rat hippocampal neurons [80].
On developmental exposure to 1500 ppm of Pb2+, a decrease in the amount of phosphorylated CREB was observed in both the hippocampus (25% decrease) and the cerebral cortex (25% decrease) but there were no significant changes in unphosphorylated CREB levels [81]. Also, significant changes in the binding kinetics of CREB to CRE were observed in the hippocampus. The K d and B max both were decreased by 38% and 30%, respectively, in the hippocampus but no significant changes in binding kinetics were observed in the cortex [244].
4.3. Summary of Long-Term Effects
Metals affect various mediators of synaptic plasticity. Calmodulin (CaM) activity is affected by aluminum, cadmium, and inorganic lead. Both aluminum and cadmium inhibited CaM activity [20, 58, 60–63] whereas inorganic lead first increased CaM activity at lower concentrations (possibly by mimicking calcium), but then at higher concentrations it decreased CaM activity [59, 232]. CaM is the central modulator of NMDAR-mediated synaptic plasticity and a majority of the regulators of synaptic plasticity depend on CaM for their activation. Thus, interference with CaM function will indirectly affect the function of numerous LTP- and LTD-related proteins such as adenylyl cyclase, Ca2+/Calmodulin kinases, nitric oxide synthase, and Ca2+ channels [226]. Al3+, Pb2+, and MeHg affect PKC activity, which is involved in the formation of LTP-1. In vitro studies demonstrated that in rat brain, Pb2+ inhibited PKC activity at low concentrations but increased PKC activity at higher concentrations. However, when recombinant human PKC-γ was used, an opposite trend was observed [70]. Exposure to Pb2+ in vivo resulted in a decrease in protein expression whereas MeHg decreased the activity of PKC. Al3+ inhibited PKC activity in vitro but ironically, oral administration lead to increase in PKC activity [20]. Arsenic trioxide (As2O3) decreases the expression of Ca2+/calmodulin kinase IV (CaMKIV) with a greater decrease in the β-subunit than the α-subunit in vivo [77]. Nitric oxide synthase (NOS) activity is decreased by various metals such as Al3+, As2+, Cd2+, and Pb2+ [71–73]. The components of the transcription pathway, p38 MAPK and ERK1/2 were phosphorylated more when incubated with Pb2+ and Cd2+. As discussed earlier, P38 MAPK is involved in the induction of LTD whereas ERK1/2 is involved in induction of LTP-3 [79, 80, 229].
5. Discussion and Conclusion
Most metals act on multiple modulators of synaptic transmission. Heavy metals such as mercury, lead, and arsenic interfere with normal functioning of molecules both presynaptically and postsynaptically. They also target molecules involved in synaptic plasticity. As discussed above in vivo and in vitro studies have shown that metals inappropriately inhibit or activate various molecules involved in synaptic transmission and synaptic plasticity. Even though the current pool of the literature gives us valuable insights into the mechanisms of metal toxicity at the synapse, there are many limitations of the current studies.
Firstly, there is hardly any sufficient information with regard to the effect of metals at different stages of development. Postsynaptically, there is a strong suggestion that the different effects on development are due to different subunit expression. As discussed in Section 3.2, Pb2+ was a more potent inhibitor of Glu-activated currents in NMDAR expressing NR2A or NR2B compared to receptors expressing both these subunits [157]. At the same time, Pb2+ showed high- and low-affinity components for its inhibition in PN14 and PN21 hippocampal membranes. These data suggested that the high-affinity Pb2+-sensitive site was associated with receptors expressing NR2A or NR2B subunits, while the low-affinity site was associated with receptors expressing both subunits (to see the effects on other brain areas, review [153]). The support to this hypothesis came from studies showing that the developmental pattern of NR2A and NR2B mRNA in the hippocampus was similar to that in the data presented [256].
This developmental aspect is not well studied for most of the metals and also for AMPA/kainate and GABA receptors. The effects of divalent metals on AMPA/kainate receptors seem to be dependent upon the subunit composition as well, particularly the presence of GluR2 subunit which rendered the channel impermeable to Ca2+. Zn2+ and Co2+ both had dual effects on AMPA-Rs: at micromolar concentrations they enhanced AMPA receptor responses whereas at millimolar concentrations, they had inhibitory actions. Various metals caused inhibition of Ca2+ impermeable AMPA-R, the inhibition was fast, reversible, and voltage independent. The rank order of activities was Ni2+ > Zn2+> Co2+> Ca2+> Mn2+> Mg2+ [190]. The proposed mechanism of action was that complexes of AMPAR agonists and divalent cations compete with the free agonists for the binding sites.
Prenatal exposure to heavy metals also leads to various changes in the LTP machinery in the developing brain as discussed before. Lead, for instance, changed the expression pattern for NMDAR subunits, and decreased the expression and activity of PKC-γ, CaMKII β, and nNOS in various areas of the brain. These results could explain why the development exposure to some metals causes cognitive deficits in children.
Moreover, a majority of the studies were done in vitro, and the in vivo studies were done in rats. In most in vitro studies, either brain homogenates or purified target molecules were incubated with a given metal. These studies therefore may not accurately depict the physiologic effects of the metals since they do not undergo the physiological process of absorption from the gut as in the human body, and the alterations that may occur in the blood before the metals reach the target tissue. Also, the solutions used for the preservation of the cells may interfere with the experimental results rendering them inaccurate. Moreover, the contents of the media and the forms of metals used were inconsistent between studies. Consequently properties such as solubility of metals in the media and presence of anions and pH were variable and beyond the scope of this study to discuss. It is definitely a limitation of in vitro studies, which makes it difficult to compare the different experiments, even when an identical concentration of the same metals was used. However, the ease of carrying out the experiment and lack of requirement for storage space for animals make them a likely choice for most researchers. In vivo studies may be closer to the physiologic processes; however it becomes harder than to vary the concentrations of metals and monitor them at the selected site of interest.
In addition, most of the findings presented in this review both in vivo and in vitro were based on studies done using rats. Even though it is easy to measure concentrations of metals, levels of proteins and enzymes in rats, elucidating the clinical manifestations in animal models can be challenging. Also, it is hard to find whether the effects in rats are similar to those in humans and if the effects in rats are representative of the effects in humans. Higher cognitive functions in humans might alter the presentation of the toxicity in ways that cannot be adjusted for because much of the mechanisms of the functioning of the human nervous system are not fully understood today.
Another important limitation of the currently available literature is that the majority of studies discussed were on effects of inorganic metals on the brain cells or brain molecules, and very few centered around organic metals which are perhaps even more significant than inorganic metal toxicity since in some cases the organic forms are more toxic than the inorganic forms, as for mercury [257]. There is a rapidly growing body of evidence that the majority of metals may actually be methylated to their organic form as the body attempts to detoxify metals. For arsenic, in the past it was believed that conversion of arsenic to monomethyl arsenic and dimethyl-arsenic was a method of detoxification; however, the view has changed since then with the recognition that methylated metabolites of trivalent arsenic are carcinogenic [258]. Antimony, mercury, lead, tin, and selenium are known to cause public health problems in their methylated forms. Cadmium, cobalt, mercury, and nickel reportedly undergo biomethylation; however, the effects of biomethylation have been studied more in unicellular organisms rather than plants and animals, therefore it has been suggested that although biomethylation does occur in plants and animals, the rates are likely to vary on the basis of the animal and the metal, and its concentration [259].
There are some situations that have not been considered in most experimental designs. One such issue that arises with the study of metals is the problem that most studies are not reflective with regard to the actual exposure in nature where humans are simultaneously exposed to more than one metal. Very few studies have targeted this issue, most likely due to the complicated nature of conducting an experiment with many variables and determining the contribution of each. One study by Platt and Büsselberg who examined the effects of combinations of Pb2+, Zn2+, and Al3+, on voltage-activated calcium channels by simultaneous application of various combinations of two metals determined that regardless of the order in which the metals were added, the actions were in fact additive [260]. Whether this is the case with other metals is not certain and there is not enough data in the literature describing effects of combinations of metals. Another limitation of the study is that some targets may not be as relevant as others in causing the clinical symptoms of metal toxicity; however, it is not possible to know at this stage the exact contribution of each target.
Finally, the most important objective is to put these effects of metals into practical use. This can be done by using the data of the toxic concentrations of metals to make a meaningful decision in regard to their acceptable blood levels. There is evidence to suggest that currently accepted levels for some metals are still not “safe” levels. Even at the currently accepted blood lead levels of 10 μg/dL, it is causative of preterm labor and adverse pregnancy outcome [261]. Therefore, there is a need to reevaluate the accepted blood concentrations of metals in light of the newer evidence as it appears.
Metal neurotoxicity is a field, which is abounding with the literature and excellent research; however, in the current literature some metals are highlighted while for other metals (or metal compounds) hardly any data are available. There is an emphasis on certain metals such as lead, whose harmful effects are well known while there is very little known about certain metal groups such as lanthanides and actinides.
There are certain targets where metal actions have been excessively examined such as voltage-activated calcium channels while there is little known about the effects of metals on parts of signaling pathways such as phosphodiesterases and IP3. This raises the need to evaluate new targets for metals, which have not been studied before, which may prove to have a groundbreaking effect in the field of neurotoxicity.
To summarize, exposure to different metals occurs due to industrial activities, environmental, and food chain contamination. This paper elucidated the various targets of metals in synaptic transmission and synaptic plasticity. Exposure to metals had varied effects on different synaptic targets, which were dependent on the form of metal, the concentration of metal, route of exposure (in vitro or in vivo), the medium used, and even the duration of exposure in some cases.
Authors' Contribution
S. Sadiq, Z. Ghazala and A. Chowdhury contributed equally.
Abbreviations
- [Ca2+]i:
Intracellular calcium concentration
- ABD:
Agonist-binding domain
- AC:
Adenylate cyclase
- Ach:
Acetylcholine
- Al:
Aluminum
- AlCl3:
Aluminum trichloride
- AMPA:
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- As:
Arsenic
- As2O3:
Arsenic trioxide
- ATD:
Amino terminal domain
- ATP:
Adenosine triphosphate
- ATPase:
Adenosine triphosphatase
- Ba:
Barium
- BAPTA:
1,2-bis(2-aminophenoxy)ethane-N,N,N9,N9-tetracetic acid tetrakis(acetoxymethyl) ester
- Bmax:
Maximal binding
- Ca:
Calcium
- CaM:
Calmodulin
- CaMK:
Ca2+/calmodulin-dependent protein kinase
- cAMP:
Cyclic adenosine monophosphate
- Cd:
Cadmium
- CdCl2:
Cadmium chloride
- CDDP:
cis-Diammine-dichloroplatin
- CH3Hg:
Methylmercury
- CNS:
Central nervous system
- Co:
Cobalt
- Cr:
Chromium
- CRE:
cAMP response elements
- CREB:
Ca+2/cAMP response element-binding protein
- Cu:
Copper
- DAG:
Diacylglycerol
- DDW:
Distilled deionized water
- DOC2:
Double C2 domain
- DOPAC:
3.4-dihydroxyphenylacetic acid
- DOPAC:
3.4-dihydroxyphenylacetic acid
- DRG:
Dorsal root ganglion
- E-LTP:
Early-phase LTP
- ED:
Embryonic day
- eNOS:
Eendothelial NOS
- EPP:
End-plate potential
- ERK1/2:
Extracellular signal-related kinase 1/2
- Et3Pb:
Triethyl lead
- Et3Sn:
Triethyl-tin
- Fe:
Iron
- GABA:
γ-Aminobutyric acid
- GAP:
GTPase-activating protein
- GEF:
Guanyl nucleotide exchange factors
- Gi/o:
Inhibitory Sguanine nucleotide binding protein/other guanine nucleotide-binding protein
- Glu:
Glutamate
- GluR:
Glutamate Receptor
- Gly:
Glycine
- GPCR:
G-protein-coupled Receptors
- Gpp(NH)p:
5′ Guanylylimidodiphosphate
- Gs:
Stimulatory guanine nucleotide binding protein
- GTP:
Guanosine-5′-triphosphate
- Hg:
Mercury
- HVA:
Homovanillic acid
- IC50:
Concentration for 50% inhibition
- iNOS:
Inducible NOS
- IP3:
Inositol triphosphate
- Inositol triphosphate:
Inhibitory postsynaptic currents
- IQ:
Intelligence quotient
- JNK:
c-JUN-N-terminal kinase
- K:
Potassium
- KCl:
Potassium chloride
- Kd:
Dissociation constant
- L-LTP:
Late-phase LTP
- La:
Lanthanum
- Li:
Lithium
- LTD:
Long-term depression
- LTP:
Long-term potentiation
- mAb:
Monoclonal antibody
- MAPK:
Mitogen-activated protein kinase
- Me3Pb:
Trimethyl lead
- Me3Sn:
Trimethyl-tin
- MeHg:
Methylmercury
- MEK:
MAPK/ERK kinase
- MEPP:
Miniature end-plate potential
- Met:
Methionine
- Mg:
Magnesium
- mGluR:
Metabotropic glutamate receptors
- mIPSC:
Miniature inhibitory postsynaptic currents
- Mn:
Manganese
- mRNA:
Messenger ribo nucleic acid
- Na:
Sodium
- NADPH:
Reduced nicotinamide adenine dinucleotide phosphate
- Ni:
Nickel
- NMDA:
N-methyl-D-aspartate
- NMDAR:
N-methyl-D-aspartate receptors
- nNOS:
Neuronal NOS
- NOS:
Nitric oxide synthase
- NT:
Neurotransmitter
- Pb:
Lead
- PKA:
Protein kinase A
- PLC:
Phospholipase C
- PND:
Postnatal day
- Ras:
Rat sarcoma family
- RBC:
Red blood cells
- Sn:
Tin
- SNAP-25:
Synaptosome-associated protein 25 kDa
- SNARE:
SNAP and NSF attachment receptor
- SnCl2:
Stannous chloride
- Sr:
Strontium
- STP:
Short-term potentiation
- Thr:
Threonine
- VGCC:
Voltage-gated calcium Channel
- Vm:
Membrane voltage
- Zn:
Zinc.
References
- 1.Craig PJ, Eng G, Jenkins RO. Occurrence and pathways of organometallic compounds in the environment—general considerations. In: Craig PJ, editor. Organometallic Compounds in the Environment. Chichester, UK: John Wiley & Sons; 2003. pp. 1–55. [Google Scholar]
- 2.Waldron HA. Lead poisoning in the ancient world. Medical History. 1973;17(4):391–399. doi: 10.1017/s0025727300019013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crapper DR, Krishnan SS, Quittkat S. Aluminium, neurofibrillary degeneration and Alzheimer’s disease. Brain. 1976;99(1):67–80. doi: 10.1093/brain/99.1.67. [DOI] [PubMed] [Google Scholar]
- 4.Michalke B, Halbach S, Nischwitz V. JEM spotlight: metal speciation related to neurotoxicity in humans. Journal of Environmental Monitoring. 2009;11(5):939–954. doi: 10.1039/b817817h. [DOI] [PubMed] [Google Scholar]
- 5.Needleman H. Low level lead exposure: history and discovery. Annals of Epidemiology. 2009;19(4):235–238. doi: 10.1016/j.annepidem.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Swandulla D, Armstrong CM. Calcium channel block by cadmium in chicken sensory neurons. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(5):1736–1740. doi: 10.1073/pnas.86.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Florea AM, Dopp E, Büsselberg D. Elevated Ca2+i transients induced by trimethyltin chloride in HeLa cells: types and levels of response. Cell Calcium. 2005;37(3):251–258. doi: 10.1016/j.ceca.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity—a review. Human and Experimental Toxicology. 2007;26(10):823–832. doi: 10.1177/0960327107084539. [DOI] [PubMed] [Google Scholar]
- 9.Wong PCL, Lai JCK, Lim L, Davison AN. Selective inhibition of L-glutamate and gammaaminobutyrate transport in nerve ending particles by aluminium, manganese, and cadmium chloride. Journal of Inorganic Biochemistry. 1981;14(3):253–260. doi: 10.1016/s0162-0134(00)80005-7. [DOI] [PubMed] [Google Scholar]
- 10.Rodrigues ALS, Regner A, Rubin MA, Souza DO. Effects of lead on adenylate cyclase activity in rat cerebral cortex. Neurochemical Research. 1999;24(8):1037–1042. doi: 10.1023/a:1021008910900. [DOI] [PubMed] [Google Scholar]
- 11.Chow RH. Cadmium block of squid calcium currents. Macroscopic data and a kinetic model. Journal of General Physiology. 1991;98(4):751–770. doi: 10.1085/jgp.98.4.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minami A, Takeda A, Nishibaba D, Takefuta S, Oku N. Cadmium toxicity in synaptic neurotransmission in the brain. Brain Research. 2001;894(2):336–339. doi: 10.1016/s0006-8993(01)02022-4. [DOI] [PubMed] [Google Scholar]
- 13.Marchetti C, Gavazzo P. NMDA receptors as targets of heavy metal interaction and toxicity. Neurotoxicity Research. 2005;8(3-4):245–258. doi: 10.1007/BF03033978. [DOI] [PubMed] [Google Scholar]
- 14.Celentano JJ, Gyenes M, Gibbs TT, Farb DH. Negative modulation of the γ-aminobutyric acid response by extracellular zinc. Molecular Pharmacology. 1991;40(5):766–773. [PubMed] [Google Scholar]
- 15.Florea AM, Büsselberg D. Occurrence, use and potential toxic effects of metals and metal compounds. BioMetals. 2006;19(4):419–427. doi: 10.1007/s10534-005-4451-x. [DOI] [PubMed] [Google Scholar]
- 16.Ragan CI. Metal ions in neuroscience. Metal-Based Drugs. 1997;4(3):125–132. doi: 10.1155/MBD.1997.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomaszewski A, Büsselberg D. Cisplatin modulates voltage gated channel currents of dorsal root ganglion neurons of rats. NeuroToxicology. 2007;28(1):49–58. doi: 10.1016/j.neuro.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Purves D, et al. Neuroscience. Sunderland, UK: Sinauer Associates; 2008. Synaptic transmission; pp. 85–118. [Google Scholar]
- 19.Busselberg D, Platt B, Michael D, Carpenter DO, Haas HL. Mammalian voltage-activated calcium channel currents are blocked by Pb2+, Zn2+, and Al3+ Journal of Neurophysiology. 1994;71(4):1491–1497. doi: 10.1152/jn.1994.71.4.1491. [DOI] [PubMed] [Google Scholar]
- 20.Julka D, Gill KD. Altered calcium homeostasis: a possible mechanism of aluminium-induced neurotoxicity. Biochimica et Biophysica Acta. 1996;1315(1):47–54. doi: 10.1016/0925-4439(95)00100-x. [DOI] [PubMed] [Google Scholar]
- 21.Kumar V, Gill KD. Aluminium neurotoxicity: neurobehavioural and oxidative aspects. Archives of Toxicology. 2009;83(11):965–978. doi: 10.1007/s00204-009-0455-6. [DOI] [PubMed] [Google Scholar]
- 22.Welling A. Voltage-dependent calcium channels. Biotrend Reviews. 2009:1–2009. [Google Scholar]
- 23.Busselberg D, Evans ML, Rahmann H, Carpenter DO. Lead and zinc block a voltage-activated calcium channel of Aplysia neurons. Journal of Neurophysiology. 1991;65(4):786–795. doi: 10.1152/jn.1991.65.4.786. [DOI] [PubMed] [Google Scholar]
- 24.Busselberg D, Pekel M, Michael D, Platt B. Mercury (Hg2+) and zinc (Zn2+): two divalent cations with different actions on voltage-activated calcium channel currents. Cellular and Molecular Neurobiology. 1994;14(6):675–686. doi: 10.1007/BF02088676. [DOI] [PubMed] [Google Scholar]
- 25.Evans ML, Busselberg D, Carpenter DO. Pb2+ blocks calcium currents of cultured dorsal root ganglion cells. Neuroscience Letters. 1991;129(1):103–106. doi: 10.1016/0304-3940(91)90730-h. [DOI] [PubMed] [Google Scholar]
- 26.Hajela RK, Peng SQ, Atchison WD. Comparative effects of methylmercury and Hg2+ on human neuronal N- and R-type high-voltage activated calcium channels transiently expressed in human embryonic kidney 293 cells. Journal of Pharmacology and Experimental Therapeutics. 2003;306(3):1129–1136. doi: 10.1124/jpet.103.049429. [DOI] [PubMed] [Google Scholar]
- 27.Busselberg D, Evans ML, Haas HL, Carpenter DO. Blockade of mammalian and invertebrate calcium channels by lead. NeuroToxicology. 1993;14(2-3):249–258. [PubMed] [Google Scholar]
- 28.Audesirk G, Audesirk T. The effects of inorganic lead on voltage-sensitive calcium channels differ among cell types and among channel subtypes. NeuroToxicology. 1993;14(2-3):259–266. [PubMed] [Google Scholar]
- 29.Reuveny E, Narahashi T. Potent blocking action of lead on voltage-activated calcium channels in human neuroblastoma cells SH-SY5Y. Brain Research. 1991;545(1-2):312–314. doi: 10.1016/0006-8993(91)91304-j. [DOI] [PubMed] [Google Scholar]
- 30.Audesirk G, Audesirk T. Effects of inorganic lead on voltage-sensitive calcium channels in N1E-115 neuroblastoma cells. NeuroToxicology. 1991;12(3):519–528. [PubMed] [Google Scholar]
- 31.Busselberg D, Michael D, Evans ML, Carpenter DO, Haas HL. Zinc (Zn2+) blocks voltage gated calcium channels in cultured rat dorsal root ganglion cells. Brain Research. 1992;593(1):77–81. doi: 10.1016/0006-8993(92)91266-h. [DOI] [PubMed] [Google Scholar]
- 32.Schild L, Moczydlowski E. Competitive binding interaction between Zn2+ and saxitoxin in cardiac Na+ channels. Evidence for a sulfhydryl group in the Zn2+ saxitoxin binding site. Biophysical Journal. 1991;59(3):523–537. doi: 10.1016/S0006-3495(91)82269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gawrisch E, Leonhardt R, Büsselberg D. Voltage-activated calcium channel currents of rat dorsal root ganglion cells are reduced by trimethyl lead. Toxicology Letters. 1997;92(2):117–122. doi: 10.1016/s0378-4274(97)00054-4. [DOI] [PubMed] [Google Scholar]
- 34.Hattori T, Maehashi H. Stannous chloride-induced increase in calcium entry into motor nerve terminals of the frog. European Journal of Pharmacology. 1989;166(3):527–530. doi: 10.1016/0014-2999(89)90369-5. [DOI] [PubMed] [Google Scholar]
- 35.Alexander SPH, Mathie A, Peters JA. Ion channels. British Journal of Pharmacology. 2004;141(S1):S71–S91. doi: 10.1038/sj.bjp.0705672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sue Marty M, Atchison WD. Pathways mediating Ca2+ entry in rat cerebellar granule cells following in vitro exposure to methyl mercury. Toxicology and Applied Pharmacology. 1997;147(2):319–330. doi: 10.1006/taap.1997.8262. [DOI] [PubMed] [Google Scholar]
- 37.Goldin AL, Barchi RL, Caldwell JH, et al. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28(2):365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 38.Sirois JE, Atchison WD. Methylmercury affects multiple subtypes of calcium channels in rat cerebellar granule cells. Toxicology and Applied Pharmacology. 2000;167(1):1–11. doi: 10.1006/taap.2000.8967. [DOI] [PubMed] [Google Scholar]
- 39.Shafer TJ, Atchison WD. Methylmercury blocks N- and L-type Ca++ channels in nerve growth factor-differentiated pheochromocytoma (PC12) cells. Journal of Pharmacology and Experimental Therapeutics. 1991;258(1):149–157. [PubMed] [Google Scholar]
- 40.Pekel M, Platt B, Busselberg D. Mercury (Hg2+) decreases voltage-gated calcium channels currents in rat DRG and Aplysia neurons. Brain Research. 1993;632(1-2):121–126. doi: 10.1016/0006-8993(93)91146-j. [DOI] [PubMed] [Google Scholar]
- 41.Tomaszewski A, Büsselberg D. SnCl2 reduces voltage-activated calcium channel currents of dorsal root ganglion neurons of rats. NeuroToxicology. 2008;29(6):958–963. doi: 10.1016/j.neuro.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Frelin C, Cognard C, Vigne P, Lazdunski M. Tetrodotoxin-sensitive and tetrodotoxin-resistant Na+ channels differ in their sensitivity to Cd2+ and Zn2+ European Journal of Pharmacology. 1986;122(2):245–250. doi: 10.1016/0014-2999(86)90109-3. [DOI] [PubMed] [Google Scholar]
- 43.Györi J, Platoshyn O, Carpenter DO, Salánki J. Effect of inorganic and organic tin compounds on ACh- and voltage- activated Na currents. Cellular and Molecular Neurobiology. 2000;20(5):591–604. doi: 10.1023/a:1007016012520. [DOI] [PubMed] [Google Scholar]
- 44.Ravindran A, Schild L, Moczydlowski E. Divalent cation selectivity for external block of voltage-dependent Na+ channels prolonged by batrachotoxin. Zn2+ induces discrete substates in crdiac Na+ channels. Journal of General Physiology. 1991;97(1):89–115. doi: 10.1085/jgp.97.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Annals of the New York Academy of Sciences. 1999;868(1):233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- 46.Huang RC, Peng YW, Yau KW. Zinc modulation of a transient potassium current and histochemical localization of the metal in neurons of the suprachiasmatic nucleus. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(24):11806–11810. doi: 10.1073/pnas.90.24.11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo CC, Chen FP. Zn2+ modulation of neuronal transient K+ current: fast and selective binding to the deactivated channels. Biophysical Journal. 1999;77(5):2552–2562. doi: 10.1016/s0006-3495(99)77090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu TX, Gong N, Xu TL. Divalent cation modulation of A-type potassium channels in acutely dissociated central neurons from wide-type and mutant Drosophila. Journal of Neurogenetics. 2005;19(2):87–107. doi: 10.1080/01677060591007182. [DOI] [PubMed] [Google Scholar]
- 49.Watkins CS, Mathie A. Modulation of the gating of the transient outward potassium current of rat isolated cerebellar granule neurons by lanthanum. Pflugers Archiv: European Journal of Physiology. 1994;428(3-4):209–216. doi: 10.1007/BF00724499. [DOI] [PubMed] [Google Scholar]
- 50.Mayer ML, Sugiyama K. A modualtory action of divalent cations on transient outward current in cultured rat sensory neurones. Journal of Physiology. 1988;396:417–433. doi: 10.1113/jphysiol.1988.sp016970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leonhardt R, Haas H, Büsselberg D. Methyl mercury reduces voltage-activated currents of rat dorsal root ganglion neurons. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1996;354(4):532–538. doi: 10.1007/BF00168447. [DOI] [PubMed] [Google Scholar]
- 52.de Jong AP, Verhage M. Presynaptic signal transduction pathways that modulate synaptic transmission. Current Opinion in Neurobiology. 2009;19(3):245–253. doi: 10.1016/j.conb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Sandhir R, Gill KD. Calmodulin and cAMP dependent synaptic vesicle protein phosphorylation in rat cortex following lead exposure. International Journal of Biochemistry. 1994;26(12):1383–1389. doi: 10.1016/0020-711x(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 54.Ewers U, Erbe R. Effects of lead, cadmium and mercury on brain adenylate cyclase. Toxicology. 1980;16(3):227–237. doi: 10.1016/0300-483x(80)90119-5. [DOI] [PubMed] [Google Scholar]
- 55.Narahashi T, Ma JY, Arakawa O, Reuveny E, Nakahiro M. GABA receptor-channel complex as a target site of mercury, copper, zinc, and lanthanides. Cellular and Molecular Neurobiology. 1994;14(6):599–621. doi: 10.1007/BF02088671. [DOI] [PubMed] [Google Scholar]
- 56.Nihei MK, Guilarte TR. NNMDAR-2A subunit protein expression is reduced in the hippocampus of rats exposed to Pb2+ during development. Molecular Brain Research. 1999;66(1-2):42–49. doi: 10.1016/s0169-328x(99)00005-4. [DOI] [PubMed] [Google Scholar]
- 57.Guilarte TR, McGlothan JL. Hippocampal NMDA receptor MRNA undergoes subunit specific changes during developmental lead exposure. Brain Research. 1998;790(1-2):98–107. doi: 10.1016/s0006-8993(98)00054-7. [DOI] [PubMed] [Google Scholar]
- 58.Vig PJS, Ravi K, Nath R. Interaction of metals with brain calmodulin purified from normal and cadmium exposed rats. Drug and Chemical Toxicology. 1991;14(1-2):207–218. doi: 10.3109/01480549109017877. [DOI] [PubMed] [Google Scholar]
- 59.Sandhir R, Gill KD. Lead perturbs calmodulin dependent cyclic AMP metabolism in rat central nervous system. Biochemistry and Molecular Biology International. 1994;33(4):729–742. [PubMed] [Google Scholar]
- 60.Siegel N, Haug A. Aluminum interaction with calmodulin. Evidence for altered structure and function from optical and enzymatic studies. Biochimica et Biophysica Acta. 1983;744(1):36–45. doi: 10.1016/0167-4838(83)90337-0. [DOI] [PubMed] [Google Scholar]
- 61.Cox JL, Harrison SD. Correlation of metal toxicity with in vitro calmodulin inhibition. Biochemical and Biophysical Research Communications. 1983;115(1):106–111. doi: 10.1016/0006-291x(83)90975-0. [DOI] [PubMed] [Google Scholar]
- 62.Levi R, Wolf T, Fleminger G, Solomon B. Immuno-detection of aluminium and aluminium induced conformational changes in calmodulin-implications in Alzheimer’s disease. Molecular and Cellular Biochemistry. 1998;189(1-2):41–46. doi: 10.1023/a:1006887809463. [DOI] [PubMed] [Google Scholar]
- 63.Ohtani-Kaneko R, Tazawa H, Yokosuka M, Yoshida M, Satoh M, Watanabe C. Suppressive effects of cadmium on neurons and affected proteins in cultured developing cortical cells. Toxicology. 2008;253(1-3):110–116. doi: 10.1016/j.tox.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 64.Rajanna B, Chetty CS, Rajanna S, Hall E, Fail S, Yallapragada PR. Modulation of protein kinase C by heavy metals. Toxicology Letters. 1995;81(2-3):197–203. doi: 10.1016/0378-4274(95)03433-1. [DOI] [PubMed] [Google Scholar]
- 65.Xu SZ, Bullock L, Shan CJ, Cornelius K, Rajanna B. PKC isoforms were reduced by lead in the developing rat brain. International Journal of Developmental Neuroscience. 2005;23(1):53–64. doi: 10.1016/j.ijdevneu.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 66.Inoue Y, Saijoh K, Sumino K. Action of mercurials on activity of partially purified soluble protein kinase C from mice brain. Pharmacology and Toxicology. 1988;62(5):278–281. doi: 10.1111/j.1600-0773.1988.tb01887.x. [DOI] [PubMed] [Google Scholar]
- 67.Saijoh K, Fukunaga T, Katsuyama H, Myeong Jin Lee, Sumino K. Effects of methylmercury on protein kinase A and protein kinase C in the mouse brain. Environmental Research. 1993;63(2):264–273. doi: 10.1006/enrs.1993.1146. [DOI] [PubMed] [Google Scholar]
- 68.Nihei MK, McGlothan JL, Toscano CD, Guilarte TR. Low level Pb2+ exposure affects hippocampal protein kinase Cγ gene and protein expression in rats. Neuroscience Letters. 2001;298(3):212–216. doi: 10.1016/s0304-3940(00)01741-9. [DOI] [PubMed] [Google Scholar]
- 69.Johnson GVW, Cogdill KW, Jope RS. Oral alumimum alters in vitro protein phosphorylation and kinase activities in rat brain. Neurobiology of Aging. 1990;11(3):209–216. doi: 10.1016/0197-4580(90)90547-d. [DOI] [PubMed] [Google Scholar]
- 70.Coppi AA, Lesniak J, Zieba D, Schanne FAX. The effects of lead on PKC isoforms. Annals of the New York Academy of Sciences. 2000;919:304–306. doi: 10.1111/j.1749-6632.2000.tb06891.x. [DOI] [PubMed] [Google Scholar]
- 71.Hermenegildo C, Sáez R, Minoia C, Manzo L, Felipo V. Chronic exposure to aluminium impairs the glutamate-nitric oxide-cyclic GMP pathway in the rat in vivo. Neurochemistry International. 1999;34(3):245–253. doi: 10.1016/s0197-0186(99)00010-8. [DOI] [PubMed] [Google Scholar]
- 72.Zarazúa S, Pérez-Severiano F, Delgado JM, Martínez LM, Ortiz-Pérez D, Jiménez-Capdeville ME. Decreased nitric oxide production in the rat brain after chronic arsenic exposure. Neurochemical Research. 2006;31(8):1069–1077. doi: 10.1007/s11064-006-9118-7. [DOI] [PubMed] [Google Scholar]
- 73.Mittal CK, Harrell WB, Mehta CS. Interaction of heavy metal toxicants with brain constitutive nitric oxide synthase. Molecular and Cellular Biochemistry. 1995;149-150:263–265. doi: 10.1007/BF01076586. [DOI] [PubMed] [Google Scholar]
- 74.García-Arenas G, Ramírez-Amaya V, Balderas I, et al. Cognitive deficits in adult rats by lead intoxication are related with regional specific inhibition of cNOS. Behavioural Brain Research. 2004;149(1):49–59. doi: 10.1016/s0166-4328(03)00195-5. [DOI] [PubMed] [Google Scholar]
- 75.Chetty CS, Reddy GR, Murthy KS, Johnson J, Sajwan K, Desaiah D. Perinatal lead exposure alters the expression of neuronal nitric oxide synthase in rat brain. International Journal of Toxicology. 2001;20(3):113–120. doi: 10.1080/109158101317097692. [DOI] [PubMed] [Google Scholar]
- 76.Llansola M, Miñana MD, Montoliu C, et al. Prenatal exposure to aluminum reduces expression of neuronal nitric oxide synthase and of soluble guanylate cyclase and impairs glutamatergic neurotransmission in rat cerebellum. Journal of Neurochemistry. 1999;73(2):712–718. doi: 10.1046/j.1471-4159.1999.0730712.x. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y, Li S, Piao F, Hong Y, Liu P, Zhao Y. Arsenic down-regulates the expression of Camk4, an important gene related to cerebellar LTD in mice. Neurotoxicology and Teratology. 2009;31(5):318–322. doi: 10.1016/j.ntt.2009.04.064. [DOI] [PubMed] [Google Scholar]
- 78.Toscano CD, O’Callaghan JP, Guilarte TR. Calcium/calmodulin-dependent protein kinase II activity and expression are altered in the hippocampus of Pb2+-exposed rats. Brain Research. 2005;1044(1):51–58. doi: 10.1016/j.brainres.2005.02.076. [DOI] [PubMed] [Google Scholar]
- 79.Rigon AP, Cordova FM, Oliveira CS, et al. Neurotoxicity of cadmium on immature hippocampus and a neuroprotective role for p38MAPK. NeuroToxicology. 2008;29(4):727–734. doi: 10.1016/j.neuro.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 80.Cordova FM, Rodrigues ALS, Giacomelli MBO, et al. Lead stimulates ERK1/2 and p38MAPK phosphorylation in the hippocampus of immature rats. Brain Research. 2004;998(1):65–72. doi: 10.1016/j.brainres.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 81.Toscano CD, Hashemzadeh-Gargari H, McGlothan JL, Guilarte TR. Developmental Pb2+ exposure alters NMDAR subtypes and reduces CREB phosphorylation in the rat brain. Developmental Brain Research. 2002;139(2):217–226. doi: 10.1016/s0165-3806(02)00569-2. [DOI] [PubMed] [Google Scholar]
- 82.Klein C, Sunahara RK, Hudson TY, Heyduk T, Howlett AC. Zinc inhibition of cAMP signaling. Journal of Biological Chemistry. 2002;277(14):11859–11865. doi: 10.1074/jbc.M108808200. [DOI] [PubMed] [Google Scholar]
- 83.Klein C, Heyduk T, Sunahara RK. Zinc inhibition of adenylyl cyclase correlates with conformational changes in the enzyme. Cellular Signalling. 2004;16(10):1177–1185. doi: 10.1016/j.cellsig.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 84.Ziv NE, Garner CC. Cellular and molecular mechanisms of presynaptic assembly. Nature Reviews Neuroscience. 2004;5(5):385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]
- 85.Suszkiw JB. Presynaptic disruption of transmitter release by lead. NeuroToxicology. 2004;25(4):599–604. doi: 10.1016/j.neuro.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 86.Slotkin TA, Seidler FJ. Protein kinase C is a target for diverse developmental neurotoxicants: transcriptional responses to chlorpyrifos, diazinon, dieldrin and divalent nickel in PC12 cells. Brain Research. 2009;1263(C):23–32. doi: 10.1016/j.brainres.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rizo J, Südhof TC. Snares and munc18 in synaptic vesicle fusion. Nature Reviews Neuroscience. 2002;3(8):641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- 88.Splettstoesser F, Florea AM, Büsselberg D. IP3 receptor antagonist, 2-APB, attenuates cisplatin induced Ca2+-influx in HeLa-S3 cells and prevents activation of calpain and induction of apoptosis. British Journal of Pharmacology. 2007;151(8):1176–1186. doi: 10.1038/sj.bjp.0707335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sarafian TA. Methyl mercury increases intracellular Ca2+ and inositol phosphate levels in cultured cerebellar granule neurons. Journal of Neurochemistry. 1993;61(2):648–657. doi: 10.1111/j.1471-4159.1993.tb02169.x. [DOI] [PubMed] [Google Scholar]
- 90.Aggarwal SK. A histochemical approach to the mechanism of action of cisplatin and its analogues. Journal of Histochemistry and Cytochemistry. 1993;41(7):1053–1073. doi: 10.1177/41.7.8515048. [DOI] [PubMed] [Google Scholar]
- 91.Gemba M, Nakatani E, Teramoto M, Nakano S. Effect of cisplatin on calcium uptake by rat kidney cortical mitochondria. Toxicology Letters. 1987;38(3):291–297. doi: 10.1016/0378-4274(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 92.Sue Marty M, Atchison WD. Elevations of intracellular Ca2+ as a probable contributor to decreased viability in cerebellar granule cells following acute exposure to methylmercury. Toxicology and Applied Pharmacology. 1998;150(1):98–105. doi: 10.1006/taap.1998.8383. [DOI] [PubMed] [Google Scholar]
- 93.DeLorenzo RJ. The calmodulin hypothesis of neurotransmission. Cell Calcium. 1981;2(4):365–385. doi: 10.1016/0143-4160(81)90026-9. [DOI] [PubMed] [Google Scholar]
- 94.Angle CR, McIntire MS. Red cell lead, whole blood lead, and red cell enzymes. Environmental Health Perspectives. 1974;7:133–137. doi: 10.1289/ehp.747133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aschner M, Aschner JL. Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neuroscience and Biobehavioral Reviews. 1990;14(2):169–176. doi: 10.1016/s0149-7634(05)80217-9. [DOI] [PubMed] [Google Scholar]
- 96.Antonio MT, Corredor L, Leret ML. Study of the activity of several brain enzymes like markers of the neurotoxicity induced by perinatal exposure to lead and/or cadmium. Toxicology Letters. 2003;143(3):331–340. doi: 10.1016/s0378-4274(03)00194-2. [DOI] [PubMed] [Google Scholar]
- 97.Aschner M, Aschner JL. Cellular and molecular effects of trimethyltin and triethyltin: relevance to organotin neurotoxicity. Neuroscience and Biobehavioral Reviews. 1992;16(4):427–435. doi: 10.1016/s0149-7634(05)80184-8. [DOI] [PubMed] [Google Scholar]
- 98.Haeffner EW, Zimmermann HP, Hoffmann CJK. Influence of triethyl lead on the activity of enzymes of the ascites tumor cell plasma membrane and its microviscosity. Toxicology Letters. 1984;23(2):183–188. doi: 10.1016/0378-4274(84)90124-3. [DOI] [PubMed] [Google Scholar]
- 99.Stine KE, Reiter LW, Lemasters JJ. Alkyltin inhibition of ATPase activities in tissue homogenates and subcellular fractions from adults and neonatal rats. Toxicology and Applied Pharmacology. 1988;94(3):394–406. doi: 10.1016/0041-008x(88)90280-3. [DOI] [PubMed] [Google Scholar]
- 100.Südhof TC, Rizo J. Synaptotagmins: C2-domain proteins that regulate membrane traffic. Neuron. 1996;17(3):379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- 101.Bouton CMLS, Frelin LP, Forde CE, Godwin HA, Pevsner J. Synaptotagmin I is a molecular target for lead. Journal of Neurochemistry. 2001;76(6):1724–1735. doi: 10.1046/j.1471-4159.2001.00168.x. [DOI] [PubMed] [Google Scholar]
- 102.Rajalingam D, Kumar TKS, Yu C. The C2A domain of synaptotagmin exhibits a high binding affinity for copper: implications in the formation of the multiprotein FGF release complex. Biochemistry. 2005;44(44):14431–14442. doi: 10.1021/bi051387r. [DOI] [PubMed] [Google Scholar]
- 103.Kathir KM, Gao L, Rajalingam D, et al. NMR characterization of copper and lipid interactions of the C2B domain of synaptotagmin I-relevance to the non-classical secretion of the human acidic fibroblast growth factor (hFGF-1) Biochimica et Biophysica Acta. 2010;1798(2):297–302. doi: 10.1016/j.bbamem.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Brock TO, O’Callaghan JP. Quantitative changes in the synaptic vesicle proteins synapsin I and p38 and the astrocyte-specific protein glial fibrillary acidic protein are associated with chemical-induced injury to the rat central nervous system. Journal of Neuroscience. 1987;7(4):931–942. doi: 10.1523/JNEUROSCI.07-04-00931.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang J, Rahman MF, Duhart HM, et al. Expression changes of dopaminergic system-related genes in PC12 cells induced by manganese, silver, or copper nanoparticles. NeuroToxicology. 2009;30(6):926–933. doi: 10.1016/j.neuro.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 106.Verstraeten SV, Aimo L, Oteiza PI. Aluminium and lead: molecular mechanisms of brain toxicity. Archives of Toxicology. 2008;82(11):789–802. doi: 10.1007/s00204-008-0345-3. [DOI] [PubMed] [Google Scholar]
- 107.Mailman RB, Krigman MR, Frye GD, Hanin I. Effects of postnatal trimethyltin or triethyltin treatment on CNS catecholamine, GABA, and acetylcholine systems in the rat. Journal of Neurochemistry. 1983;40(5):1423–1429. doi: 10.1111/j.1471-4159.1983.tb13585.x. [DOI] [PubMed] [Google Scholar]
- 108.Hattori T, Maehashi H. Acceleration by stannous ion of the evoked release of transmitter from motor nerve endings in the frog. Brain Research. 1988;473(1):157–160. doi: 10.1016/0006-8993(88)90328-9. [DOI] [PubMed] [Google Scholar]
- 109.Braga MFM, Pereira EFR, Marchioro M, Albuquerque EX. Lead increases tetrodotoxin-insensitive spontaneous release of glutamate and GABA from hippocampal neurons. Brain Research. 1999;826(1):10–21. doi: 10.1016/s0006-8993(99)01193-2. [DOI] [PubMed] [Google Scholar]
- 110.Doctor SV, Costa LG, Kendall DA, Murphy SD. Trimethyltin inhibits uptake of neurotransmitters into mouse forebrain synaptosomes. Toxicology. 1982;25(2-3):213–221. doi: 10.1016/0300-483x(82)90031-2. [DOI] [PubMed] [Google Scholar]
- 111.Seidman BC, Verity MA. Selective inhibition of synaptosomal γ-aminobutyric acid uptake by triethyllead: role of energy transduction and chloride ion. Journal of Neurochemistry. 1987;48(4):1142–1149. doi: 10.1111/j.1471-4159.1987.tb05639.x. [DOI] [PubMed] [Google Scholar]
- 112.Skilleter DN. The decrease of mitochondrial substrate uptake caused by trialkyltin and trialkyl lead compounds in chloride media and its relevance to inhibition of oxidative phosphorylation. Biochemical Journal. 1975;146(2):465–471. doi: 10.1042/bj1460465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vahidnia A, Romijn F, Tiller M, Van Der Voet GB, De Wolff FA. Arsenic-induced toxicity: effect on protein composition in sciatic nerve. Human and Experimental Toxicology. 2006;25(11):667–674. doi: 10.1177/0960327106070671. [DOI] [PubMed] [Google Scholar]
- 114.Zimmermann HP, Doenges KH, Roderer G. Interaction of triethyl lead chloride with microtubules in vitro and in mammalian cells. Experimental Cell Research. 1985;156(1):140–152. doi: 10.1016/0014-4827(85)90268-x. [DOI] [PubMed] [Google Scholar]
- 115.Faulstich H, Stournaras C, Doenges KH, Zimmermann HP. The molecular mechanism of interaction of Et3Pb+ with tubulin. FEBS Letters. 1984;174(1):128–131. doi: 10.1016/0014-5793(84)81090-x. [DOI] [PubMed] [Google Scholar]
- 116.Stournaras C, Weber G, Zimmermann HP. High cytotoxicity and membrane permeability of Et3Pb+ in mammalian and plant cells. Cell Biochemistry and Function. 1984;2(4):213–216. doi: 10.1002/cbf.290020406. [DOI] [PubMed] [Google Scholar]
- 117.Zimmermann HP, Plagens U, Traub P. Influence of triethyl lead on neurofilaments in vivo and in vitro. NeuroToxicology. 1987;8(4):569–578. [PubMed] [Google Scholar]
- 118.Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10(5):432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- 119.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacological Reviews. 1999;51(1):7–61. [PubMed] [Google Scholar]
- 120.Paoletti P, Neyton J. NMDA receptor subunits: function and pharmacology. Current Opinion in Pharmacology. 2007;7(1):39–47. doi: 10.1016/j.coph.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 121.Hollmann M, Boulter J, Maron C, et al. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron. 1993;10(5):943–954. doi: 10.1016/0896-6273(93)90209-a. [DOI] [PubMed] [Google Scholar]
- 122.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent Zinc inhibition of NMDA receptors by the NR1 subunit. Journal of Neuroscience. 1998;18(16):6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gavazzo P, Guida P, Zanardi I, Marchetti C. Molecular determinants of multiple effects of nickel on NMDA receptor channels. Neurotoxicity Research. 2009;15(1):38–48. doi: 10.1007/s12640-009-9003-7. [DOI] [PubMed] [Google Scholar]
- 124.Schoepfer R, Monyer H, Sommr B, et al. Molecular biology of glutamate receptors. Progress in Neurobiology. 1994;42(2):353–357. doi: 10.1016/0301-0082(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 125.Mayer ML, Westbrook GL, Buthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 126.Nowak L, Bregestovski P, Ascher P. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307(5950):462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 127.Ault B, Evans RH, Francis AA, Oakes DJ, Watkins JC. Selective depression of excitatory amino acid induced depolarizations by magnesium ions in isolated spinal cord preparations. Journal of Physiology. 1980;307:413–428. doi: 10.1113/jphysiol.1980.sp013443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wollmuth LP, Kuner T, Sakmann B. Adjacent asparagines in the NR2-subunit of the NMDA receptor channel control the voltage-dependent block by extracellular Mg2+ Journal of Physiology. 1998;506(1):13–32. doi: 10.1111/j.1469-7793.1998.013bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang LY, MacDonald JF. Modulation by magnesium of the affinity of NMDA receptors for glycine in murine hippocampal neurones. Journal of Physiology. 1995;486(1):83–95. doi: 10.1113/jphysiol.1995.sp020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Paoletti P, Neyton J, Ascher P. Glycine-independent and subunit-specific potentiation of NMDA responses by extracellular Mg2+ Neuron. 1995;15(5):1109–1120. doi: 10.1016/0896-6273(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 131.Kumamoto E. Neuromodulation by Mg2+ and polyamines of excitatory amino acid currents in rodent neurones in culture. Magnesium Research. 1996;9(4):317–327. [PubMed] [Google Scholar]
- 132.Benveniste M, Mayer ML. Multiple effects of spermine on N-methyl-D-aspartic acid receptor responses of rat cultured hippocampal neurones. Journal of Physiology. 1993;464:131–163. doi: 10.1113/jphysiol.1993.sp019627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mony L, Kew JNC, Gunthorpe MJ, Paoletti P. Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. British Journal of Pharmacology. 2009;157(8):1301–1317. doi: 10.1111/j.1476-5381.2009.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158(1):126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- 135.Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB. Importance of zinc in the central nervous system: the zinc-containing neuron. Journal of Nutrition. 2000;130(5):1471S–1483S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 136.Mayer ML, Vyklicky L, Westbrook GL. Modulation of excitatory amino acid receptors by group IIB metal cations in cultured mouse hippocampal neurones. Journal of Physiology. 1989;415:329–350. doi: 10.1113/jphysiol.1989.sp017724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328(6131):640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 138.Christine CW, Choi DW. Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. Journal of Neuroscience. 1990;10(1):108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Legendre P, Westbrook GL. The inhibition of single N-methyl-D-aspartate-activated channels by zinc ions on cultured rat neurones. Journal of Physiology. 1990;429:429–449. doi: 10.1113/jphysiol.1990.sp018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paoletti P, Perin-Dureau F, Fayyazuddin A, Le Goff A, Callebaut I, Neyton J. Molecular organization of a zinc binding N-terminal modulatory domain in a NMDA receptor subunit. Neuron. 2000;28(3):911–925. doi: 10.1016/s0896-6273(00)00163-x. [DOI] [PubMed] [Google Scholar]
- 141.Herin GA, Aizenman E. Amino terminal domain regulation of NMDA receptor function. European Journal of Pharmacology. 2004;500(1–3):101–111. doi: 10.1016/j.ejphar.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 142.Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1-NR2A receptors. Journal of Neuroscience. 1997;17(15):5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gielen M, Le Goff A, Stroebel D, Johnson JW, Neyton J, Paoletti P. Structural rearrangements of NR1/NR2A NMDA receptors during allosteric inhibition. Neuron. 2008;57(1):80–93. doi: 10.1016/j.neuron.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Erreger K, Traynelis SF. Zinc inhibition of rat NR1/NR2A N-methyl-d-aspartate receptors. Journal of Physiology. 2008;586(3):763–778. doi: 10.1113/jphysiol.2007.143941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rachline J, Perin-Dureau F, Le Goff A, Neyton J, Paoletti P. The micromolar zinc-binding domain on the NMDA receptor subunit NR2B. Journal of Neuroscience. 2005;25(2):308–317. doi: 10.1523/JNEUROSCI.3967-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, Neyton J. Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron. 2000;25(3):683–694. doi: 10.1016/s0896-6273(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 147.Busselberg D, Michael D, Platt B. Pb2+ reduces voltage- and N-methyl-D-aspartate (NMDA)-activated calcium channel currents. Cellular and Molecular Neurobiology. 1994;14(6):711–722. doi: 10.1007/BF02088679. [DOI] [PubMed] [Google Scholar]
- 148.Alkondon M, Costa ACS, Radhakrishnan V, Aronstam RS, Albuquerque EX. Selective blockade of NMDA-activated channel currents may be implicated in learning deficits caused by lead. FEBS Letters. 1990;261(1):124–130. doi: 10.1016/0014-5793(90)80652-y. [DOI] [PubMed] [Google Scholar]
- 149.Ishihara K, Alkondon M, Montes JG, Albuquerque EX. Ontogenically related properties of N-methyl-D-aspartate receptors in rat hippocampal neurons and the age-specific sensitivity of developing neurons to lead. Journal of Pharmacology and Experimental Therapeutics. 1995;273(3):1459–1470. [PubMed] [Google Scholar]
- 150.Ujihara H, Albuquerque EX. Developmental change of the inhibition by lead of NMDA-activated currents in cultured hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics. 1992;263(2):868–875. [PubMed] [Google Scholar]
- 151.Marchetti C. Molecular targets of lead in brain neurotoxicity. Neurotoxicity Research. 2003;5(3):221–235. doi: 10.1007/BF03033142. [DOI] [PubMed] [Google Scholar]
- 152.Guilarte TR, Miceli RC. Age-dependent effects of lead on [3H]MK-801 binding to the NMDA receptor-gated ionophore: In vitro and in vivo studies. Neuroscience Letters. 1992;148(1-2):27–30. doi: 10.1016/0304-3940(92)90796-a. [DOI] [PubMed] [Google Scholar]
- 153.Guilarte TR. Pb2+ inhibits NMDA receptor function at high and low affinity sites: developmental and regional brain expression. NeuroToxicology. 1997;18(1):43–52. [PubMed] [Google Scholar]
- 154.Guilarte TR, Miceli RC, Jett DA. Biochemical evidence of an interaction of lead at the zinc allosteric sites of the NMDA receptor complex: effects of neuronal development. NeuroToxicology. 1995;16(1):63–71. [PubMed] [Google Scholar]
- 155.Alfano DP, Petit TL. Neonatal lead exposure alters the dendritic development of hippocampal dentate granule cells. Experimental Neurology. 1982;75(2):275–288. doi: 10.1016/0014-4886(82)90160-1. [DOI] [PubMed] [Google Scholar]
- 156.Petit TL, Alfano DP, LeBoutillier JC. Early lead exposure and the hippocampus: a review and recent advances. NeuroToxicology. 1983;4(1):79–94. [PubMed] [Google Scholar]
- 157.Omelchenko IA, Nelson CS, Marino JL, Allen CN. The sensitivity of N-methyl-D-aspartate receptors to lead inhibition is dependent on the receptor subunit composition. Journal of Pharmacology and Experimental Therapeutics. 1996;278(1):15–20. [PubMed] [Google Scholar]
- 158.Yamada Y, Ujihara H, Sada H, Ban T. Pb2+ reduces the current from NMDA receptors expressed in Xenopus oocytes. FEBS Letters. 1995;377(3):390–392. doi: 10.1016/0014-5793(95)01402-0. [DOI] [PubMed] [Google Scholar]
- 159.Omelchenko IA, Nelson CS, Allen CN. Lead inhibition of N-methyl-D-aspartate receptors containing NR2A, NR2C and NR2D subunits. Journal of Pharmacology and Experimental Therapeutics. 1997;282(3):1458–1464. [PubMed] [Google Scholar]
- 160.Lasley SM, Gilbert ME. Lead inhibits the rat N-methyl-D-aspartate receptor channel by binding to a site distinct from the zinc allosteric site. Toxicology and Applied Pharmacology. 1999;159(3):224–233. doi: 10.1006/taap.1999.8743. [DOI] [PubMed] [Google Scholar]
- 161.Guilarte TR, Miceli RC, Jett DA. Neurochemical aspects of hippocampal and cortical Pb2+ neurotoxicity. NeuroToxicology. 1994;15(3):459–466. [PubMed] [Google Scholar]
- 162.Schulte S, Muller WE, Friedberg KD. In vitro and in vivo effects of lead on specific 3H-MK-801 binding to NMDA-receptors in the brain of mice. NeuroToxicology. 1995;16(2):309–318. [PubMed] [Google Scholar]
- 163.Marchetti C, Gavazzo P. Subunit-dependent effects of nickel on NMDA receptor channels. Molecular Brain Research. 2003;117(2):139–144. doi: 10.1016/s0169-328x(03)00293-6. [DOI] [PubMed] [Google Scholar]
- 164.Low CM, Zheng F, Lyuboslavsky P, Traynelis SF. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-D-aspartate NR1/NR2A receptors. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):11062–11067. doi: 10.1073/pnas.180307497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hung YH, Bush AI, Cherny RA. Copper in the brain and Alzheimer’s disease. Journal of Biological Inorganic Chemistry. 2010;15(1):61–76. doi: 10.1007/s00775-009-0600-y. [DOI] [PubMed] [Google Scholar]
- 166.Bush AI, Tanzi RE. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics. 2008;5(3):421–432. doi: 10.1016/j.nurt.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Barnea A, Hartter DE, Cho G, Bhasker KR, Katz BM, Edwards MD. Further characterization of the process of in vitro uptake of radiolabeled copper by the rat brain. Journal of Inorganic Biochemistry. 1990;40(2):103–110. doi: 10.1016/0162-0134(90)80043-w. [DOI] [PubMed] [Google Scholar]
- 168.Kardos J, Kovacs I, Hajos F, Kalman M, Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neuroscience Letters. 1989;103(2):139–144. doi: 10.1016/0304-3940(89)90565-x. [DOI] [PubMed] [Google Scholar]
- 169.Hopt A, Korte S, Fink H, et al. Methods for studying synaptosomal copper release. Journal of Neuroscience Methods. 2003;128(1-2):159–172. doi: 10.1016/s0165-0270(03)00173-0. [DOI] [PubMed] [Google Scholar]
- 170.Hartter DE, Barnea A. Evidence for release of copper in the brain: depolarization-induced release of newly taken-up 67copper. Synapse. 1988;2(4):412–415. doi: 10.1002/syn.890020408. [DOI] [PubMed] [Google Scholar]
- 171.Wong PY, Fritze K. Determination by neutron activation of copper, manganese, and zinc in the pineal body and other areas of brain tissue. Journal of Neurochemistry. 1969;16(8):1231–1234. doi: 10.1111/j.1471-4159.1969.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 172.Donaldson J, Pierre TS, Minnich JL, Barbeau A. Determination of Na+, K+, Mg2+, Cu2+, Zn2+, and Mn2+ in rat brain regions. Canadian Journal of Biochemistry. 1973;51(1):87–92. doi: 10.1139/o73-010. [DOI] [PubMed] [Google Scholar]
- 173.Weiser T, Wienrich M. The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Research. 1996;742(1-2):211–218. doi: 10.1016/s0006-8993(96)01009-8. [DOI] [PubMed] [Google Scholar]
- 174.Vlachová V. Copper modulation of NMDA responses in mouse and rat cultured hippocampal neurons. European Journal of Neuroscience. 1996;8(11):2257–2264. doi: 10.1111/j.1460-9568.1996.tb01189.x. [DOI] [PubMed] [Google Scholar]
- 175.Larsen NA, Pakkenberg H, Damsgaard E, Heydorn K. Topographical distribution of arsenic, manganese, and selenium in the normal human brain. Journal of Neurosurgical Sciences. 1979;42(3):407–416. doi: 10.1016/0022-510x(79)90173-4. [DOI] [PubMed] [Google Scholar]
- 176.Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. Journal of Physiology. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Guilarte TR, Chen MK. Manganese inhibits NMDA receptor channel function: implications to psychiatric and cognitive effects. NeuroToxicology. 2007;28(6):1147–1152. doi: 10.1016/j.neuro.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hashemzadeh-Gargari H, Guilarte TR. Divalent cations modulate N-methyl-D-aspartate receptor function at the glycine site. Journal of Pharmacology and Experimental Therapeutics. 1999;290(3):1356–1362. [PubMed] [Google Scholar]
- 179.Gu Y, Huang LYM. Modulation of glycine affinity for NMDA receptors by extracellular Ca2+ in trigeminal neurons. Journal of Neuroscience. 1994;14(7):4561–4570. doi: 10.1523/JNEUROSCI.14-07-04561.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Marchioro M, Swanson KL, Aracava Y, Albuquerque EX. Glycine- and calcium-dependent effects of lead on N-methyl-D-aspartate receptor function in rat hippocampal neurons. Journal of Pharmacology and Experimental Therapeutics. 1996;279(1):143–153. [PubMed] [Google Scholar]
- 181.Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 182.Platt SR. The role of glutamate in central nervous system health and disease—a review. Veterinary Journal. 2007;173(2):278–286. doi: 10.1016/j.tvjl.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 183.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glial cells. Neuron. 1991;7(3):509–518. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 184.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurones. Journal of Physiology. 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. Journal of Nutrition. 2000;130(4):1007S–1015S. doi: 10.1093/jn/130.4.1007S. [DOI] [PubMed] [Google Scholar]
- 186.Buldakova SL, Bolshakov KV, Tikhonov DB, Magazanik LG. Ca2+-dependent desensitization of AMPA receptors. NeuroReport. 2000;11(13):2937–2941. doi: 10.1097/00001756-200009110-00021. [DOI] [PubMed] [Google Scholar]
- 187.Rassendren FA, Lory P, Pin JP, Nargeot J. Zinc has opposite effects on NMDA and non-NMDA receptors expressed in Xenopus oocytes. Neuron. 1990;4(5):733–740. doi: 10.1016/0896-6273(90)90199-p. [DOI] [PubMed] [Google Scholar]
- 188.Dreixler JC, Leonard JP. Subunit-specific enhancement of glutamate receptor responses by zinc. Molecular Brain Research. 1994;22(1–4):144–150. doi: 10.1016/0169-328x(94)90042-6. [DOI] [PubMed] [Google Scholar]
- 189.Dreixler JC, Leonard JP. Effects of external calcium on zinc modulation of AMPA receptors. Brain Research. 1997;752(1-2):170–174. doi: 10.1016/s0006-8993(96)01439-4. [DOI] [PubMed] [Google Scholar]
- 190.Dorofeeva NA, Tikhonov DB, Barygin OI, Tikhonova TB, Salnikov YI, Magazanik L. Action of extracellular divalent cations on native α-amino-3-hydroxy- 5-methylisoxazole-4-propionate (AMPA) receptors. Journal of Neurochemistry. 2005;95(6):1704–1712. doi: 10.1111/j.1471-4159.2005.03533.x. [DOI] [PubMed] [Google Scholar]
- 191.Zhu WJ, Wang JF, Corsi L, Vicini S. Lanthanum-mediated modification of GABA(A) receptor deactivation, desensitization and inhibitory synaptic currents in rat cerebellar neurons. Journal of Physiology. 1998;511(3):647–661. doi: 10.1111/j.1469-7793.1998.647bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Olsen RW, Tobin AJ. Molecular biology of GABA(A) receptors. FASEB Journal. 1990;4(5):1469–1480. doi: 10.1096/fasebj.4.5.2155149. [DOI] [PubMed] [Google Scholar]
- 193.Schwartz RD. The GABA(A) receptor-gated ion channel: biochemical and pharmacological studies of structure and function. Biochemical Pharmacology. 1988;37(18):3369–3375. doi: 10.1016/0006-2952(88)90684-3. [DOI] [PubMed] [Google Scholar]
- 194.Sieghart W. Structure and pharmacology of γ-aminobutyric acid(A) receptor subtypes. Pharmacological Reviews. 1995;47(2):181–234. [PubMed] [Google Scholar]
- 195.Ma JY, Narahashi T. Differential modulation of GABA(A) receptor-channel complex by polyvalent cations in rat dorsal root ganglion neurons. Brain Research. 1993;607(1-2):222–232. doi: 10.1016/0006-8993(93)91510-y. [DOI] [PubMed] [Google Scholar]
- 196.Barberis A, Cherubini E, Mozrzymas JW. Zinc inhibits miniature GABAergic currents by allosteric modulation of GABA(A) receptor gating. Journal of Neuroscience. 2000;20(23):8618–8627. doi: 10.1523/JNEUROSCI.20-23-08618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Buhl EH, Otis TS, Mody I. Zinc-induced collapse of augmented inhibition by GABA in a temporal lobe epilepsy model. Science. 1996;271(5247):369–373. doi: 10.1126/science.271.5247.369. [DOI] [PubMed] [Google Scholar]
- 198.Legendre P, Westbrook GL. Noncompetitive inhibition of γ-aminobutyric acid(A) channels by Zn. Molecular Pharmacology. 1991;39(3):267–274. [PubMed] [Google Scholar]
- 199.Smart TG. A novel modulatory binding site for zinc on the GABA(A) receptor complex in cultured rat neurones. Journal of Physiology. 1992;447:587–625. doi: 10.1113/jphysiol.1992.sp019020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kilic G, Moran O, Cherubini E. Currents activated by GABA and their modulation by Zn2+ in cerebellar granule cells in culture. European Journal of Neuroscience. 1993;5(1):65–72. doi: 10.1111/j.1460-9568.1993.tb00206.x. [DOI] [PubMed] [Google Scholar]
- 201.Sieghart W. GABA(A) receptors: ligand-gated Cl- ion channels modulated by multiple drug-binding sites. Trends in Pharmacological Sciences. 1992;13(12):446–450. doi: 10.1016/0165-6147(92)90142-s. [DOI] [PubMed] [Google Scholar]
- 202.Draguhn A, Verdorn TA, Ewert M, Seeburg PH, Sakmann B. Functional and molecular distinction between recombinant rat GABA(A) receptor subtypes by Zn2+ Neuron. 1990;5(6):781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- 203.Saxena NC, Macdonald RL. Assembly of GABA(A) receptor subunits: role of the δ subunit. Journal of Neuroscience. 1994;14(11):7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fisher JL, Macdonald RL. The role of an α subtype M2-M3 his in regulating inhibition of GABA(A) receptor current by zinc and other divalent cations. Journal of Neuroscience. 1998;18(8):2944–2953. doi: 10.1523/JNEUROSCI.18-08-02944.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Saxena NC, Macdonald RL. Properties of putative cerebellar γ-aminobutyric acidA receptor isoforms. Molecular Pharmacology. 1996;49(3):567–579. [PubMed] [Google Scholar]
- 206.Knoflach F, Benke D, Wang Y, et al. Pharmacological modulation of the diazepam-insensitive recombinant γ- aminobutyric acid(A) receptors α4β2γ2 and α6β2γ2. Molecular Pharmacology. 1996;50(5):1253–1261. [PubMed] [Google Scholar]
- 207.White G, Gurley DA. α subunits influence Zn block of γ2 containing GABA(A) receptor currents. NeuroReport. 1995;6(3):461–464. doi: 10.1097/00001756-199502000-00014. [DOI] [PubMed] [Google Scholar]
- 208.Moss SJ, Smart TG. Constructing inhibitory synapses. Nature Reviews Neuroscience. 2001;2(4):240–250. doi: 10.1038/35067500. [DOI] [PubMed] [Google Scholar]
- 209.Sharonova IN, Vorobjev VS, Haas HL. High-affinity copper block of GABAA receptor-mediated currents in acutely isolated cerebellar Purkinje cells of the rat. European Journal of Neuroscience. 1998;10(2):522–528. doi: 10.1046/j.1460-9568.1998.00057.x. [DOI] [PubMed] [Google Scholar]
- 210.Trombley PQ, Shepherd GM. Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. Journal of Neurophysiology. 1996;76(4):2536–2546. doi: 10.1152/jn.1996.76.4.2536. [DOI] [PubMed] [Google Scholar]
- 211.Barilà B, Cupello A, Robello M. Modulation by lanthanum ions of γ-aminobutyric acidA receptors of rat cerebellum granule cells in culture: clues on their subunit composition. Neuroscience Letters. 2001;298(1):13–16. doi: 10.1016/s0304-3940(00)01727-4. [DOI] [PubMed] [Google Scholar]
- 212.Boldyreva AA. Lanthanum potentiates GABA-activated currents in rat pyramidal neurons of CA1 hippocampal field. Bulletin of Experimental Biology and Medicine. 2005;140(4):403–405. doi: 10.1007/s10517-005-0503-z. [DOI] [PubMed] [Google Scholar]
- 213.Saxena NC, Neelands TR, Macdonald RL. Contrasting actions of lanthanum on different recombinant γ- aminobutyric acid receptor isoforms expressed in l929 fibroblasts. Molecular Pharmacology. 1997;51(2):328–335. doi: 10.1124/mol.51.2.328. [DOI] [PubMed] [Google Scholar]
- 214.Arakawa O, Nakahiro M, Narahashi T. Mercury modulation of GABA-activated chloride channels and non-specific cation channels in rat dorsal root ganglion neurons. Brain Research. 1991;551(1-2):58–63. doi: 10.1016/0006-8993(91)90913-g. [DOI] [PubMed] [Google Scholar]
- 215.Krüger K, Diepgrond V, Ahnefeld M, et al. Blockade of glutamatergic and GABAergic receptor channels by trimethyltin chloride. British Journal of Pharmacology. 2005;144(2):283–292. doi: 10.1038/sj.bjp.0706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wasserman GA, Liu X, Parvez F, et al. Water arsenic exposure and children’s intellectual function in Araihazar, Bangladesh. Environmental Health Perspectives. 2004;112(13):1329–1333. doi: 10.1289/ehp.6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Calderón J, Navarro ME, Jimenez-Capdeville ME, et al. Exposure to arsenic and lead and neuropsychological development in Mexican children. Environmental Research. 2001;85(2):69–76. doi: 10.1006/enrs.2000.4106. [DOI] [PubMed] [Google Scholar]
- 218.Gilbert SG, Weiss B. A rationale for lowering the blood lead action level from 10 to 2 μg/dL. NeuroToxicology. 2006;27(5):693–701. doi: 10.1016/j.neuro.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Téllez-Rojo MM, Bellinger DC, Arroyo-Quiroz C, et al. Longitudinal associations between blood lead concentrations lower than 10 μg/dL and neurobehavioral development in environmentally exposed children in Mexico City. Pediatrics. 2006;118(2):e323–e330. doi: 10.1542/peds.2005-3123. [DOI] [PubMed] [Google Scholar]
- 220.Carpenter DO. Effects of metals on the nervous system of humans and animals. International Journal of Occupational Medicine and Environmental Health. 2001;14(3):209–218. [PubMed] [Google Scholar]
- 221.Platt B, Carpenter DO, Busselberg D, Reymann KG, Riedel G. Aluminum impairs hippocampal long-term potentiation in rats in vitro and in vivo. Experimental Neurology. 1995;134(1):73–86. doi: 10.1006/exnr.1995.1038. [DOI] [PubMed] [Google Scholar]
- 222.Chen J, Wang M, Ruan D, She J. Early chronic aluminium exposure impairs long-term potentiation and depression to the rat dentate gyrus in vivo. Neuroscience. 2002;112(4):879–887. doi: 10.1016/s0306-4522(02)00138-0. [DOI] [PubMed] [Google Scholar]
- 223.Ahmed SS, Santosh W. Metallomic profiling and linkage map analysis of early Parkinson’s disease: a new insight to aluminum marker for the possible diagnosis. PloS One. 2010;5(6) doi: 10.1371/journal.pone.0011252. Article ID e11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Raymond CR. LTP forms 1, 2 and 3: different mechanisms for the ’long’ in long-term potentiation. Trends in Neurosciences. 2007;30(4):167–175. doi: 10.1016/j.tins.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 225.Reymann KG, Frey JU. The late maintenance of hippocampal LTP: requirements, phases, ’synaptic tagging’, ‘late-associativity’ and implications. Neuropharmacology. 2007;52(1):24–40. doi: 10.1016/j.neuropharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 226.Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nature Reviews Neuroscience. 2005;6(4):267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- 227.Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. Journal of Pharmacological Sciences. 2006;100(5):433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- 228.Izumi Y, Tokuda K, Zorumski CF. Long-term potentiation inhibition by low-level N-methyl-D-aspartate receptor activation involves calcineurin, nitric oxide, and p38 mitogen-activated protein kinase. Hippocampus. 2008;18(3):258–265. doi: 10.1002/hipo.20383. [DOI] [PubMed] [Google Scholar]
- 229.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature Reviews Neuroscience. 2004;5(3):173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 230.Prichard L, Deloulme JC, Storm DR. Interactions between neurogranin and calmodulin in vivo. Journal of Biological Chemistry. 1999;274(12):7689–7694. doi: 10.1074/jbc.274.12.7689. [DOI] [PubMed] [Google Scholar]
- 231.Baudier J, Deloulme JC, Van Dorsselaer A, Black D, Matthes HWD. Purification and characterization of a brain-specific protein kinase C substrate, neurogranin (p17). Identification of a consensus amino acid sequence between neurogranin and neuromodulin (GAP43) that corresponds to the protein kinase C phosphorylation site and the calmodulin-binding domain. Journal of Biological Chemistry. 1991;266(1):229–237. [PubMed] [Google Scholar]
- 232.Habermann E, Crowell K, Janicki P. Lead and other metals can substitute for Ca2+ in calmodulin. Archives of Toxicology. 1983;54(1):61–70. doi: 10.1007/BF00277816. [DOI] [PubMed] [Google Scholar]
- 233.Ramakers GMJ, Pasinelli P, Hens JJH, Gispen WH, De Graan PNE. Protein kinase C in synaptic plasticity: changes in the in situ phosphorylation state of identified pre- and postsynaptic substrates. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 1997;21(3):455–486. doi: 10.1016/s0278-5846(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 234.Cremin JD, Jr., Smith DR. In vitro vs in vivo Pb effects on brain protein kinase C activity. Environmental Research. 2002;90(3):191–199. doi: 10.1016/s0013-9351(02)00007-5. [DOI] [PubMed] [Google Scholar]
- 235.Lee SJR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458(7236):299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin- dependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. Journal of Biological Chemistry. 1996;271(49):31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- 237.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285(5435):1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 238.Anderson KA, Kane CD. Ca2+/calmodulin-dependent protein kinase IV and calcium signaling. BioMetals. 1998;11(4):331–343. doi: 10.1023/a:1009276932076. [DOI] [PubMed] [Google Scholar]
- 239.Mayer B, John M, Heinzel B, et al. Brain nitric oxide synthase is a biopterin- and flavin-containing multi-functional oxido-reductase. FEBS Letters. 1991;288(1-2):187–191. doi: 10.1016/0014-5793(91)81031-3. [DOI] [PubMed] [Google Scholar]
- 240.Lohse MJ, Forstermann U, Schmidt HHHW. Pharmacology of NO: cGMP signal transduction. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1998;358(1):111–112. doi: 10.1007/pl00005230. [DOI] [PubMed] [Google Scholar]
- 241.Prast H, Philippu A. Nitric oxide as modulator of neuronal function. Progress in Neurobiology. 2001;64(1):51–68. doi: 10.1016/s0301-0082(00)00044-7. [DOI] [PubMed] [Google Scholar]
- 242.Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. Journal of Neuroscience. 1999;19(23):10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods in Molecular Biology. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 244.Toscano CD, McGlothan JL, Guilarte TR. Lead exposure alters cyclic-AMP response element binding protein phosphorylation and binding activity in the developing rat brain. Developmental Brain Research. 2003;145(2):219–228. doi: 10.1016/j.devbrainres.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 245.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35(4):605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 246.Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365(6449):855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 247.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16(1):89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 248.Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends in Neurosciences. 2003;26(2):81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- 249.Montminy MR, Bilezikjian LM. Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature. 1987;328(6126):175–178. doi: 10.1038/328175a0. [DOI] [PubMed] [Google Scholar]
- 250.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annual Review of Neuroscience. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 251.Bolshakov VY, Carboni L, Cobb MH, Siegelbaum SA, Belardetti F. Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nature Neuroscience. 2000;3(11):1107–1112. doi: 10.1038/80624. [DOI] [PubMed] [Google Scholar]
- 252.Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annual Review of Pharmacology and Toxicology. 2004;44:87–110. doi: 10.1146/annurev.pharmtox.44.101802.121424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Current Opinion in Neurobiology. 2001;11(3):327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 254.Xu YZ, Ruan DY, Wu Y, et al. Nitric oxide affects LTP in area CA1 and CA3 of hippocampus in low-level lead-exposed rat. Neurotoxicology and Teratology. 1998;20(1):69–73. doi: 10.1016/s0892-0362(97)00072-x. [DOI] [PubMed] [Google Scholar]
- 255.Altmann L, Weinsberg F, Sveinsson K, Lilienthal H, Wiegand H, Winneke G. Impairment of long-term potentiation and learning following chronic lead exposure. Toxicology Letters. 1993;66(1):105–112. doi: 10.1016/0378-4274(93)90085-c. [DOI] [PubMed] [Google Scholar]
- 256.Zhong J, Carrozza DP, Williams K, Pritchett DB, Molinoff PB. Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. Journal of Neurochemistry. 1995;64(2):531–539. doi: 10.1046/j.1471-4159.1995.64020531.x. [DOI] [PubMed] [Google Scholar]
- 257.Faro LRF, Durán R, Do Nascimento JLM, Alfonso M, Picanço-Diniz CW. Effects of methyl mercury on the in vivo release of dopamine and its acidic metabolites DOPAC and HVA from striatum of rats. Ecotoxicology and Environmental Safety. 1997;38(2):95–98. doi: 10.1006/eesa.1997.1567. [DOI] [PubMed] [Google Scholar]
- 258.Stýblo M, Drobná Z, Jaspers I, Lin S, Thomas DJ. The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environmental Health Perspectives. 2002;110(supplement 5):767–771. doi: 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Thayer JS. Biological methylation of less-studied elements. Applied Organometallic Chemistry. 2002;16(12):677–691. [Google Scholar]
- 260.Platt B, Busselberg D. Combined actions of Pb2+, Zn2+, and Al3+ on voltage-activated calcium channel currents. Cellular and Molecular Neurobiology. 1994;14(6):831–840. doi: 10.1007/BF02088688. [DOI] [PubMed] [Google Scholar]
- 261.Vigeh M, Yokoyama K, Seyedaghamiri Z, et al. Blood lead at currently acceptable levels may cause preterm labour. Occupational and Environmental Medicine. 2011;68(3):231–234. doi: 10.1136/oem.2009.050419. [DOI] [PubMed] [Google Scholar]