

Abstract
Introduction
The World Health Organisation defines PEComa's as “a mesenchymal tumour composed of histologically and immunohistochemically distinctive perivascular cells”.1 These ubiquitous tumours show distinctive perivascular epithelioid cell differentiation and arise most commonly at visceral and abdominopelvic sites.
Presentation of case
We present a case of a forty-two year old man presenting to accident and emergency department with upper gastro-intestinal bleeding. He had a palpable epigatric mass on examination. He underwent a CT Scan Abdomen which displayed a tumour arising from the gastric wall. Upper GI endoscopy and biopsy was carried out and biopsied were taken for histological analysis. A primary gastric PEComa was diagnosed and the patient underwent distal polya gastrectomy and gastrojejunostomy. This is believed to be the first reported case of a Primary malignant gastric PEComa.
Discussion
Perivascular epithelioid carcinomas were first described in 1943 as an abnormal myoblast in a case of renal angiomyolipoma. PEComas display a strong female predominance with a typical benign course. There are approximately 100 reported cases of PEComa to date, with 55 of which were malignant. PEComa's may be subdivided into benign, uncertain malignant potential and malignant. Their natural history can be very aggressive leading to multiple metastases and death as expected with a high-grade sarcoma.
Conclusion
This case depicts the aggressive nature of malignant gastric PEComa's. The majority of PEComa's are benign in nature and have a better prognosis. We display here the challenges in ascertaining a definitive diagnosis and management of such patients due to limited clinical studies.
Keywords: PEComa, Gastric, Diagnostic, Desmin, Melan-A
1. Introduction
In 1991 Bonetti et al. suggested the term perivascular epithelioid cell (PEC) to describe a characteristic cell type found in three unusual mesenchymal lesions, lymphangiomyomatosis, clear cell sugar tumour of the lung and angiomyolipoma of the liver and kidney after noting the consistent morphological, immunophenotypic, genetic and ultrastructural features.2 In 1996 Zamboni et al. subsequently employed the term PEComa to amalgamate this family of lesions conveying this perivascular epithelioid cell differentiation after noting the overlapping features of a benign clear cell sugar tumour of the lung and a PEComa of the pancreas, indicating the possibility that similar tumours could possibly arise in many if not all locations.3 Thus the term PEComa was introduced to include all similar lesions outside the lung. To date, there have been fifty-five reported malignant cases, with only three presentations noted within the gastro-intestinal tract, none of whom were gastric in origin.
Immunohistochemically, nearly all PEComas show reactivity for melanocytic (HMB-45 and/or Melan-A) and smooth muscle (actin and/or desmin) markers.4 Also noted is a consistent theme within PEComa's during immunohistochemistry is the typical perivascular location. A genetic predispostition to renal angiomyolipoma has been documented in individuals with an alteration to the tuberous sclerosis complex located in the TSC1 and TSC2 genes on chromosomes 9q and 16p. There is no known normal physiological counterpart to the perivascular epithelioid cell however a number of hypotheses have been proposed including the derivation from undifferentiated neural crest cells, a possible molecular alteration from a myoblastic smooth muscle origin or evolution from a pericytic origin.5
2. Case
We present the case of a forty-two year old male who presented with epigastric pain, melaena and weight loss. He had a palpable epigastric mass which was fixed, solid and irregular on examination. He underwent an abdominal CT scan which disclosed a 10 cm × 7 cm mass obstructing the pylorus of the stomach, associated with metastatic liver disease and retroperitoneal lymphadenopathy (Fig. 1). He proceeded to upper GI endoscopy which displayed a large fungating mass occupying the distal 1/3 of his stomach (Fig. 2).
Fig. 1.
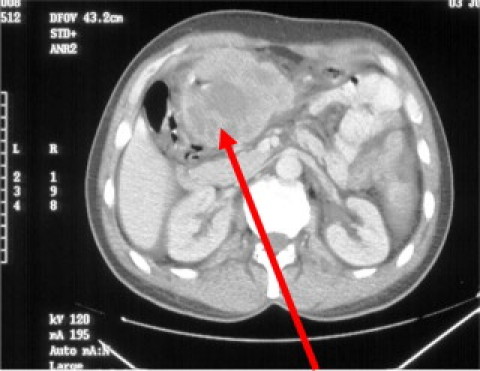
CT abdomen displaying large mass obstructing the gastric pylorus.
Fig. 2.
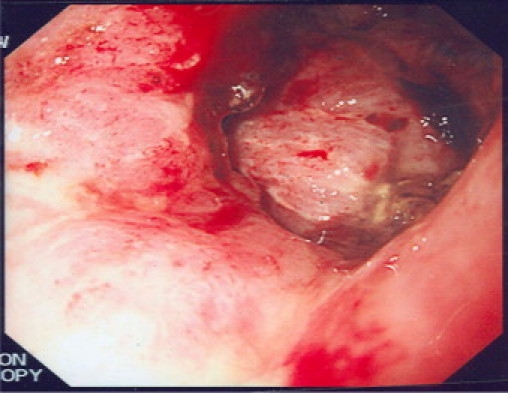
Large fungating mass in the pylorus on Upper GI endoscopy.
Histology demonstrated a large cell malignant tumour which was negative for all epithelial markers excluding carcinoma and negative c-kit excluding GIST. The melanoma marker Melan-A was positive. This prompted consideration of metastatic malignant melanoma. Other Melan-A positive tumours were considered – these are few in number; primarily Adreno-cortical carcinoma and gonadal Serolti\Leydig cell tumours. However, both these tumours consistently express the immuno marker Inhibin, which was negative in this case thus virtually excluding these two tumours from the differential.
Finally as the tumour expressed the muscle marker desmin, in addition to Melan-A, indicating myo-melanocytic differentiation the possibility of a PEComa was considered (Fig. 3, Fig. 4).
Fig. 3.
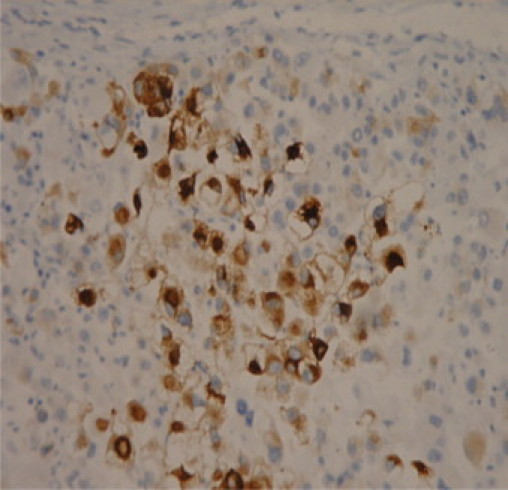
Histological slide – melanoma marker – Melan A.
Fig. 4.
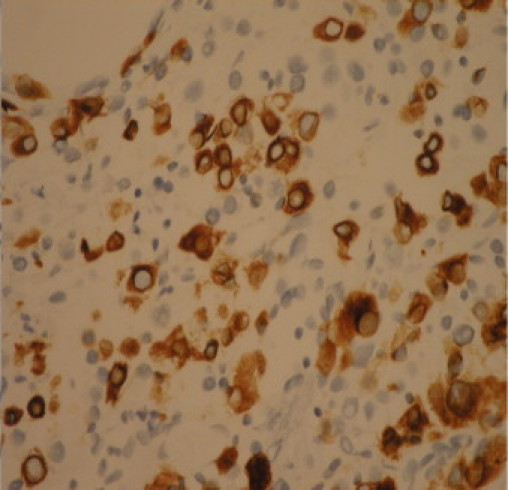
Muscle marker – desmin positive.
The patient underwent a distal polya gastrectomy and gastrojejunostomy due to recurrent symptomatic upper gastrointestinal haemorrhage and obstruction (Fig. 5). Following gastrectomy, a definitive histological diagnosis was made a malignant PEComa of gastric origin was confirmed, due to the presence of large epithelioid and polygonal cells associated with positive immunohistochemical stains for desmin, Melan-A and EMA (Fig. 6).
Fig. 5.
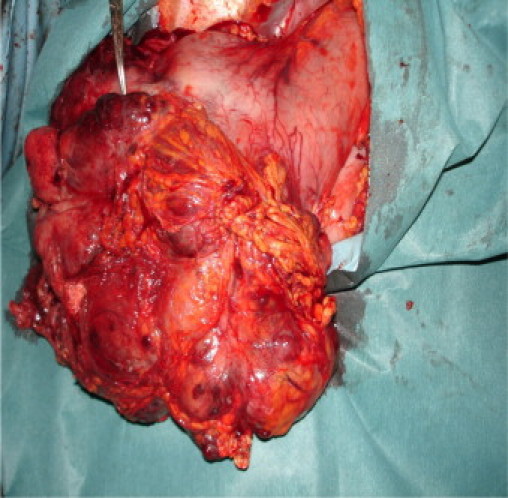
Gross intra-operative specimen – tumour involving distal greater curvature of the stomach.
Fig. 6.
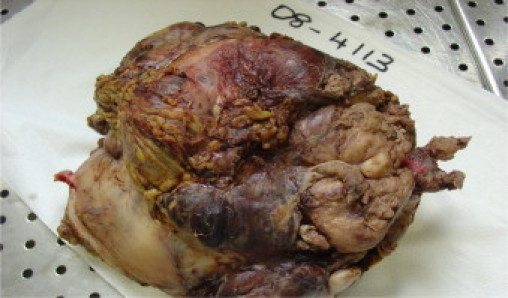
Final gross histological specimen post resection.
3. Discussion
PEComa or perivascular epithelioid cell tumours are a family of related mesenchymal neoplasms composed of histologically and immunohistochemically distinctive perivascular epithelioid cells that include angiomyolipoma, lymphangiomyomatosis, clear cell sugar tumours of the lung and a group of rare visceral, retroperitoneal and abdominopelvic tumours.1 They were first described in 1943 as an abnormal myoblast in a case of renal angiomyolipoma. PEComa's display a strong female predominance with a typical benign course. Recurrent chromosomal alterations involving the tuberous sclerosis complex have been demonstrated in the perivascular epithelioid cell. There are approximately 100 reported cases of PEComa, with 55 of whom were malignant.
PEComa's may be subdivided into benign, uncertain malignant potential and malignant. Guidelines regarding the classification criteria for malignant cases of PEComa's have not yet been internationally agreed upon due to the rarity of cases, however it is agreed that they pursue an consistently aggressive clinical course. However, after review and follow up of PEComa's arising at visceral and somative sites Folpe et al. hypothesised that the criteria for malignancy should include tumour size greater than 70 mm, a mitotic index of greater than 1 per 50 high power field, infiltrative growth patterns and marked hypercellularity, pleomorphism, necrosis and nuclear atypia.6
Their natural history can be very aggressive leading to multiple metastases and death as expected with a high-grade sarcoma. It is estimated that the mean survival time of patients with malignant PEComas affecting the terminal Ileum & Caecum is 28 months and Mesentery and Colon, 27 months and 38 months, respectively. A recent case by Slevaggi et al. reported 25 day mortality in a 42 year old male with a malignant PEComa which was hepatic in origin.7 This has reflected our own experience in the case outlined here with our patient not surviving greater than 3 months since diagnosis.
The sparity of PEComa diagnosis and restricted clinical follow up time in aggressive malignant cases is limited throughout the literature. Due to this, issues regarding the origin of this unusual cell differentiation, clinical behaviour and patterns of disease progression remains elusive. Longer clinical follow up of known cases and the evaluation of any additional patients is required. A recent small case series and a separate case report have shown improved clinical and radiological responses to the mTOR inhibitors, sirolimus and temsirolimus.8 This may well represent a promising targeted therapeutic approach. Therefore a diagnosis of PEComa introduces a number of dilemmas with regard to optimum management and treatment of the primary and any subsequent metastatic disease. The need for implementation of guidelines for favourable screening programmes and the role of adjuvant therapies for local and metastatic disease bulk are required.
Conflicts of interest statement
Nil.
Funding
Nil.
Ethical approval
Written consent was obtained by the patient's next of kin and is available on request.
Author contributions
Peadar Waters: Primary Author,
David Mitchell & Ruth Murphy: Data collection and processing of images,
Michael McKenna: Review of the pathology surrounding the case with review and corrections made within the case report prior to submission,
Ronan Waldron: review and correction of the final manuscript prior to submission.
References
- 1.Folpe A.L., Fletcher C.D.M., Unni K.K., Epstein J., Mertens F. IARC Press; Lyon: 2002. Neoplasms with perivascular epithelioid cell differentiation (PEComas). Pathology and genetics of tumours of soft tissue and bone. Series: WHO classification of tumours. p. 221–2. [Google Scholar]
- 2.Bonetti F., Martignoni G., Colato C., Manfrin E., Gambacorta M., Faleri M., et al. Abdominopelvic sarcoma of perivascular epithelioid cells. Report of four cases in young women, one with tuberous sclerosis. Mod Pathol. 2001;14:563–568. doi: 10.1038/modpathol.3880351. [DOI] [PubMed] [Google Scholar]
- 3.Zamboni G., Pea M., Martignoni G., Zancanaro C., Faccioli G., Gilioli E., et al. Clear cell “sugar” tumor of the pancreas. A novel member of the family of lesions characterized by the presence of perivascular epithelioid cells. Am J Surg Pathol. 1996;20:722–730. doi: 10.1097/00000478-199606000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Jungbluth A.A., Busam K.J., Gerald W.L., Stockert E., Coplan K.A., Iversen K., et al. A103: an anti-melan-a monoclonal antibody for the detection of malignant melanoma in paraffin-embedded tissues. Am J Surg Pathol. 1998;22:595–602. doi: 10.1097/00000478-199805000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Stone C.H., Lee M.W., Amin M.B., Yaziji H., Gown A.M., Ro J.Y., et al. Renal angiomyolipoma: further immunophenotypic characterization of an expanding morphologic spectrum. Arch Pathol Lab Med. 2001;125:751–758. doi: 10.5858/2001-125-0751-RA. [DOI] [PubMed] [Google Scholar]
- 6.Folpe A.L., Mentzel T., Lehr H.A., Fisher C., Balzer B.L., Weiss S.W. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29:1558–1575. doi: 10.1097/01.pas.0000173232.22117.37. [DOI] [PubMed] [Google Scholar]
- 7.Selvaggi F., Risio D., Claudi R., Cianci R., Angelucci D., Pulcini D., et al. Malignant PEComa: a case report with emphasis on clinical and morphological criteria. BMC Surg. 2011;11 doi: 10.1186/1471-2482-11-3. 1471-2482/11/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner A.J., Malinowska-Kolodziej I., Morgan J.A., Qin W., Fletcher C.D., Vena N., et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010;28(February 10 (5)):835–840. doi: 10.1200/JCO.2009.25.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]