

Abstract
Multiple new small molecules such as tyrosine kinase, mammalian target of rapamycin (mTOR) and proteasome inhibitors have been approved in the last decade and are a considerable progress for cancer therapy. Drug transporters are important determinants of drug concentrations in the systemic circulation. Moreover, expression of drug transporters in blood–tissue barriers (e.g. blood–brain barrier) can limit access of small molecules to the tumour (e.g. brain tumour). Finally, transporter expression and (up)regulation in the tumour itself is known to affect local drug concentrations in the tumour tissue contributing to multidrug resistance observed for multiple anticancer agents. This review summarizes the current knowledge on: (i) small molecules as substrates of uptake and efflux transporters; (ii) the impact of transporter deficiency in knockout mouse models on plasma and tissue concentrations; (iii) small molecules as inhibitors of uptake and efflux transporters with possible consequences for drug–drug interactions and the reversal of multidrug resistance; and (iv) on clinical studies investigating the association of polymorphisms in genes encoding drug transporters with pharmacokinetics, outcome and toxicity during treatment with the small molecules.
Keywords: drug transport, drug metabolism, P-glycoprotein, BCRP, MRP, MATE, OCT, OATP, drug–drug interaction, tyrosine kinase inhibitor, imatinib, sorafenib, sunitinib
Introduction
The development of new small molecules such as tyrosine kinase, mammalian target of rapamycin (mTOR) and proteasome inhibitors is a significant progress for cancer therapy. In order to achieve desired therapeutic effects, these small molecules must reach sufficient concentrations within the target cells (tumour cells, endothelial cells of tumour vessels) to block intracellular signal transduction pathways (Krause and Van Etten, 2005).
Most new small molecules are substrates of the major drug metabolizing enzyme cytochrome P450 3A4 (CYP3A4), which is expressed in small intestine and liver (Table 1, Hartmann et al., 2009; van Erp et al., 2009b; Duckett and Cameron, 2010). In addition to drug metabolizing enzymes, drug transporters are now well-recognized determinants of drug disposition and effects (Ho and Kim, 2005; Funk, 2008; Zolk and Fromm, 2011). Transporters affect drug disposition and effects via different mechanisms. First, due to their expression in the small intestine, liver and kidney, they are important determinants for systemic plasma concentrations, as they influence the extent of drug absorption from the gastrointestinal (GI) tract, of hepatic drug metabolism and of biliary as well as of renal drug elimination (Figure 1; Ho and Kim, 2005; Funk, 2008). Second, drug transporters affect drug penetration into certain tissues (e.g. brain) due to their expression in blood–tissue barriers (e.g. blood–brain barrier). Third, drug transporters are expressed in tumour cells and are considered as important determinants of drug concentrations at the site of action of these drugs. One well-known example for the last mentioned mechanism is the overexpression of the ABCB1[multidrug resistance 1 (MDR1)] gene product P-glycoprotein in tumour cells as one reason for the development of resistance against certain anticancer agents. Finally, concomitant administration of two or more drugs can lead to transporter-mediated drug–drug interactions via induction or inhibition of drug transporters (Shitara et al., 2005; Müller and Fromm, 2011). For example, concomitantly administered drugs used for treatment of non-tumour diseases (e.g. infections by rifampin or macrolides, depression by St John's wort, epilepsy by phenytoin or carbamazepine) can influence transporter expression or function (Ho and Kim, 2005), and therefore disposition of the transporter substrate such as small molecules used for cancer treatment.
Table 1.
Metabolism and excretion of small molecules used for cancer treatment. All data are derived from the US Food and Drug Administration drug labels and the German summary of product characteristics (‘Fachinformation’)
| Bioavailability (%) | Metabolism | Elimination (% of radioactivity) | |||||
|---|---|---|---|---|---|---|---|
| Compound | Major | Additional | In Urine | In faeces | Unchanged | Brand name | |
| Dasatinib | CYP3A4 | UGT | 4 | 85 | 0.1 (in urine), 19 (in faeces) | Sprycel® | |
| Erlotinib | 59 | CYP3A4 | CYP1A2 | 8 | 83 | 0.3 (in urine), 1 (in faeces) | Tarceva® |
| Gefitinib | 59 | CYP3A4, CYP2D6 | <4 | 86 | Iressa® | ||
| Imatinib | 98 | CYP3A4 | CYP1A2, CYP2D6, CYP2C9, CYP2C19 | 13 | 68 | 5 (in urine), 20 (in faeces) | Glivec® |
| Lapatinib | CYP3A4, CYP3A5 | CYP2C19, CYP2C8 | <2 | 27 (in faeces) | Tykerb® | ||
| Nilotinib | 30 | CYP3A4 | CYP2C8 | 94 | 69 | Tasigna® | |
| Pazopanib | CYP3A4 | CYP1A2, CYP2C8 | <4 | Votrient® | |||
| Sorafenib | 38–49 (relative) | CYP3A4 | UGT1A9 | 19 | 77 | 51 (in faeces) | Nexavar® |
| Sunitinib | CYP3A4 | 16 | 61 | Sutent® | |||
| Everolimus | CYP3A4 | 5 | 80 | 0 | Afinitor® | ||
| Temsirolimus | CYP3A4 | 4.6 | 78 | Torisel® | |||
| Bortezomib | CYP3A4, CYP2C19, CYP1A2 | CYP2D6, CYP2C9 | NC | NC | NC | Velcade® | |
NC, not characterized in humans.
Figure 1.
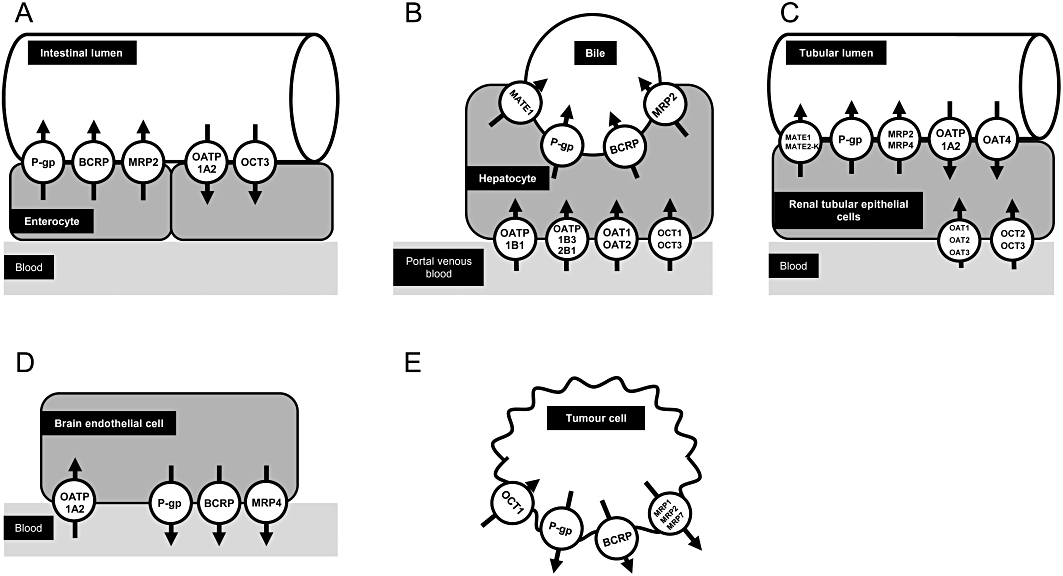
Tissue expression of selected drug transporters, which are involved in disposition or effects of small molecules used for cancer treatment. (A) Enterocyte. (B) Hepatocyte. (C) Renal tubular epithelial cell. (D) Brain endothelial cell. (E) Tumour cell. P-gp, P-glycoprotein.
This review focuses on the interaction of currently approved major small molecule drugs with drug transporters. Particular attention is given to in vitro data on small molecules as substrates and inhibitors of drug transporters as well as on clinical studies linking transporter expression or function (e.g. determined by genetic polymorphisms) with treatment outcome.
Overview on major drug transporters
Functionally, drug transporters can be categorized into two groups. The first group mediates uptake of drugs into the cells, the second group transports its substrates from the intracellular compartment out of the cells (Figure 1). The major uptake transporters are organic anion transporting polypeptide (OATP) family members [e.g. protein name: OATP1B1, respective gene name: SLCO1B1; OATP1B3, SLCO1B3; OATP2B1, SLCO2B1; solute carrier gene family encoding for OATPs (SLCO)], organic anion transporters (OATs; e.g. SLC22A6) and the organic cation transporter 1 (OCT1; SLC22A1), which are localized in the basolateral membrane of hepatocytes (Figure 1) and mediate drug uptake from the portal venous blood into the hepatocytes (König, 2011; Niemi et al., 2011; Nies et al., 2011). OCT2 (SLC22A2) is an uptake transporter localized in the basolateral membrane of renal proximal tubular cells mediating the first step of renal excretion of certain drugs into urine [Figure 1 (Nies et al., 2011)].
ATP-binding cassette (ABC) transporters are a group of the efflux transporter family. P-glycoprotein (gene name: ABCB1), breast cancer resistance protein (protein name: BCRP, gene name: ABCG2) and multidrug resistance protein 2 (MRP2; ABCC2) are localized in the apical (luminal) membrane of enterocytes, the canalicular membrane of hepatocytes and the apical membrane of renal proximal tubular cells thereby reducing drug absorption from the GI tract and mediating drug efflux into bile and urine respectively (Fromm, 2004; Keppler, 2011; zu Schwabedissen and Kroemer, 2011). Efflux transporters for cationic compounds in the liver and kidney are multidrug and toxin extrusion protein 1 (MATE1; SLC47A1) and 2-K, [MATE2-K; SLC47A2; Figure 1 (Minematsu and Giacomini, 2011; Yonezawa and Inui, 2011)].
Expression of drug transporters in tumour cells
The expression and localization of the above mentioned drug transporters in healthy tissues are well characterized. The expression and regulation of drug transporters in tumours is with some exceptions (e.g. P-glycoprotein) much less well studied. It should be considered that changes in expression levels occur during the course of the tumour disease per se. Moreover, treatment of the tumour (e.g. by drugs, radiotherapy) is likely to have an effect on certain transporters.
For example, the tyrosine kinase inhibitor imatinib is a substrate of OCT1, BCRP and P-glycoprotein. Mononuclear cells of patients with chronic myeloid leukaemia (CML) express these three transporters and clinical studies showed an association between OCT1 tumour cell expression or function and antitumour effects of imatinib in patients with CML (for review see Eechoute et al., 2011b).
Individual small molecules and drug transporters
The subsequent paragraphs on the individual small molecules highlight the most relevant, known interactions of these compounds with drug transporters. The following sections are structured into a presentation of the interaction of small molecule kinase inhibitors (in alphabetical order) with drug transporters, followed by sections on the mTOR inhibitors everolimus and temsirolimus and the proteasome inhibitor bortezomib. An overview of the pharmacokinetic properties of the small molecules is given in Table 1. Tables 2 and 3 summarize the available information on small molecules as substrates and inhibitors of drug transporters respectively. The impact of polymorphisms in genes encoding for drug transporters on pharmacokinetics and effects of the small molecules are summarized in Table 4. All chapters on individual drugs in this review are structured in the same way and provide information (if available) in the following, identical order: (i) small molecule drug as substrate of uptake transporters; (ii) as substrate of efflux transporters; (iii) as inhibitor of uptake transporters; (iv) as inhibitor of efflux transporters; and (v) pharmacogenomic data from clinical studies in humans.
Table 2.
Overview of small molecules used for cancer therapy as substrates of drug transporters
| Drug | Substrate of transporter | In vitro | Animal models |
|---|---|---|---|
| Dasatinib | P-glycoprotein (ABCB1) | CCRF-CEM cells (Hiwase et al., 2008), K562 cells (Hiwase et al., 2008; Hegedus et al., 2009; Haouala et al., 2010), MDCKII cells (Chen et al., 2009; Lagas et al., 2009) | Abcb1a/1b(–/–) mice (Lagas et al., 2009), Abcb1a/1b(–/–) Abcg2(–/–) mice (Chen et al., 2009; Lagas et al., 2009) |
| BCRP (ABCG2) | K562 cells (Hiwase et al., 2008; Hegedus et al., 2009), MDCKII cells (Chen et al., 2009; Lagas et al., 2009), murine fibroblast cell line MEF3.8 (Hiwase et al., 2008) | Abcb1a/1b(–/–) Abcg2(–/–) mice (Chen et al., 2009; Lagas et al., 2009) | |
| Erlotinib | OAT3 (SLC22A8) | HEK293 cells (Elmeliegy et al., 2011) | |
| OCT2 (SLC22A2) | HEK293 cells (Elmeliegy et al., 2011) | ||
| P-glycoprotein (ABCB1) | LLC-PK1 cells (Marchetti et al., 2008; Kodaira et al., 2010), MDCKII cells (Marchetti et al., 2008) | Abcb1a/1b(–/–) mice (Kodaira et al., 2010), Abcb1a/1b(–/–) Abcg2(–/–) mice (Marchetti et al., 2008; de Vries et al., 2010; Kodaira et al., 2010) | |
| BCRP (ABCG2) | HEK293 cells (Li et al., 2007), MDCKII cells (Marchetti et al., 2008; Kodaira et al., 2010), Saos2 cells (Elmeliegy et al., 2011) | Abcg2(–/–) mice (Kodaira et al., 2010; Elmeliegy et al., 2011), Abcb1a/1b(–/–) Abcg2(–/–) mice (Marchetti et al., 2008; de Vries et al., 2010; Kodaira et al., 2010; Elmeliegy et al., 2011) | |
| Gefitinib | P-glycoprotein (ABCB1) | MDCKII cells (Agarwal et al., 2010) | Abcb1a/1b(–/–) Abcg2(–/–) mice (Kawamura et al., 2009; Agarwal et al., 2010) |
| BCRP (ABCG2) | HEK293 cells (Li et al., 2007), MDCKII cells (Agarwal et al., 2010) | Abcb1a/1b(–/–) Abcg2(–/–) mice (Kawamura et al., 2009; Agarwal et al., 2010) | |
| Imatinib | OATP1A2 (SLC21A3) | HeLa cells (Eechoute et al., 2011a), Xenopus laevis oocytes (Hu et al., 2008; Yamakawa et al., 2011; Eechoute et al., 2011a) | |
| OATP1B3 (SLC21A8) | Xenopus laevis oocytes (Hu et al., 2008) | ||
| OCT1 (SLC22A1) | CEM cells (Thomas et al., 2004), CML cells (White et al., 2006), HEK293 cells (Hu et al., 2008), CML cells (Wang et al., 2008) | ||
| OCTN2 (SLC22A5) | HEK293 cells (Hu et al., 2008) | ||
| P-glycoprotein (ABCB1) | K562 cells (Mahon et al., 2003), LLC-PK1 cells (Hu et al., 2008), MDCKII cells (Dai et al., 2003; Thomas et al., 2004) | Abcb1a/1b(–/–) mice (Dai et al., 2003; Zhou et al., 2009), Abcb1a/1b(–/–) Abcg2(–/–) mice (Zhou et al., 2009) | |
| MRP4 (ABCC4) | Saos2 cells (Hu et al., 2008) | ||
| BCRP (ABCG2) | HEK293 cells (Burger et al., 2004), MCF7 cells (Burger et al., 2004), MDCKII cells (Breedveld et al., 2005), Saos2 cells (Hu et al., 2008) | Abcg2(–/–) mice (Zhou et al., 2009), Abcb1a/1b(–/–) Abcg2(–/–) mice (Zhou et al., 2009) | |
| Lapatinib | P-glycoprotein (ABCB1) | MDCKII cells (Polli et al., 2008) | Abcb1a/1b(–/–) mice (Polli et al., 2009), Abcb1a/1b(–/–) Abcg2(–/–) mice (Polli et al., 2009) |
| BCRP (ABCG2) | MDCKII cells (Polli et al., 2008) | Abcb1a/1b(–/–) Abcg2(–/–) mice (Polli et al., 2009) | |
| Nilotinib | P-glycoprotein (ABCB1) | K562 cells (Mahon et al., 2008) | |
| BCRP (ABCG2) | K562 cells (Hegedus et al., 2009) | ||
| Pazopanib | P-glycoprotein (ABCB1) | (FDA, 2010b) | |
| BCRP (ABCG2) | (FDA, 2010b) | ||
| Sorafenib | P-glycoprotein (ABCB1) | Human epithelial colorectal adenocarcinoma cells (Caco-2, Gnoth et al., 2010), K562 cells (Haouala et al., 2010), LLC-PK1 cells (Hu et al., 2009; Gnoth et al., 2010), MDCKII cells (Lagas et al., 2010) | Abcb1a/1b(–/–) mice (Hu et al., 2009; Gnoth et al., 2010), Abcb1a/1b(–/–) Abcg2(–/–) mice (Lagas et al., 2010; Asakawa et al., 2011) |
| MRP2 (ABCC2) | LLC-PK1 cells (Shibayama et al., 2011) | ||
| BCRP (ABCG2) | MDCKII cells (Lagas et al., 2010; Agarwal et al., 2011) | Abcg2(–/–) mice (Lagas et al., 2010; Agarwal et al., 2011), Abcb1a/1b(–/–) Abcg2(–/–) mice (Lagas et al., 2010; Agarwal et al., 2011; Asakawa et al., 2011) | |
| Sunitinib | P-glycoprotein (ABCB1) | K562 cells (Haouala et al., 2010), LLC-PK1 cells (Hu et al., 2009), MDCKII cells (Tang et al., 2011) | Abcb1a/1b(–/–) mice (Hu et al., 2009; Tang et al., 2011), Abcb1a/1b(–/–) Abcg2(–/–) mice (Tang et al., 2011) |
| BCRP (ABCG2) | MDCKII cellls (Tang et al., 2011) | Abcb1a/1b(–/–) Abcg2(–/–) mice (Tang et al., 2011) | |
| Everolimus | P-glycoprotein (ABCB1) | Caco-2 cells (Crowe and Lemaire, 1998) | Abcb1a/1b(–/–) mice (Chu et al., 2009) |
| Temsirolimus | P-glycoprotein (ABCB1) | Caco-2 cells (Crowe and Lemaire, 1998) | |
| Bortezomib | P-glycoprotein (ABCB1) | K562 cells (Rumpold et al., 2007) |
FDA, US Food and Drug Administration.
Table 3.
Overview of small molecules used for cancer therapy as inhibitors of drug transporters
| Drug | Inhibition of transporter | Substrate | In vitro/animal model |
|---|---|---|---|
| Dasatinib | P-glycoprotein (ABCB1) | Calcein-AM | K562 cells (Hegedus et al., 2009) |
| BCRP (ABCG2) | Hoechst 33342 | K562 cells (Hegedus et al., 2009) | |
| Erlotinib | OCT1 (SLC22A1) | Metformin (IC50 = 0.356 µM) | HEK293 cells (Minematsu and Giacomini, 2011) |
| MATE2-K (SLC47A2) | Metformin (IC50 = 3.45 µM) | HEK293 cells (Minematsu and Giacomini, 2011) | |
| P-glycoprotein (ABCB1) | Vincristine (IC50 = 2 µM) | K562 cells (Noguchi et al., 2009) | |
| MRP7 (ABCC10) | Paclitaxel | HEK293 cells (Kuang et al., 2010) | |
| BCRP (ABCG2) | E217βG, methotrexate, mitoxantrone | HEK293 (Shi et al., 2007), K562 cells (Noguchi et al., 2009) | |
| Gefitinib | OCT1 (SLC22A1) | MPP+ | HEK293 cells (Galetti et al., 2010) |
| OCT2 (SLC22A2) | MPP+ | HEK293 cells (Galetti et al., 2010) | |
| MATE2-K (SLC47A2) | Metformin (IC50 = 0.194 µM) | HEK293 cells (Minematsu and Giacomini, 2011) | |
| P-glycoprotein (ABCB1) | Calcein-AM, docetaxel, doxorubicin, paclitaxel, rhodamine-123, topotecan | CL1 cells (Yang et al., 2005), LLC-PK1 cells (Leggas et al., 2006), MCF7 cells (Kitazaki et al., 2005; Yang et al., 2005), human small cell lung cancer cell line PC-6 (Kitazaki et al., 2005) | |
| BCRP (ABCG2) | Hoechst 33342, estrone-3-sulfate (IC50 = 1.0 µM), mitoxantrone, topotecan (Ki = 1.0 µM) | CL1 cells (Yang et al., 2005), HL60 cells (Ozvegy-Laczka et al., 2004), K562 cells (Yanase et al., 2004), MCF7 cells (Yang et al., 2005), PC-6 cells (Nakamura et al., 2005), Saos2 cells (Leggas et al., 2006) | |
| Imatinib | OCT1 (SLC22A1) | Metformin (IC50 = 1.47 µM) | HEK293 cells (Minematsu and Giacomini, 2011) |
| MATE1 (SLC47A1) | Metformin (IC50 = 0.048 µM) | HEK293 cells (Minematsu and Giacomini, 2011) | |
| MATE2-K (SLC47A2) | Metformin (IC50 = 0.478 µM) | HEK293 cells (Minematsu and Giacomini, 2011) | |
| P-glycoprotein (ABCB1) | Calcein-AM (Ki = 18.3 µM) | LLC-PK1 cells (Hamada et al., 2003) | |
| MRP7 (ABCC10) | Paclitaxel | HEK293 cells (Shen et al., 2009) | |
| BCRP (ABCG2) | Mitoxantrone, SN-38, topotecan | HEK293 cells (Burger et al., 2004), MCF7 cells (Burger et al., 2004), Saos2 cells (Houghton et al., 2004) | |
| Lapatinib | OATP1B1 (SLCO21A6) | E217βG | CHO cells (Polli et al., 2008; Fachinformation, 2010) |
| P-glycoprotein (ABCB1) | Topotecan | CHO cells (Molina et al., 2008) | |
| MRP7 (ABCC10) | Paclitaxel | HEK293 cells (Kuang et al., 2010) | |
| BCRP (ABCG2) | E217βG, methotrexate, SN-38, topotecan | HEK293 cells (Dai et al., 2008), H1975 cells (Perry et al., 2010), H358 cells (Perry et al., 2010), MDA-MB-231 cells (Molina et al., 2008; Perry et al., 2010), Susa S/R cells (Perry et al., 2010) | |
| Nilotinib | OCT1 (SLC22A1) | Metformin | CML cells (Davies et al., 2009), HEK293 cells (Minematsu and Giacomini, 2011) |
| OCT3 (SLC22A3) | Metformin (IC50 = 0.35 µM) | HEK293 cells (Minematsu and Giacomini, 2011) | |
| P-glycoprotein (ABCB1) | Calcein-AM, dasatinib, rhodamine | CML cells (Davies et al., 2009; Hiwase et al., 2010), HEK293 cells (Dohse et al., 2010), K562 cells (Hegedus et al., 2009) | |
| MRP7 (ABCC10) | Paclitaxel | HEK293 cells (Shen et al., 2009) | |
| BCRP (ABCG2) | E217βG, Hoechst 33342, methotrexate (Ki = 0.69 µM), pheophorbide A | CML cells (Davies et al., 2009), HEK293 cells (Tiwari et al., 2009; Dohse et al., 2010), HSC cells (Brendel et al., 2007), K562 cells (Hegedus et al., 2009) | |
| Pazopanib | OATP1B1 (SLC21A6) | (FDA, 2010b; Keisner and Shah, 2011) | |
| P-glycoprotein (ABCB1) | (FDA, 2010b) | ||
| BCRP (ABCG2) | (FDA, 2010b) | ||
| Sorafenib | P-glycoprotein (ABCB1) | Calcein-AM, vinblastine | LLC-PK1 (Hu et al., 2009), MDCKII cells (Agarwal et al., 2011) |
| MRP2 (ABCC2) | Docetaxel | Saos2 cells (Hu et al., 2009) | |
| MRP4 (ABCC4) | PMEA | Saos2 cells (Hu et al., 2009) | |
| BCRP (ABCG2) | Hoechst 33342 | Saos2 cells (Hu et al., 2009) | |
| Sunitinib | P-glycoprotein (ABCB1) | Calcein-AM, rhodamine 123, vincristine (Ki = 7.6 µM) | HEK293 cells (Shukla et al., 2009), K562 cells (Kawahara et al., 2010), LLC-PK1 cells (Hu et al., 2009) |
| MRP2 (ABCC2) | Docetaxel | Saos2 cells (Hu et al., 2009) | |
| MRP4 (ABCC4) | PMEA | Saos2 cells (Hu et al., 2009) | |
| BCRP (ABCG2) | Estrone-3-sulfate (Ki = 0.32 µM), Hoechst 33342, methotrexate, pheophorbide A, rhodamine 123 | HEK293 cells (Dai et al., 2009; Shukla et al., 2009), K562 cells (Kawahara et al., 2010), Saos2 cells (Hu et al., 2009), S1-M1-80 cells (Dai et al., 2009) | |
| Everolimus | OATP1A2 (SLC21A3) | Estrone-3-sulfate (IC50 = 4.2 µM) | Picard et al., 2011 |
| OATP1B1 (SLC21A6) | Estrone-3-sulfate (IC50 = 4.1 µM) | Picard et al., 2011 | |
| OATP1B3 (SLC21A8) | Mycophenolic acid 7-O-glucuronide (IC50 = 4.3 µM) | Picard et al., 2011 | |
| Temsirolimus | OATP1A2 (SLC21A3) | Estrone-3-sulfate (IC50 = 11.9 µM) | Picard et al., 2011 |
| OATP1B1 (SLC21A6) | Estrone-3-sulfate (IC50 = 9.8 µM) | Picard et al., 2011 | |
| OATP1B3 (SLC21A8) | Mycophenolic acid 7-O-glucuronide (IC50 = 1.3 µM) | Picard et al., 2011 | |
| P-glycoprotein (ABCB1) | Fachinformation, 2011 | ||
| Bortezomib | NC | NC | NC |
FDA, US Food and Drug Administration; NC, not characterized in humans.
Table 4.
Pharmacogenomics of small molecules used for cancer therapy. The influence of drug uptake and efflux transporters on pharmacokinetics (PK) and/or pharmacodynamics (PD) are shown
| Drug | Transporter (gene) | Genotype/polymorphism | Effect on PK | Effect on PD (outcome, toxicity) | Reference |
|---|---|---|---|---|---|
| Erlotinib | BCRP (ABCG2) | c.421C>A | Clearance reduced by 24% (head and neck squamous cell carcinoma) | (Thomas et al., 2009) | |
| c.1143/−15622 diplotypes (CC/TT) or (TT/TT) | Higher AUC and Cmax (non-small-cell lung cancer, head and neck cancer, and ovarian cancer) | No influence on diarhea or skin toxicity | (Rudin et al., 2008) | ||
| P-glycoprotein (ABCB1) | c.2677G>T/A | No influence on pharmacokinetics (head and neck squamous cell carcinoma) | (Thomas et al., 2009) | ||
| c.3435C>T | No influence on pharmacokinetics (head and neck squamous cell carcinoma) | (Thomas et al., 2009) | |||
| Gefitinib | BCRP (ABCG2) | c.421C>A | Trend to higher Css, min (n.s., trough concentrations at steady state; malignant solid tumours) | (Li et al., 2007) | |
| c.421C>A | Increased drug accumulation (ratio of trough concentrations at steady state to day 1 trough concentration; malignant solid tumours) | (Li et al., 2007) | |||
| c.421C>A | Increased risk of diarrhea (non-small-cell lung cancer) | (Cusatis et al., 2006) | |||
| c.421C>A | No influence on risk of skin toxicity (non-small-cell lung cancer) | (Cusatis et al., 2006) | |||
| c.421C>A | No effect on adverse side effects for example, diarrhea, interstitial pneumonia (non-small-cell lung cancer) | (Akasaka et al., 2010) | |||
| −15622TT | Increased risk of diarrhea (non-small-cell lung cancer) | (Lemos et al., 2011) | |||
| haplotype TT (c.1143C>T, −15622C>T) | Increased risk of higher grade diarrhea (non-small-cell lung cancer) | (Lemos et al., 2011) | |||
| P-glycoprotein (ABCB1) | c.3435C>T | No influence on pharmacokinetics | (Li et al., 2007) | ||
| c.3435C>T | No influence on risk of diarrhea (non-small-cell lung cancer) | (Cusatis et al., 2006) | |||
| c.3435C>T | No influence on risk of skin toxicity (non-small-cell lung cancer) | (Cusatis et al., 2006) | |||
| Imatinib | OATP1A2 (SLCO1A2) | −361GG | Reduced imatinib clearance (CML) | (Yamakawa et al., 2011) | |
| OATP1B3 (SLCO1B3) | c.334T>G | No influence on plasma trough concentrations (CML) | Higher rates of major molecular response (CML) | (Takahashi et al., 2010) | |
| c.334T>G | Higher intracellular concentrations of imatinib in leukocytes (chronic phase CML) | (Nambu et al., 2011) | |||
| c.334T>G | Higher intracellular (leukocytes) to plasma ratio of imatinib (chronic phase CML) | (Nambu et al., 2011) | |||
| OCT1 (SLC22A1) | c.156T>C | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | |
| c.286C>T | No change in oral imatinib clearance | (Hu et al., 2008) | |||
| c.480GG | Increased risk for imatinib resistance due to loss of response and treatment failure (CML) | (Kim et al., 2009) | |||
| c.480C>G | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| c.1022C>T | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| c.1222GG | No influence on plasma trough concentrations (CML) | Higher rates of major molecular response (CML) | (Takahashi et al., 2010) | ||
| c.1498C>A | No change in oral imatinib clearance | (Hu et al., 2008) | |||
| BCRP (ABCG2) | c.34GG | Decreased major and complete cytogenetic response (CML) | (Kim et al., 2009) | ||
| c.421CC | Decreased complete molecular response (CML) | (Kim et al., 2009) | |||
| c.421C>A | No influence on oral clearance (GIST) | (Gardner et al., 2006) | |||
| c.421C>A | Increased plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| P-glycoprotein (ABCB1) | c.3435TT | Decreased overall survival (univariate analysis, CML) | (Kim et al., 2009) | ||
| c.3435C>T | No influence on the oral clearance (GIST) | (Gardner et al., 2006) | |||
| c.1236TT; c.2677TT; c.3435TT | Increased oral clearance (GIST) | (Gurney et al., 2007) | |||
| c.1236C>T | Higher major molecular response (CML) | (Dulucq et al., 2008) | |||
| c.1236C>T | Higher imatinib trough concentrations (CML) | (Dulucq et al., 2008) | |||
| c.1236TT | Increased resistance [cytogenetic resistance or relapse after major cytogenetic response (MCyR, CML)] | (Ni et al., 2011) | |||
| c.1236C>T | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| c.2677TT / TA | Higher major molecular response (CML) | (Dulucq et al., 2008) | |||
| c.2677GT | Increased resistance (cytogenetic resistance or relapse after MCyR) | (Ni et al., 2011) | |||
| c.2677G>T/A | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| c.2677AG / AT / AA | Lower resistance (cytogenetic resistance or relapse after MCyR) | (Ni et al., 2011) | |||
| c.3435CC | Lower resistance (cytogenetic resistance or relapse after MCyR) | (Ni et al., 2011) | |||
| c.3435C>T | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | ||
| haplotype TC or TT (c.1236C>T; c.3435C>T | Higher imatinib trough concentrations (CML) | (Dulucq et al., 2008) | |||
| c.1236C; c.2677G; c.3435C | Lower major molecular response (CML) | (Dulucq et al., 2008) | |||
| MRP2 (ABCC2) | −24C>T | No influence on plasma trough concentrations (CML) | No influence on major molecular response (CML) | (Takahashi et al., 2010) | |
| Sunitinib | BCRP (ABCG2) | c.34G>A | Trend for increased progression free survival (n.s., clear-cell metastatic renal cell carcinoma) | (van der Veldt et al., 2011) | |
| 422AA | Higher blood concentrations (renal cell carcinoma) | (Mizuno et al., 2010) | |||
| haplotype TT; −15622C>T, c.1143C>T | Increased risk for toxicity (>grade 2 CTCAE, metastatic renal cell carcinoma, GIST) | (van Erp et al., 2009a) | |||
| P-glycoprotein (ABCB1) | haplotype TCG (c.3435C>T; c.1236C>T; c.2677G>T) | Increased progression free survival (clear-cell metastatic renal cell carcinoma) | (van der Veldt et al., 2011) | ||
| haplotype TTT (c.1236C>T, c.2677G>T, c.3435C>T) | Increased risk for hand-foot syndrome (metastatic renal cell carcinoma, GIST) | (van Erp et al., 2009a) |
GIST, gastrointestinal stromal tumours.
All compounds discussed in this review are extensively metabolized by CYP3A4 (van Erp et al., 2009b; Hartmann et al., 2009; Duckett and Cameron, 2010). It should be noted that multiple drug–drug-interactions are reported between small molecules and the CYP3A4 and P-glycoprotein inducer rifampicin or the CYP3A4 and P-glycoprotein inhibitor ketoconazole. However, the contribution of transporters such as BCRP and P-glycoprotein to induction or inhibition of CYP3A4 for these drug–drug interactions is still not completely understood.
Dasatinib
In humans, after a single oral dose of radiolabelled dasatinib, 85% of radioactivity was recovered in faeces and 4% in urine (Table 1; Brave et al., 2008). Coadministration of dasatinib with the CYP3A4 inducer rifampin decreased the dasatinib area under the curve (AUC) by ∼82%, and coadministration with the CYP3A4 inhibitor ketoconazole increased the dasatinib AUC fivefold (Brave et al., 2008).
OCT1 does not play a significant role for dasatinib uptake (Giannoudis et al., 2008; Hiwase et al., 2008). In vitro data indicate that dasatinib is a substrate of the efflux transporters BCRP and P-glycoprotein [Table 2 (Hiwase et al., 2008; Chen et al., 2009; Hegedus et al., 2009; Lagas et al., 2009; Haouala et al., 2010)]. Data from P-glycoprotein- and Bcrp-deficient mice indicate that P-glycoprotein, but not Bcrp, limits dasatinib absorption after oral drug administration [Table 2 (Lagas et al., 2009)]. Moreover, dasatinib brain concentrations were considerably higher in P-glycoprotein-deficient Abcb1a/1b knockout mice, but not in Bcrp-deficient mice compared with wild-type mice (Chen et al., 2009; Lagas et al., 2009). Interestingly, Abcb1a/1b Abcg2 knockout mice accumulated considerably more dasatinib in the brain compared with Abcb1a/1b knockout mice, indicating that Bcrp can partly take over P-glycoprotein function in the absence of P-glycoprotein (Chen et al., 2009; Lagas et al., 2009).
The inhibition of the organic cation transporters OCT1, OCT2, OCT3, MATE1 and MATE2-K by dasatinib in relation to the estimated portal venous and systemic plasma concentrations was relatively poor (Minematsu and Giacomini, 2011).
Erlotinib
In vitro experiments showed that erlotinib and its metabolite OSI-420 are substrates of the uptake transporters OAT3 and OCT2 (Elmeliegy et al., 2011). ABCG2-transfected cells exhibited lower intracellular accumulation of erlotinib than cells lacking ABCG2, indicating that erlotinib is a substrate of BCRP (Li et al., 2007; Elmeliegy et al., 2011). In vitro, erlotinib was transported by mouse and human P-glycoprotein and by Bcrp/BCRP (Li et al., 2007; Marchetti et al., 2008; Kodaira et al., 2010; Elmeliegy et al., 2011). No active transport of erlotinib by MRP2 was observed using Madin-Darby canine kidney cell (MDCKII)-MRP2 monolayers (Marchetti et al., 2008). Several studies investigated erlotinib disposition in mice deficient for P-glycoprotein and/or Bcrp (Marchetti et al., 2008; de Vries et al., 2010; Kodaira et al., 2010; Elmeliegy et al., 2011). Marchetti et al. (2008) reported that calculated apparent oral bioavailability of erlotinib was significantly increased in Abcb1a/1b Abcg2 knockout mice (60.4%) compared with wild-type mice (40.0%; P = 0.02). The absence of P-glycoprotein or the simultaneous absence of Bcrp and P-glycoprotein had greater effects than the absence of Bcrp alone on brain and testis concentrations of erlotinib in the knockout mouse models as reported by Kodaira et al. (2010) and de Vries et al. (2010). This is in contrast to the report by Elmeliegy et al. (2011), who concluded that Bcrp is the major efflux transporter preventing erlotinib penetration into mouse brain.
Erlotinib inhibits the organic cation transporters MATE2-K and OCT1 at potentially clinical relevant concentrations [Table 3 (Minematsu and Giacomini, 2011)]. In vitro, erlotinib reverses BCRP-mediated multidrug resistance (Shi et al., 2007; Noguchi et al., 2009). Modulation of P-glycoprotein-mediated drug resistance by erlotinib appears to be substrate dependent (Shi et al., 2007; Noguchi et al., 2009). In addition, Kuang et al. reported that erlotinib potently reverses MRP7-mediated multidrug resistance (Kuang et al., 2010).
Thomas et al. (2009) reported population pharmacokinetics in erlotinib-treated patients with head and neck squamous cell carcinoma. Among other factors, the association of polymorphisms in ABCB1, ABCG2 and CYP3A5 with erlotinib clearance was investigated. Interestingly, patients with at least one ABCG2 variant allele (c.421A) had a significant 24% decrease in erlotinib clearance, whereas no association was found with the polymorphisms in ABCB1 and CYP3A5 (Thomas et al., 2009). In a study on determinants of erlotinib disposition and toxicity in 80 patients, Rudin et al. showed that a diplotype of two polymorphic loci in the ABCG2 promoter involving −15622C>T and 1143C>T was associated with a higher erlotinib AUC (Rudin et al., 2008). In contrast to the study by Thomas et al. (2009), in this study the ABCG2 c.421C>A polymorphism was not associated with erlotinib disposition (Rudin et al., 2008).
Gefitinib
Gefitinib is not a substrate of OCT1 and OCT2, which was shown using transporter-overexpressing HEK293 cells (Galetti et al., 2010). In vitro studies using MDCKII cells showed that human P-glycoprotein effectively transports gefitinib (Agarwal et al., 2010). Gefitinib was also efficiently transported by mouse Bcrp in MDCK-Bcrp monolayers (Agarwal et al., 2010). Stewart et al. (2004) and Nakamura et al. (2005) reported that gefitinib is not a substrate of human BCRP, whereas Li et al. (2007) detected a significantly lower gefitinib accumulation in BCRP overexpressing HEK cells at lower concentrations. In vivo studies in knockout mice or using P-glycoprotein/Bcrp inhibitors revealed that transport of gefitinib across the blood–brain barrier is significantly limited by P-glycoprotein and Bcrp (Kawamura et al., 2009; Agarwal et al., 2010). Steady-state brain-to-plasma concentration ratios were 70-fold higher in the Abcb1a/1b(–/–) Abcg2(–/–) mice than in wild-type mice (Agarwal et al., 2010). Brain-to-plasma concentration ratios after oral administration of gefitinib were also significantly higher in P-glycoprotein-deficient, Bcrp expressing animals compared with wild-type animals, whereas the absence of Bcrp alone did not affect gefitinib brain-to-plasma concentration ratios (Agarwal et al., 2010).
Among the organic cation transporters gefitinib inhibited the MATE2-K-mediated transport of metformin with the greatest potency (Minematsu and Giacomini, 2011). Moreover, gefitinib inhibits OCT1- and OCT2-mediated 1-methyl-4-phenylpyridinium (MPP+) uptake (Galetti et al., 2010). Kitazaki et al. showed that gefitinib reverses the P-glycoprotein-mediated resistance to paclitaxel and docetaxel in a dose-dependent manner, indicating that gefitinib inhibits P-glycoprotein (Kitazaki et al., 2005). Moreover, gefitinib inhibited the BCRP-mediated topotecan transport in inside-out membrane vesicles of PC-6/SN2-5H cells with an inhibition constant (Ki) value of 1.0 µM (Nakamura et al., 2005). In human BCRP-transfected erythromyeloblastoid leukaemia cells (K562), gefitinib inhibited the BCRP-mediated transport of estrone-3-sulfate (Yanase et al., 2004). In addition, gefitinib increased the accumulation of topotecan in K562/BCRP cells (Yanase et al., 2004). Yang et al. reported that gefitinib reverses the resistance to paclitaxel in human lung adenocarcinoma cells (CL1/Pac) and to doxorubicin in human breast adenocarcinoma cells (MCF7/Adr) by inhibition of P-glycoprotein and to topotecan in MCF7/TPT and CL1/Tpt cells by inhibition of BCRP (Yang et al., 2005). In vivo, gefitinib increased the oral bioavailability of irinotecan after concomitant administration in mice (Stewart et al., 2004). Additionally, gefitinib coadministration further decreased the systemic clearance of topotecan in Abcb1a/1b(–/–) and Abcg2(–/–) mice, indicating that additional transporters were inhibited (Leggas et al., 2006). It also increased topotecan brain penetration in a mouse model (Zhuang et al., 2006). Furthermore, Ozvegy-Laczka et al. (Ozvegy-Laczka et al., 2004) used human myelomonocytic cells (HL60/PLB) overexpressing BCRP, P-glycoprotein or MRP1 to determine mitoxantrone accumulation with and without addition of gefitinib. The tyrosine kinase inhibitor led to a significant increase in mitoxantrone accumulation in the BCRP-expressing HL60/PLB cells, whereas the effect was considerably smaller with the P-glycoprotein- and MRP1-expressing HL60/PLB cells (Ozvegy-Laczka et al., 2004). Very recently, Huang et al. (2011) reported that nuclear translocation of the epidermal growth factor receptor by AKT-dependent phosphorylation enhances BCRP expression in gefitinib-resistant cells, thus providing insights into one potential molecular mechanism contributing to gefitinib-resistance via BCRP expression.
In humans, dose-normalized plasma concentrations following multiple doses of gefitinib were significantly higher in patients heterozygous for the ABCG2 c.421C>A polymorphism, whereas no significant effects were observed for the ABCB1 c.3435C>T polymorphism (Li et al., 2007). In a recent study in 94 patients with non-small-cell lung cancer treated with gefitinib ABCG2 polymorphisms were not associated with outcome (Lemos et al., 2011). However, the ABCG2 polymorphism −15622C>T and the ABCG2 (c.1143C>T, −15622C>T) haplotype were associated with gefitinib-dependent, moderate-to-severe diarrhea (Lemos et al., 2011). In another study, an association between the ABCG2 c.421C>A polymorphism and diarrhea in patients with locally advanced or metastatic non-small-cell lung cancer treated with gefitinib was reported (Cusatis et al., 2006). In contrast, Akasaka et al. did not find any association between ABCG2 polymorphisms (c.376C>T, c.421C>A) and gefitinib-induced adverse events in Japanese patients with non-small-cell lung cancer (Akasaka et al., 2010).
Imatinib
Among all drugs discussed in this review, there is the largest amount of data available for imatinib, which is also discussed in detail in a recent review by Eechoute et al., (2011b). It was reported by Thomas et al. that the uptake of imatinib is mediated by OCT1, because inhibitors of OCT1 significantly decreased imatinib uptake into human leukaemic lymphoblast cells (CEM, Thomas et al., 2004). In addition, the intracellular uptake and retention of imatinib was 20% higher in transfected HEK293 cells overexpressing OCT1, indicating that imatinib is only moderately transported by OCT1 (Hu et al., 2008). Furthermore, Wang et al. showed that OCT1 transported imatinib in human chronic myelogenous leukaemia (CML) cells (KCL-22) overexpressing OCT1 (Wang et al., 2008). Ex vivo, the addition of prazosin, a potent inhibitor of OCT1, reduced the intracellular uptake of imatinib into mononuclear cells (White et al., 2006). OCT2 and OCT3 did not transport imatinib in vitro (Thomas et al., 2004). In contrast, significantly higher uptake rates for imatinib were found in cells transfected with OATP1A2 (Xenopus laevis oocytes), OATP1B3 (Xenopus laevis oocytes) and OCTN2 (HEK293 cells; Hu et al., 2008). Imatinib was not transported by OATP1B1 (Xenopus laevis oocytes), OCT2, OCT3, OAT1, OAT2, OAT3 and OCTN1 [all expressed in HEK293 cells (Hu et al., 2008)]. Recently, Eechoute et al. showed that imatinib is transported in Xenopus laevis oocytes and HeLa cells expressing OATP1A2 and this transport could be inhibited by rosuvastatin (Eechoute et al., 2011a,b). First evidence that imatinib is a substrate of P-glycoprotein was provided by Hegedus et al. in 2002 (Hegedus et al., 2002). Mahon et al. showed that a K562/DOX cell line overexpressing P-glycoprotein exhibited a reduced sensitivity to imatinib compared with the parental K562 cells (Mahon et al., 2003). Studies using transfected MDCK cell lines revealed an active efflux component for imatinib attributable to P-glycoprotein (Thomas et al., 2004). Hu et al. reported a weak but statistically significant interaction between imatinib and MRP4 (Hu et al., 2008). In addition, imatinib was transported by BCRP in BCRP-overexpressing HEK293 and MCF7/MR cells (Burger et al., 2004). Furthermore, Breedveld et al. showed that imatinib is transported by mouse Bcrp in MDCKII cells (Breedveld et al., 2005). For imatinib, the brain-to-plasma ratios in Abcg2(–/–) mice were comparable with those in wild-type mice, whereas the brain-to-plasma ratios in Abcb1a/1b(–/–) and Abcb1a/1b(–/–) Abcg2(–/–) mice were more than 4- and 28-fold of those in wild-type mice, respectively (Zhou et al., 2009). Dai et al. showed that the brain-to-plasma ratio in the Abcb1a/1b(–/–) mice was approximately sevenfold greater than that of wild-type mice, indicating that imatinib is a substrate of P-glycoprotein (Dai et al., 2003).
Imatinib inhibited metformin uptake by MATE1, MATE2-K and OCT1 at potentially clinically relevant concentrations (Minematsu and Giacomini, 2011). The Ki value for the inhibition of P-glycoprotein function by imatinib was estimated to be 18.3 µM using a calcein-AM efflux assay in P-glycoprotein overexpressing pig kidney epithelial cells (LLC-PK1, Hamada et al., 2003). Houghton et al. showed that imatinib significantly reversed BCRP-mediated resistance to topotecan and SN-38 and significantly increased accumulation of topotecan only in BCRP-expressing human osteosarcoma cells (Saos2, Houghton et al., 2004). Imatinib inhibited BCRP-mediated mitoxantrone efflux in MCF7 and HEK293 cell lines overexpressing BCRP (Burger et al., 2004). Furthermore, imatinib inhibited the MRP7-mediated efflux of paclitaxel in HEK293 cells (Shen et al., 2009). The cellular uptake of nilotinib was increased by coadministration of imatinib in vitro due to P-glycoprotein and BCRP inhibition (White et al., 2007b).
Multiple studies have been performed to elucidate associations between polymorphisms in genes encoding for drug metabolizing enzymes and drug transporters and pharmacokinetic parameters and/or clinical outcome (Table 4). It should be noted that in several studies the mean plasma concentrations or median minimum or ‘trough’ concentration (Cmin) of imatinib were higher in responders compared with non-responders in the treatment of CML and GI stromal tumours [GIST; for review see (Eechoute et al., 2011b)]. This indicates that the systemic concentrations of imatinib are correlated with the treatment outcome.
Very recently, Yamakawa et al. (2011) reported a significantly lower clearance of imatinib in patients with CML with SLCO1A2−361GG genotype compared with patients with −361 GA or AA genotypes. The pharmacogenetic data for the association of SLC22A1 polymorphisms (SLC22A1 encodes for OCT1) and pharmacodynamics of imatinib are inconsistent. One study demonstrated an increased risk for imatinib resistance due to loss of response and treatment failure in patients with CML, who are carriers of the c.480GG genotype in SLC22A1 (Kim et al., 2009), whereas a second study did not find any influence of this polymorphism on the major molecular response (Takahashi et al., 2010). In the later study the c.1222GG genotype in SLC22A1 was associated with higher rates of major molecular response in the treatment of CML with imatinib (Takahashi et al., 2010).
In the last years different approaches were chosen to predict determinants of outcome in imatinib-treated patients. OCT1 was identified as a promising factor influencing the clinical outcome in the treatment with imatinib. A study by White et al. (2006) indicated that the intrinsic activity (defined as the in vitro concentration of drug required to reduce the phosphorylation of the adaptor protein Crkl by 50%) of newly diagnosed patients with CML to imatinib correlates with the molecular response. The intrinsic activity was mainly dependent from the intracellular uptake and retention of imatinib (White et al., 2006). The uptake of imatinib into mononuclear cells was attributed to OCT1, because the uptake and retention was reduced by the OCT1 inhibitor prazosin (White et al., 2006). Subsequent studies showed that the function or mRNA expression of OCT1 was associated with response to imatinib in patients with CML (White et al., 2007a, 2010a,b; Wang et al., 2008). It was consistently shown that a high OCT1 expression or function is related to a better response to imatinib treatment in CML patients compared with patients with a lower OCT1 expression or function. These studies suggest an association between the OCT1 expression and function with the prognosis in CML patients treated with imatinib. However, the underlying mechanism seems to be still not completely clarified, because a study by Hu and colleagues indicated that imatinib is only marginally transported by OCT1 (Hu et al., 2008). This study also showed that the SLC22A1 expression in leukaemia cell lines was interrelated with SLCO1A2, ABCB1 and ABCG2 mRNA expression (Hu et al., 2008). Further studies are necessary to completely clarify the mechanism of OCT1-associated response to treatment with imatinib.
As mentioned above, imatinib is a substrate of P-glycoprotein and BCRP. Therefore, polymorphisms in the ABCB1 and/or ABCG2 genes could influence the intestinal absorption and elimination pathways. For BCRP, two studies revealed inconclusive associations between the c.421C>A polymorphism and clinical endpoints in the treatment of CML. In one study the c.421CC genotype was associated with a decreased complete molecular response, whereas a second study showed no relationship between this polymorphism and major molecular response (Takahashi et al., 2010). In a further study with patients taking imatinib for the treatment of GIST no influence of the c.421C>A polymorphism on the oral clearance of imatinib was observed (Gardner et al., 2006). Due to these results it can be concluded that additional studies enrolling a higher number of patients are needed in order to clarify the clinical relevance of this ABCG2 c.421C>A polymorphism for the treatment of CML and GIST with imatinib.
The pharmacogenetic studies with regard to ABCB1 polymorphisms and treatment outcome in CML patients are difficult to compare because the clinical endpoints and the investigated polymorphisms differ. Nevertheless, two studies revealed comparable results with respect to the c.3435C>T polymorphism (Kim et al., 2009; Ni et al., 2011). In a univariate analysis the c.3435TT genotype was associated with a decreased overall survival in patients with CML (Kim et al., 2009). The second study showed a lower resistance to imatinib for the c.3435CC genotype (Ni et al., 2011). Even though the analyses were performed either with patients with c.3435 wild-type genotype or homozygous carriers of the polymorphism (c.3435TT) and different clinical endpoints, both studies indicate that the c.3435C>T polymorphism impairs the response to imatinib treatment in CML.
Lapatinib
In vitro, lapatinib is a substrate of the efflux transporters P-glycoprotein and BCRP (Polli et al., 2008). Based on a GF120918-treated rat model, Polli et al. (2008) concluded that lapatinib disposition after oral administration is not affected when P-glycoprotein and Bcrp are absent. Similar to other tyrosine kinase inhibitors (e.g. dasatinib), brain penetration of lapatinib is affected by P-glycoprotein and Bcrp. It was shown using knockout mouse models that the brain-to-plasma concentration ratios in Abcb1a/1b and Abcb1a/1b Abcg2 knockout mice were three- to fourfold and 40-fold respectively, higher compared with wild-type mice, whereas there was no significant effect in Abcg2 knockout mice compared with wild-type mice (Polli et al., 2009).
In vitro data indicate that lapatinib is an inhibitor of OATP1B1 function at clinically relevant concentrations (Polli et al., 2008; Fachinformation, 2010). Currently, there are no data on the impact of lapatinib on plasma concentrations of OATP1B1 substrates in humans. In contrast, lapatinib had little effects on OAT1 to 4 and OCT1 to 3 (Polli et al., 2008; Minematsu and Giacomini, 2011). Lapatinib inhibits the efflux transporters P-glycoprotein, BCRP and MRP7 in vitro (Dai et al., 2008; Molina et al., 2008; Kuang et al., 2010; Perry et al., 2010). It was speculated that these properties might be advantageous for concomitant treatment of lapatinib with conventional chemotherapeutic drugs, whose effects are limited due to multidrug resistance mediated in part via transporter-mediated efflux. Molina et al. (2008) recently showed that the combination of lapatinib with the P-glycoprotein/BCRP substrate topotecan showed enhanced efficacy in human breast carcinoma xenografts. Moreover, lapatinib moderately reduced topotecan clearance in patients (Molina et al., 2008). In line with the inhibition of P-glycoprotein function by lapatinib in vitro, lapatinib increased the AUC after oral administration of the P-glycoprotein substrate digoxin by 80% (Fachinformation, 2010).
Nilotinib
Nilotinib is not transported by OCT1 (White et al., 2006; Davies et al., 2009). There are conflicting data whether nilotinib is a substrate of BCRP or P-glycoprotein. Mahon et al. reported by reversing the resistance of K562/DOX cells to nilotinib with verapamil or PSC833 that nilotinib is a substrate of P-glycoprotein (Mahon et al., 2008). Haouala et al. (2010), however, did not observe an impact of P-glycoprotein silencing on cellular nilotinib disposition. Hegedus et al. (2009) reported that nilotinib is a high-affinity substrate of BCRP. Brendel et al. (2007) described nilotinib as a modest BCRP substrate. Finally, the data from Davies et al. (2009) indicate that nilotinib is not transported by BCRP, MRP1 and P-glycoprotein.
OCT3-mediated metformin uptake in HEK293 cells was potently inhibited by nilotinib with an IC50 value of 0.345 µM (Minematsu and Giacomini, 2011). Nilotinib was also an inhibitor of OCT1 (Davies et al., 2009; Minematsu and Giacomini, 2011), but probably at clinically less relevant concentrations (Minematsu and Giacomini, 2011). Nilotinib inhibited the BCRP/Bcrp-mediated Hoechst 33342 dye efflux from primary human and murine haematopoietic stem cells (HSCs; Brendel et al., 2007). Several groups showed that nilotinib is an inhibitor of BCRP and P-glycoprotein (Davies et al., 2009; Tiwari et al., 2009; Dohse et al., 2010). Nilotinib was a more potent inhibitor of BCRP and P-glycoprotein compared with imatinib and dasatinib (Dohse et al., 2010). Hiwase et al. reported that inhibition of P-glycoprotein by nilotinib increased dasatinib accumulation in CML cells with potential implications for combination therapy with tyrosine kinase inhibitors (Hiwase et al., 2010). Similar to imatinib, nilotinib reversed MRP7-mediated paclitaxel resistance, most likely due to inhibition of MRP7-mediated paclitaxel efflux (Shen et al., 2009).
Pazopanib
In vitro studies indicate that pazopanib is a substrate of BCRP and P-glycoprotein [US Food and Drug Administration (FDA), 2010b]. The uptake transporter OATP1B1 is potently inhibited by pazopanib with an IC50 value of 0.79 µM and may therefore increase serum concentrations of concomitantly administered OATP1B1 substrates such as 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA) reductase inhibitors (FDA, 2010b; Keisner and Shah, 2011). Coadministration of lapatinib, a weak inhibitor of CYP3A4 and an inhibitor of P-glycoprotein and BCRP, with pazopanib resulted in a 50–60% increase in mean pazopanib AUC compared with the administration of pazopanib alone (FDA, 2010b).
Sorafenib
Sorafenib is highly permeable (Gnoth et al., 2010) and in vitro uptake of sorafenib is not affected by major OATPs, OCT1, OAT2, OAT3 and OCTNs (Hu et al., 2009). Sorafenib is a weak P-glycoprotein substrate in vitro (Hu et al., 2009; Gnoth et al., 2010; Haouala et al., 2010; Lagas et al., 2010; Agarwal et al., 2011), but is more efficiently transported by BCRP/Bcrp (Lagas et al., 2010; Agarwal et al., 2011). In knockout mouse models, plasma concentrations of sorafenib were largely unaffected in the absence of P-glycoprotein and/or Bcrp (Gnoth et al., 2010; Lagas et al., 2010; Agarwal et al., 2011). Sorafenib brain concentrations increased to some extent in P-glycoprotein deficient animals compared with wild-type mice, but the increase was considerably higher in the absence of Bcrp with the most pronounced effect in P-glycoprotein/Bcrp-deficient animals (Hu et al., 2009; Gnoth et al., 2010; Lagas et al., 2010; Agarwal et al., 2011; Asakawa et al., 2011). This observed interplay of P-glycoprotein and Bcrp in vivo was recently also observed in double-transfected MDCK-BCRP-P-glycoprotein cells (Poller et al., 2011). Sorafenib was also reported to be a substrate of MRP2 in one study (Shibayama et al., 2011), which might play a role for anticancer drug resistance to sorafenib, but no transport of sorafenib by MRP2 (and by BCRP and MRP4) was found in another study (Hu et al., 2009). Sorafenib inhibits P-glycoprotein, MRP2 and MRP4 function in vitro, whereas BCRP inhibition by sorafenib appears to be substrate-dependent (Hu et al., 2009; Agarwal et al., 2011).
Sunitinib
Similar to sorafenib, in vitro uptake of sunitinib was not mediated by major uptake transporters (OATPs, OCT1, OAT2, OAT3 and OCTNs; Hu et al., 2009). In vitro, sunitinib is a substrate of P-glycoprotein, BCRP, possibly of MRP4 and a good substrate of Bcrp, but it is not transported by MRP2 (Hu et al., 2009; Shibayama et al., 2011; Tang et al., 2011). In mice, brain sunitinib accumulation is restricted by P-glycoprotein (Hu et al., 2009) and Bcrp and could be enhanced by the dual P-glycoprotein/Bcrp inhibitor elacridar (Tang et al., 2011).
Sunitinib inhibits P-glycoprotein and BCRP function in vitro with possible consequences for bioavailability of coadministered drugs and for reversing efflux transporter-mediated multidrug resistance in humans (Dai et al., 2009; Hu et al., 2009; Shukla et al., 2009; Kawahara et al., 2010). Interestingly, a germ-line mutation in ABCG2 (c.1291T>C) is almost insensitive to sunitinib-mediated inhibition in a cell proliferation assay (Kawahara et al., 2010).
In a small study it was reported that the ABCG2 c.421AA genotype, which is associated with higher plasma concentrations of several drugs (Poguntke et al., 2010), was also associated with higher sunitinib concentrations in a patient with renal cell carcinoma compared with patients having the CA or CC genotype (Mizuno et al., 2010). van der Veldt et al. (2011) reported in a recent retrospective pharmacogenetic association study in 136 patients with clear-cell metastatic renal cell carcinoma that the TCG haplotype of ABCB1 (c.3435C>T, c.1236C>T, c.2677G>T) was together with variants in CYP3A5 and NR1I3 significantly associated with improved progression-free survival. In addition there was a trend for improved outcome in the presence of the A allele of the ABCG2 c.34G>A variant. Moreover, van Erp et al. (2009a) reported in a study with 219 sunitinib treated patients that prevalence of any toxicity higher than grade 2 according to the National Cancer Institute Common Toxicity Criteria was increased in patients with a copy of TT in ABCG2 (-15622C>T, c.1143C>T) haplotype (OR = 2.63, P = 0.016). Moreover, the prevalence of hand-foot syndrome was increased when a copy of TTT in the ABCB1 (c.3435C>T, c.1236C>T, c.2677G>T) haplotype (OR = 2.56; P < 0.035) was present (van Erp et al., 2009a).
Everolimus
The mTOR inhibitor everolimus is a substrate of CYP3A4 and of P-glycoprotein (Crowe and Lemaire, 1998; FDA, 2011), but not of OATP uptake transporters (OATP1A2, OATP1B1, OATP1B3; Picard et al., 2011). AUC after oral administration of everolimus to P-glycoprotein-deficient mice was increased 1.3-fold compared with wild-type animals (Chu et al., 2009). In P-glycoprotein expressing mice the tyrosine kinase inhibitor lapatinib increased everolimus AUC 2.6-fold, in part due to reducing intestinal P-glycoprotein expression (Chu et al., 2009).
In vitro, everolimus was an inhibitor of OATP1A2, OATP1B1 and OATP1B3 function determined by uptake of prototypical substrates such as estrone sulphate and mycophenolic acid 7-O-glucuronide with IC50 values in the low micromolar range (Picard et al., 2011). In healthy volunteers a single oral dose of 2 mg everolimus had no influence on the AUC (Kovarik et al., 2002) of the OATP1B1 and OATP1B3 substrate pravastatin (20 mg; Seithel et al., 2007; Fahrmayr et al., 2010). It should be noted, however, that the recommended daily dose of everolimus in cancer patients is 10 mg per day, that is, it cannot be excluded that everolimus at this higher dose used in cancer patients (and during steady-state) has effects on pharmacokinetics of pravastatin and potentially of other OATP substrates.
Temsirolimus
Temsirolimus and sirolimus, its principal metabolite in humans after intravenous administration, are substrates of CYP3A4 (FDA, 2010a). Sirolimus is a substrate of P-glycoprotein (Crowe and Lemaire, 1998). Similar to everolimus, sirolimus is an inhibitor, but not a substrate of OATP uptake transporters (OATP1A2, OATP1B1, OATP1B3; Picard et al., 2011).
In vitro data indicate that temsirolimus is an inhibitor of P-glycoprotein (Fachinformation, 2011). Zimmerman et al. reported that sirolimus did not significantly affect plasma concentrations of the P-glycoprotein substrate digoxin in healthy volunteers (Zimmerman, 2004). Currently, there are no data available regarding the impact of temsirolimus on plasma concentrations of digoxin or of the OATP substrate pravastatin in humans.
Bortezomib
Very limited data are currently available regarding the interaction of the proteasome inhibitor bortezomib with drug transporters. In vitro data from Rumpold et al. (2007) indicate that bortezomib is a moderate substrate of P-glycoprotein. In a subgroup of patients with advanced multiple myeloma treated with bortezomib alone, no association between outcome and ABCB1 or ABCC1 polymorphisms was found (Buda et al., 2010).
Conclusions
In vitro data indicate that most of the small molecules discussed in this review are substrates of the efflux transporters BCRP and/or P-glycoprotein. The relevance of Bcrp and P-glycoprotein for brain concentrations was clearly highlighted using knockout mouse models. Intracellular concentrations of some tyrosine kinase inhibitors might also depend on uptake transporters. In vitro data also indicate that the majority of the small molecules inhibit uptake and/or efflux transporters with potential consequences for the occurrence of drug–drug interactions. For some of the tyrosine kinase inhibitors pharmacogenetic association studies were conducted, showing an impact of certain polymorphisms in genes encoding drug transporters with disposition and effects. Considerably more data are necessary, to show how this interaction of small molecules with drug transporters is relevant for the clinical situation. This relates to clinical investigations of transporter-mediated drug interactions (e.g. with metformin), the importance of efflux transporter inhibition by small molecules for the reversal of multidrug resistance and on pharmacogenetic factors determining interindividual differences in efficacy and toxicity of the new small molecules.
Acknowledgments
Our work on drug transporters is supported by grants of the Deutsche Forschungsgemeinschaft (DFG Fr 1298/5–1; DFG GL 588/3–1), the German Ministry of Education and Research (BMBF 01EX1015B), the ‘Johannes and Frieda Marohn-Stiftung an der Friedrich-Alexander-Universität Erlangen-Nürnberg’ and the DOKTOR ROBERT PFLEGER-STIFTUNG Bamberg.
Glossary
- ABC
ATP-binding cassette
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- CL1
human lung adenocarcinoma cell line
- Cmin
minimum or ‘trough’ concentration
- CEM
human leukaemic lymphoblast cell line
- CML
chronic myeloid leukaemia
- CYP
cytochrome P450
- E217βG
estradiole-17β-glucuronide
- FDA
US Food and Drug Administration
- GI
gastrointestinal
- GIST
gastrointestinal stromal tumour
- HEK293
human embryonic kidney cell line
- HL60
human myelomonocytic cell line
- HMG-CoA
3-hydroxy-3-methyl-glutaryl-CoA reductase
- HSC
haematopoietic stem cell
- K562
human erythromyeloblastoid leukaemia cell line
- KCL-22
chronic myelogenous leukaemia cell line
- Ki
inhibition constant
- LLC-PK1
pig kidney epithelial cell line
- MATE
multidrug and toxin extrusion protein
- MCF7
human breast adenocarcinoma cell line
- MDCKII
Madin-Darby canine kidney cell line
- MDR1
multidrug resistance protein 1
- MPP+
1-methyl-4-phenylpyridinium
- MRP
multidrug resistance protein
- mTOR
mammalian target of rapamycin
- OAT
organic anion transporter
- OATP
organic anion transporting polypeptide
- OCT
organic cation transporter
- PC-6
human small cell lung cancer cell line
- Saos2
human osteosarcoma cell line
- SLCO
solute carrier gene family encoding for OATPs
Conflict of interest
M.F.F. received consulting fees from AstraZeneca, Bayer Schering Pharma, Boehringer Ingelheim and Merck KGaA and lecture fees from Bayer Schering Pharma, Ferring and Novartis.
References
- Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther. 2010;334:147–155. doi: 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223–233. doi: 10.1124/jpet.110.175034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasaka K, Kaburagi T, Yasuda S, Ohmori K, Abe K, Sagara H, et al. Impact of functional ABCG2 polymorphisms on the adverse effects of gefitinib in Japanese patients with non-small-cell lung cancer. Cancer Chemother Pharmacol. 2010;66:691–698. doi: 10.1007/s00280-009-1211-6. [DOI] [PubMed] [Google Scholar]
- Asakawa C, Ogawa M, Kumata K, Fujinaga M, Kato K, Yamasaki T, et al. [11C]sorafenib: radiosynthesis and preliminary PET study of brain uptake in P-gp/Bcrp knockout mice. Bioorg Med Chem Lett. 2011;21:2220–2223. doi: 10.1016/j.bmcl.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14:352–359. doi: 10.1158/1078-0432.CCR-07-4175. [DOI] [PubMed] [Google Scholar]
- Breedveld P, Pluim D, Cipriani G, Wielinga P, van Tellingen O, Schinkel AH, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005;65:2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- Brendel C, Scharenberg C, Dohse M, Robey RW, Bates SE, Shukla S, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21:1267–1275. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- Buda G, Ricci D, Huang CC, Favis R, Cohen N, Zhuang SH, et al. Polymorphisms in the multiple drug resistance protein 1 and in P-glycoprotein 1 are associated with time to event outcomes in patients with advanced multiple myeloma treated with bortezomib and pegylated liposomal doxorubicin. Ann Hematol. 2010;89:1133–1140. doi: 10.1007/s00277-010-0992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger H, van Tol H, Boersma AW, Brok M, Wiemer EA, Stoter G, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104:2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- Chen Y, Agarwal S, Shaik NM, Chen C, Yang Z, Elmquist WF. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J Pharmacol Exp Ther. 2009;330:956–963. doi: 10.1124/jpet.109.154781. [DOI] [PubMed] [Google Scholar]
- Chu C, Abbara C, Noël-Hudson MS, Thomas-Bourgneuf L, Gonin P, Farinotti R, et al. Disposition of everolimus in mdr1a-/1b- mice and after a pre-treatment of lapatinib in Swiss mice. Biochem Pharmacol. 2009;77:1629–1634. doi: 10.1016/j.bcp.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Crowe A, Lemaire M. In vitro and in situ absorption of SDZ-RAD using a human intestinal cell line (Caco-2) and a single pass perfusion model in rats: comparison with rapamycin. Pharm Res. 1998;15:1666–1672. doi: 10.1023/a:1011940108365. [DOI] [PubMed] [Google Scholar]
- Cusatis G, Gregorc V, Li J, Spreafico A, Ingersoll RG, Verweij J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–1742. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–7914. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Liang YJ, Wang YS, Tiwari AK, Yan YY, Wang F, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009;279:74–83. doi: 10.1016/j.canlet.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Dai H, Marbach P, Lemaire M, Hayes M, Elmquist WF. Distribution of STI-571 to the brain is limited by P-glycoprotein-mediated efflux. J Pharmacol Exp Ther. 2003;304:1085–1092. doi: 10.1124/jpet.102.045260. [DOI] [PubMed] [Google Scholar]
- Davies A, Jordanides NE, Giannoudis A, Lucas CM, Hatziieremia S, Harris RJ, et al. Nilotinib concentration in cell lines and primary CD34(+) chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia. 2009;23:1999–2006. doi: 10.1038/leu.2009.166. [DOI] [PubMed] [Google Scholar]
- de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs. 2010 doi: 10.1007/s10637-010-9569-1. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab Dispos. 2010;38:1371–1380. doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett DR, Cameron MD. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin Drug Metab Toxicol. 2010;6:1175–1193. doi: 10.1517/17425255.2010.506873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulucq S, Bouchet S, Turcq B, Lippert E, Etienne G, Reiffers J, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2008;112:2024–2027. doi: 10.1182/blood-2008-03-147744. [DOI] [PubMed] [Google Scholar]
- Eechoute K, Franke RM, Loos WJ, Scherkenbach LA, Boere I, Verweij J, et al. Environmental and genetic factors affecting transport of imatinib by OATP1A2. Clin Pharmacol Ther. 2011a;89:816–820. doi: 10.1038/clpt.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eechoute K, Sparreboom A, Burger H, Franke RM, Schiavon G, Verweij J, et al. Drug transporters and imatinib treatment: implications for clinical practice. Clin Cancer Res. 2011b;17:406–415. doi: 10.1158/1078-0432.CCR-10-2250. [DOI] [PubMed] [Google Scholar]
- Elmeliegy MA, Carcaboso AM, Tagen M, Bai F, Stewart CF. Role of ATP-binding cassette and solute carrier transporters in erlotinib CNS penetration and intracellular accumulation. Clin Cancer Res. 2011;17:89–99. doi: 10.1158/1078-0432.CCR-10-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinformation. 2010. (‘German Summary of Product Characteristics’) Drug label Tyverb®, 07/2010.
- Fachinformation. 2011. (‘German Summary of Product Characteristics’) Drug label Torisel®, 01/2011.
- Fahrmayr C, Fromm MF, König J. Hepatic OATP and OCT uptake transporters: their role for drug-drug interactions and pharmacogenetic aspects. Drug Metab Rev. 2010;42:380–401. doi: 10.3109/03602530903491683. [DOI] [PubMed] [Google Scholar]
- FDA. 2010a. Drug label Torisel®, 09/07/2010, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/ 022088s008lbl.pdf; last accessed 19/08/2011.
- FDA. 2010b. Drug label Votrient®, 27/04/2010, http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/ 022465s002lbl.pdf; last accessed 19/08/2011.
- FDA. 2011. Drug label Afinitor®, 05/05/2011, http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/ 022344s9s10lbl.pdf; last accessed 19/08/2011.
- Fromm MF. Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol Sci. 2004;25:423–429. doi: 10.1016/j.tips.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Funk C. The role of hepatic transporters in drug elimination. Expert Opin Drug Metab Toxicol. 2008;4:363–379. doi: 10.1517/17425255.4.4.363. [DOI] [PubMed] [Google Scholar]
- Galetti M, Alfieri RR, Cavazzoni A, La Monica S, Bonelli M, Fumarola C, et al. Functional characterization of gefitinib uptake in non-small cell lung cancer cell lines. Biochem Pharmacol. 2010;80:179–187. doi: 10.1016/j.bcp.2010.03.033. [DOI] [PubMed] [Google Scholar]
- Gardner ER, Burger H, van Schaik RH, van Oosterom AT, de Bruijn EA, Guetens G, et al. Association of enzyme and transporter genotypes with the pharmacokinetics of imatinib. Clin Pharmacol Ther. 2006;80:192–201. doi: 10.1016/j.clpt.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Giannoudis A, Davies A, Lucas CM, Harris RJ, Pirmohamed M, Clark RE. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood. 2008;112:3348–3354. doi: 10.1182/blood-2007-10-116236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnoth MJ, Sandmann S, Engel K, Radtke M. In vitro to in vivo comparison of the substrate characteristics of sorafenib tosylate toward P-glycoprotein. Drug Metab Dispos. 2010;38:1341–1346. doi: 10.1124/dmd.110.032052. [DOI] [PubMed] [Google Scholar]
- Gurney H, Wong M, Balleine RL, Rivory LP, McLachlan AJ, Hoskins JM, et al. Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clin Pharmacol Ther. 2007;82:33–40. doi: 10.1038/sj.clpt.6100201. [DOI] [PubMed] [Google Scholar]
- Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P-glycoprotein. J Pharmacol Exp Ther. 2003;307:824–828. doi: 10.1124/jpet.103.055574. [DOI] [PubMed] [Google Scholar]
- Haouala A, Rumpold H, Untergasser G, Buclin T, Ris HB, Widmer N, et al. siRNA-mediated knock-down of P-glycoprotein expression reveals distinct cellular disposition of anticancer tyrosine kinases inhibitors. Drug Metab Lett. 2010;4:114–119. doi: 10.2174/187231210791292726. [DOI] [PubMed] [Google Scholar]
- Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors – a review on pharmacology, metabolism and side effects. Curr Drug Metab. 2009;10:470–481. doi: 10.2174/138920009788897975. [DOI] [PubMed] [Google Scholar]
- Hegedus C, Ozvegy-Laczka C, Apáti A, Magócsi M, Német K, Orfi L, et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. Br J Pharmacol. 2009;158:1153–1164. doi: 10.1111/j.1476-5381.2009.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus T, Orfi L, Seprodi A, Váradi A, Sarkadi B, Kéri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta. 2002;1587:318–325. doi: 10.1016/s0925-4439(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Hiwase DK, Saunders V, Hewett D, Frede A, Zrim S, Dang P, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008;14:3881–3888. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- Hiwase DK, White D, Zrim S, Saunders V, Melo JV, Hughes TP. Nilotinib-mediated inhibition of ABCB1 increases intracellular concentration of dasatinib in CML cells: implications for combination TKI therapy. Leukemia. 2010;24:658–660. doi: 10.1038/leu.2009.242. [DOI] [PubMed] [Google Scholar]
- Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78:260–277. doi: 10.1016/j.clpt.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Houghton PJ, Germain GS, Harwood FC, Schuetz JD, Stewart CF, Buchdunger E, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Res. 2004;64:2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- Hu S, Franke RM, Filipski KK, Hu C, Orwick SJ, de Bruijn EA, et al. Interaction of imatinib with human organic ion carriers. Clin Cancer Res. 2008;14:3141–3148. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, et al. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res. 2009;15:6062–6069. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem. 2011;286:20558–20568. doi: 10.1074/jbc.M111.240796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara H, Noguchi K, Katayama K, Mitsuhashi J, Sugimoto Y. Pharmacological interaction with sunitinib is abolished by a germ-line mutation (1291T>C) of BCRP/ABCG2 gene. Cancer Sci. 2010;101:1493–1500. doi: 10.1111/j.1349-7006.2010.01539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura K, Yamasaki T, Yui J, Hatori A, Konno F, Kumata K, et al. In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [11C]gefitinib. Nucl Med Biol. 2009;36:239–246. doi: 10.1016/j.nucmedbio.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Keisner SV, Shah SR. Pazopanib: the newest tyrosine kinase inhibitor for the treatment of advanced or metastatic renal cell carcinoma. Drugs. 2011;71:443–454. doi: 10.2165/11588960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011;(201):299–323. doi: 10.1007/978-3-642-14541-4_8. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sriharsha L, Xu W, Kamel-Reid S, Liu X, Siminovitch K, et al. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin Cancer Res. 2009;15:4750–4758. doi: 10.1158/1078-0432.CCR-09-0145. [DOI] [PubMed] [Google Scholar]
- Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49:337–343. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Kodaira H, Kusuhara H, Ushiki J, Fuse E, Sugiyama Y. Kinetic analysis of the cooperation of P-glycoprotein (P-gp/Abcb1) and breast cancer resistance protein (Bcrp/Abcg2) in limiting the brain and testis penetration of erlotinib, flavopiridol, and mitoxantrone. J Pharmacol Exp Ther. 2010;333:788–796. doi: 10.1124/jpet.109.162321. [DOI] [PubMed] [Google Scholar]
- König J. Uptake transporters of the human OATP family: molecular characteristics, substrates, their role in drug-drug interactions, and functional consequences of polymorphisms. Handb Exp Pharmacol. 2011;(201):1–28. doi: 10.1007/978-3-642-14541-4_1. [DOI] [PubMed] [Google Scholar]
- Kovarik JM, Hartmann S, Hubert M, Berthier S, Schneider W, Rosenkranz B, et al. Pharmacokinetic and pharmacodynamic assessments of HMG-CoA reductase inhibitors when coadministered with everolimus. J Clin Pharmacol. 2002;42:222–228. doi: 10.1177/00912700222011148. [DOI] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- Kuang YH, Shen T, Chen X, Sodani K, Hopper-Borge E, Tiwari AK, et al. Lapatinib and erlotinib are potent reversal agents for MRP7 (ABCC10)-mediated multidrug resistance. Biochem Pharmacol. 2010;79:154–161. doi: 10.1016/j.bcp.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagas JS, van Waterschoot RA, van Tilburg VA, Hillebrand MJ, Lankheet N, Rosing H, et al. Brain accumulation of dasatinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by elacridar treatment. Clin Cancer Res. 2009;15:2344–2351. doi: 10.1158/1078-0432.CCR-08-2253. [DOI] [PubMed] [Google Scholar]
- Lagas JS, van Waterschoot RA, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther. 2010;9:319–326. doi: 10.1158/1535-7163.MCT-09-0663. [DOI] [PubMed] [Google Scholar]
- Leggas M, Panetta JC, Zhuang Y, Schuetz JD, Johnston B, Bai F, et al. Gefitinib modulates the function of multiple ATP-binding cassette transporters in vivo. Cancer Res. 2006;66:4802–4807. doi: 10.1158/0008-5472.CAN-05-2915. [DOI] [PubMed] [Google Scholar]
- Lemos C, Giovannetti E, Zucali PA, Assaraf YG, Scheffer GL, van der Straaten T, et al. Impact of ABCG2 polymorphisms on the clinical outcome and toxicity of gefitinib in non-small-cell lung cancer patients. Pharmacogenomics. 2011;12:159–170. doi: 10.2217/pgs.10.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cusatis G, Brahmer J, Sparreboom A, Robey RW, Bates SE, et al. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther. 2007;6:432–438. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101:2368–2373. doi: 10.1182/blood.V101.6.2368. [DOI] [PubMed] [Google Scholar]
- Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68:9809–9816. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- Marchetti S, de Vries NA, Buckle T, Bolijn MJ, van Eijndhoven MA, Beijnen JH, et al. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1-/-/Mdr1a/1b-/- (triple-knockout) and wild-type mice. Mol Cancer Ther. 2008;7:2280–2287. doi: 10.1158/1535-7163.MCT-07-2250. [DOI] [PubMed] [Google Scholar]
- Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10:531–539. doi: 10.1158/1535-7163.MCT-10-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Terada T, Kamba T, Fukudo M, Katsura T, Nakamura E, et al. ABCG2 421C>A polymorphism and high exposure of sunitinib in a patient with renal cell carcinoma. Ann Oncol. 2010;21:1382–1383. doi: 10.1093/annonc/mdq150. [DOI] [PubMed] [Google Scholar]
- Molina JR, Kaufmann SH, Reid JM, Rubin SD, Gálvez-Peralta M, Friedman R, et al. Evaluation of lapatinib and topotecan combination therapy: tissue culture, murine xenograft, and phase I clinical trial data. Clin Cancer Res. 2008;14:7900–7908. doi: 10.1158/1078-0432.CCR-08-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F, Fromm MF. Transporter-mediated drug-drug interactions. Pharmacogenomics. 2011;12:1017–1037. doi: 10.2217/pgs.11.44. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Oka M, Soda H, Shiozawa K, Yoshikawa M, Itoh A, et al. Gefitinib (‘Iressa’, ZD1839), an epidermal growth factor receptor tyrosine kinase inhibitor, reverses breast cancer resistance protein/ABCG2-mediated drug resistance. Cancer Res. 2005;65:1541–1546. doi: 10.1158/0008-5472.CAN-03-2417. [DOI] [PubMed] [Google Scholar]
- Nambu T, Hamada A, Nakashima R, Yuki M, Kawaguchi T, Mitsuya H, et al. Association of SLCO1B3 polymorphism with intracellular accumulation of imatinib in leukocytes in patients with chronic myeloid leukemia. Biol Pharm Bull. 2011;34:114–119. doi: 10.1248/bpb.34.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni LN, Li JY, Miao KR, Qiao C, Zhang SJ, Qiu HR, et al. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Med Oncol. 2011;28:265–269. doi: 10.1007/s12032-010-9456-9. [DOI] [PubMed] [Google Scholar]
- Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Damme K, Schwab M. Organic cation transporters (OCTs, MATEs), in vitro and in vivo evidence for the importance in drug therapy. Handb Exp Pharmacol. 2011;(201):105–167. doi: 10.1007/978-3-642-14541-4_3. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Kawahara H, Kaji A, Katayama K, Mitsuhashi J, Sugimoto Y. Substrate-dependent bidirectional modulation of P-glycoprotein-mediated drug resistance by erlotinib. Cancer Sci. 2009;100:1701–1707. doi: 10.1111/j.1349-7006.2009.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozvegy-Laczka C, Hegedus T, Várady G, Ujhelly O, Schuetz JD, Váradi A, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Mol Pharmacol. 2004;65:1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- Perry J, Ghazaly E, Kitromilidou C, McGrowder EH, Joel S, Powles T. A synergistic interaction between lapatinib and chemotherapy agents in a panel of cell lines is due to the inhibition of the efflux pump BCRP. Mol Cancer Ther. 2010;9:3322–3329. doi: 10.1158/1535-7163.MCT-10-0197. [DOI] [PubMed] [Google Scholar]
- Picard N, Levoir L, Lamoureux F, Yee SW, Giacomini KM, Marquet P. Interaction of sirolimus and everolimus with hepatic and intestinal organic anion-transporting polypeptide transporters. Xenobiotica. 2011;41:752–757. doi: 10.3109/00498254.2011.573882. [DOI] [PubMed] [Google Scholar]
- Poguntke M, Hazai E, Fromm MF, Zolk O. Drug transport by breast cancer resistance protein. Expert Opin Drug Metab Toxicol. 2010;6:1363–1384. doi: 10.1517/17425255.2010.519700. [DOI] [PubMed] [Google Scholar]
- Poller B, Wagenaar E, Tang SC, Schinkel AH. Double-transduced MDCKII cells to study human P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) interplay in drug transport across the blood-brain barrier. Mol Pharm. 2011;8:571–582. doi: 10.1021/mp1003898. [DOI] [PubMed] [Google Scholar]
- Polli JW, Humphreys JE, Harmon KA, Castellino S, O'Mara MJ, Olson KL, et al. The role of efflux and uptake transporters in N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-Chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine; GW572016. Drug Metab Dispos. 2009;37:439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Liu W, Desai A, Karrison T, Jiang X, Janisch L, et al. Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity. J Clin Oncol. 2008;26:1119–1127. doi: 10.1200/JCO.2007.13.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpold H, Salvador C, Wolf AM, Tilg H, Gastl G, Wolf D. Knockdown of PgP resensitizes leukemic cells to proteasome inhibitors. Biochem Biophys Res Commun. 2007;361:549–554. doi: 10.1016/j.bbrc.2007.07.049. [DOI] [PubMed] [Google Scholar]
- zu Schwabedissen HE, Kroemer HK. In vitro and in vivo evidence for the importance of breast cancer resistance protein transporters (BCRP/MXR/ABCP/ABCG2) Handb Exp Pharmacol. 2011;(201):325–371. doi: 10.1007/978-3-642-14541-4_9. [DOI] [PubMed] [Google Scholar]
- Seithel A, Eberl S, Singer K, Auge D, Heinkele G, Wolf NB, et al. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos. 2007;35:779–786. doi: 10.1124/dmd.106.014407. [DOI] [PubMed] [Google Scholar]
- Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, et al. Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10) PLoS One. 2009;4:e7520. doi: 10.1371/journal.pone.0007520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–11020. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- Shibayama Y, Nakano K, Maeda H, Taguchi M, Ikeda R, Sugawara M, et al. Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol Pharm Bull. 2011;34:433–435. doi: 10.1248/bpb.34.433. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Sato H, Sugiyama Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu Rev Pharmacol Toxicol. 2005;45:689–723. doi: 10.1146/annurev.pharmtox.44.101802.121444. [DOI] [PubMed] [Google Scholar]
- Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009;37:359–365. doi: 10.1124/dmd.108.024612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CF, Leggas M, Schuetz JD, Panetta JC, Cheshire PJ, Peterson J, et al. Gefitinib enhances the antitumor activity and oral bioavailability of irinotecan in mice. Cancer Res. 2004;64:7491–7499. doi: 10.1158/0008-5472.CAN-04-0096. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Miura M, Scott SA, Kagaya H, Kameoka Y, Tagawa H, et al. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet. 2010;55:731–737. doi: 10.1038/jhg.2010.98. [DOI] [PubMed] [Google Scholar]
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, et al. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer. 2011 doi: 10.1002/ijc.26000. doi: 10.1002/ijc.26000 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Thomas J, Wang L, Clark RE, Pirmohamed M. Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 2004;104:3739–3745. doi: 10.1182/blood-2003-12-4276. [DOI] [PubMed] [Google Scholar]
- Thomas F, Rochaix P, White-Koning M, Hennebelle I, Sarini J, Benlyazid A, et al. Population pharmacokinetics of erlotinib and its pharmacokinetic/pharmacodynamic relationships in head and neck squamous cell carcinoma. Eur J Cancer. 2009;45:2316–2323. doi: 10.1016/j.ejca.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR, Chen X, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009;78:153–161. doi: 10.1016/j.bcp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- van der Veldt AA, Eechoute K, Gelderblom H, Gietema J, Guchelaar HJ, van Erp NP, et al. Genetic polymorphisms associated with a prolonged progression-free survival in patients with metastatic renal cell cancer treated with sunitinib. Clin Cancer Res. 2011;17:620–629. doi: 10.1158/1078-0432.CCR-10-1828. [DOI] [PubMed] [Google Scholar]
- van Erp NP, Eechoute K, van der Veldt AA, Haanen JB, Reyners AK, Mathijssen RH, et al. Pharmacogenetic pathway analysis for determination of sunitinib-induced toxicity. J Clin Oncol. 2009a;27:4406–4412. doi: 10.1200/JCO.2008.21.7679. [DOI] [PubMed] [Google Scholar]
- van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009b;35:692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Wang L, Giannoudis A, Lane S, Williamson P, Pirmohamed M, Clark RE. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther. 2008;83:258–264. doi: 10.1038/sj.clpt.6100268. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, et al. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108:697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Dang P, Engler J, Venables A, Zrim S, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007a;110:4064–4072. doi: 10.1182/blood-2007-06-093617. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Quinn SR, Manley PW, Hughes TP. Imatinib increases the intracellular concentration of nilotinib, which may explain the observed synergy between these drugs. Blood. 2007b;109:3609–3610. doi: 10.1182/blood-2006-11-058032. [DOI] [PubMed] [Google Scholar]
- White DL, Dang P, Engler J, Frede A, Zrim S, Osborn M, et al. Functional activity of the OCT-1 protein is predictive of long-term outcome in patients with chronic-phase chronic myeloid leukemia treated with imatinib. J Clin Oncol. 2010a;28:2761–2767. doi: 10.1200/JCO.2009.26.5819. [DOI] [PubMed] [Google Scholar]
- White DL, Saunders VA, Dang P, Engler J, Hughes TP. OCT-1 activity measurement provides a superior imatinib response predictor than screening for single-nucleotide polymorphisms of OCT-1. Leukemia. 2010b;24:1962–1965. doi: 10.1038/leu.2010.188. [DOI] [PubMed] [Google Scholar]
- Yamakawa Y, Hamada A, Shuto T, Yuki M, Uchida T, Kai H, et al. Pharmacokinetic impact of SLCO1A2 polymorphisms on imatinib disposition in patients with chronic myeloid leukemia. Clin Pharmacol Ther. 2011;90:157–163. doi: 10.1038/clpt.2011.102. [DOI] [PubMed] [Google Scholar]
- Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y. Gefitinib reverses breast cancer resistance protein-mediated drug resistance. Mol Cancer Ther. 2004;3:1119–1125. [PubMed] [Google Scholar]
- Yang CH, Huang CJ, Yang CS, Chu YC, Cheng AL, Whang-Peng J, et al. Gefitinib reverses chemotherapy resistance in gefitinib-insensitive multidrug resistant cancer cells expressing ATP-binding cassette family protein. Cancer Res. 2005;65:6943–6949. doi: 10.1158/0008-5472.CAN-05-0641. [DOI] [PubMed] [Google Scholar]
- Yonezawa A, Inui KI. Importance of the multidrug and toxin extrusion MATE/SLC47A family to pharmacokinetics, pharmacodynamics/toxicodynamics and pharmacogenomics. Br J Pharmacol. 2011;164:1817–1825. doi: 10.1111/j.1476-5381.2011.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Schmidt K, Nelson FR, Zelesky V, Troutman MD, Feng B. The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab Dispos. 2009;37:946–955. doi: 10.1124/dmd.108.024489. [DOI] [PubMed] [Google Scholar]
- Zhuang Y, Fraga CH, Hubbard KE, Hagedorn N, Panetta JC, Waters CM, et al. Topotecan central nervous system penetration is altered by a tyrosine kinase inhibitor. Cancer Res. 2006;66:11305–11313. doi: 10.1158/0008-5472.CAN-06-0929. [DOI] [PubMed] [Google Scholar]
- Zimmerman JJ. Exposure-response relationships and drug interactions of sirolimus. AAPS J. 2004;6:e28. doi: 10.1208/aapsj060428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolk O, Fromm MF. Transporter-mediated drug uptake and efflux: important determinants of adverse drug reactions. Clin Pharmacol Ther. 2011;89:798–805. doi: 10.1038/clpt.2010.354. [DOI] [PubMed] [Google Scholar]