

Abstract
Human in vivo molecular imaging with positron emission tomography (PET) enables a new kind of ‘precision pharmacology’, able to address questions central to drug development. Biodistribution studies with drug molecules carrying positron-emitting radioisotopes can test whether a new chemical entity reaches a target tissue compartment (such as the brain) in sufficient amounts to be pharmacologically active. Competition studies, using a radioligand that binds to the target of therapeutic interest with adequate specificity, enable direct assessment of the relationship between drug plasma concentration and target occupancy. Tailored radiotracers can be used to measure relative rates of biological processes, while radioligands specific for tissue markers expected to change with treatment can provide specific pharmacodynamic information. Integrated application of PET and magnetic resonance imaging (MRI) methods allows molecular interactions to be related directly to anatomical or physiological changes in a tissue. Applications of imaging in early drug development can suggest approaches to patient stratification for a personalized medicine able to deliver higher value from a drug after approval. Although imaging experimental medicine adds complexity to early drug development and costs per patient are high, appropriate use can increase returns on R and D investment by improving early decision making to reduce new drug attrition in later stages. We urge that the potential value of a translational molecular imaging strategy be considered routinely and at the earliest stages of new drug development.
Keywords: drug development, molecular imaging, pharmacodynamics, pharmacokinetics, positron emission tomography (PET)
Introduction
Development of the first human positron emission tomography (PET) scanner was reported in 1975 by Michael Ter-Pogossian and Mike Phelps of Washington University in St Louis, USA. Over the next decade, applications to new therapeutics development were limited, but from the late 1980s applications began to grow at a rapid rate. Almost 3500 papers or online radiopharmaceutical reports now are accessible within PubMed (searched at http://www.ncbi.nlm.nih.gov/sites/entrez), using terms PET AND drug AND (biodistribution OR target occupancy OR pharmacodynamics).1 with almost 400 reports from 2010 alone!
Molecular imaging using PET enables a new kind of ‘precision pharmacology’ able to address questions central to drug development in humans in vivo: Does a new drug molecule reach the tissue of interest in potentially pharmacologically active concentrations? Is it interacting with the target of interest? What is the quantitative relationship between the extent of this interaction and the administered dose? What are the consequent pharmacological effects and how long do they last? Extension of answers to the latter two questions can suggest ways of stratifying clinical populations, either to speed clinical trials or to deliver higher value in the use of a new drug following registration (Table 1).
Table 1.
Selected applications of imaging in drug development
| Early phase development |
| • Molecule biodistribution studies confirming molecule reaches the target tissue and does not accumulate in non-target sites of potential toxicity |
| • Target PK (dose–target occupancy) measurements guiding dose selection |
| • Pharmacodynamic biomarkers for proof of pharmacology, stronger ‘reasons to believe’ or contributing key rationale for proof of concept |
| • Translational preclinical imaging to identify or validate new imaging biomarkers and or provide early differentiation between candidates based on target PK or PD responses |
| • In vivo measures for monitoring safety or toxicity |
| Late phase development |
| • Surrogate markers of response more sensitive than clinical measures |
| • Stratification of patients based on potential for treatment efficacy |
| • Pharmacological differentiation of asset from marketed drugs or new competitor compounds |
| Marketed drugs |
| • Differentiation between available treatments |
| • Earlier detection of disease or associated pathology: |
| • Improved disease classification/diagnosis |
| • Diagnosis of pre- symptomatic or minimally symptomatic disease |
| • Improved identification of chronic disease exacerbation/recurrence |
| • Patient stratification based on disease sub-phenotype or early treatment response |
Principles of PET
PET imaging is based on the principle that an emitted positron collides with a local electron, resulting in a mutual annihilation and the production of a pair of photons that travel at 180° to each other. The photons can be detected as coincident events by γ-detectors surrounding the subject. Knowledge of which detector pairs sense the coincident events and their precise timing enables localization of the annihilation events and reconstruction of the spatial distribution of the emitting radio-labelled molecule. Quantitative measurements of absolute concentrations of the labelled molecule over time can be made with dynamic acquisition of data, corrections to normalize sensitivity to emissions across the region of interest and application of appropriate tracer kinetic models to these data for estimation of the rates of delivery of the radiotracer and the amount retained in tissues of interest.
PET relies on the design and manufacture of radiolabelled tracers or ligands which interact selectively with a target of interest. Ligands will have the characteristics to enable the quantification of a specific binding signal, such as a suitably high ratio of specific to non-specific binding and favourable tissue kinetics. Most commonly used positron emitting radioisotopes decay with a relatively short half-life (e.g. about 20 min for 11C and 110 min for 18F), allowing administration of doses high enough to provide a strong imaging signal without substantially increasing long-term health risks associated with the ionizing radiation. However, a short half-life imposes the limitation that radiotracer production needs to be performed close enough to the PET scanner to allow injection within a few half-lives. Only microdoses of radioligands or other radio-labelled molecules need to be used. PET is exquisitely sensitive (even only picomoles of labelled material can be detected), and thus can be conducted under conditions in which the ligand occupies <5% of the target and has no pharmacologically relevant activity (‘tracer conditions’).
PET data can be co-registered with structural data from computed tomography (CT) or magnetic resonance imaging (MRI) to aid in anatomically localizing any signal. However, the spatial resolution of PET, even in modern tomographs, is lower (typically about ∼4 mm) than that achieved by CT or MRI.
Biodistribution studies applying PET molecular imaging
Pharmacological activity of a new chemical entity depends directly on the free concentration that can be achieved in the relevant tissue. Establishing the tissue free concentration with confidence can be a critical starting position for early phase development and difficult to establish confidently using conventional approaches, especially for a ‘privileged’ tissue compartment such as the central nervous system (CNS). Bengt Langstrom and colleagues including Mats Bergstrom introduced the powerful concept of determining molecule distribution and concentration in vivo in humans using PET after labelling the molecule with a positron-emitting isotope that does not change the chemical structure or properties [1].
The principles of a PET biodistribution study are straightforward. A dynamic PET scan measures the concentration–time course of the radiolabelled compound in the tissue of interest. In conjunction with associated measurements of the concentration in blood, it is possible to use bio-mathematical kinetic models to derive estimates of the clearance from plasma to tissue (a function of the blood flow and the tissue extraction of the molecule from the blood) and the ratio of the concentration of labelled drug (and drug metabolites) in tissue to blood that would be achieved at equilibrium (the tissue : blood partition coefficient). Associated HPLC analysis of blood samples ex vivo allows additional consideration of any metabolism of the radiolabelled compound.
To varying extents, different compounds will distribute in the tissue, where they will be either ‘free’ or ‘bound’ to tissue components. This binding can be either displaceable (high affinity, low capacity binding, e.g. to a receptor) or non-displaceable (low affinity, high capacity binding, as with lipophilic interactions). However, the calculation of the tissue ‘free’ concentration from the total tissue concentration is typically not feasible from PET data alone. Accurate estimation of the fraction of the non-displaceable compartment which is attributable to ‘free’ drug is performed by combining the PET estimates of the total blood : tissue partition coefficient with in vitro equilibrium dialysis assays that account for any non-specific binding in the tissue [2]. Combination of PET and equilibrium dialysis data can also allow one to infer whether the tissue uptake is by passive diffusion or by active (or facilitated) transport [3].
If it can be assumed that tissue uptake occurs by passive diffusion, the ‘free’ tissue concentration can be calculated from measurements of the ‘free’ plasma concentration of the labelled molecule at equilibrium [4]. Target occupancy (O) then can be estimated by assuming that the in vitro and in vivo KD are equivalent.
The passive diffusion assumption can be explored further by measuring changes in the tissue concentration of the drug after administration of relevant transporter inhibitors (e.g. P-glycoprotein [P-gp] antagonists [3, 5] (Figure 1) or by pre-treatment with large doses of the unlabelled compound [6].
Figure 1.
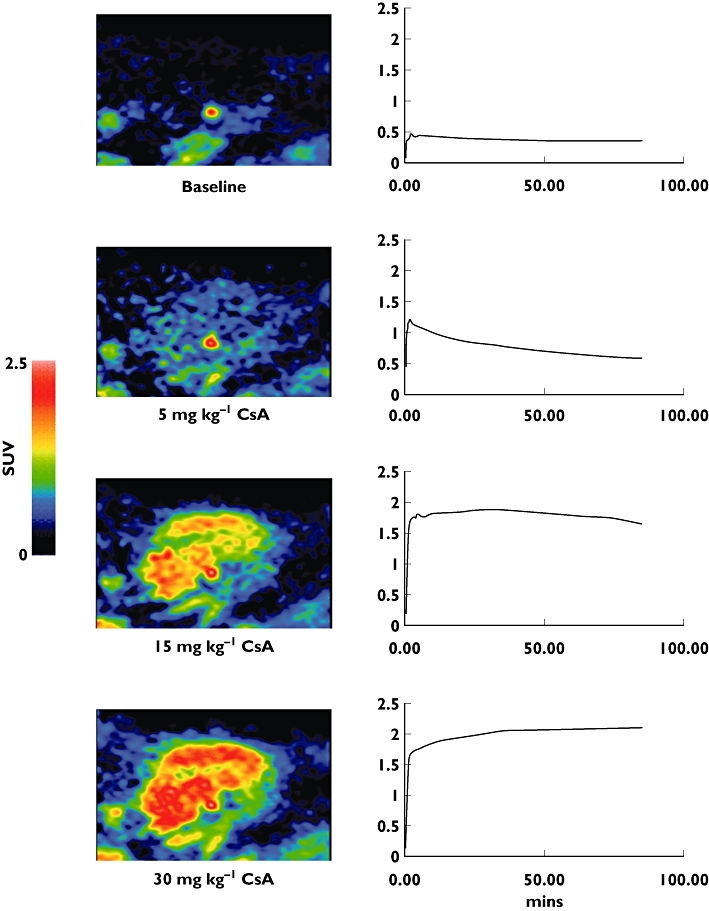
Effect of increasing doses of the competitive P-gp substrate cyclosporine A (CsA) on [11C]-loperamide uptake in porcine brain. Increasing doses of CsA lead to increased net uptake of the [11C]-loperamide with greater competition for the transporter. Note that the prominent ‘hot spot’ in the upper two images localizes to the pituitary gland, which sits outside the blood–brain barrier. Images were acquired from the same animal scanned sequentially on the same day
While the most common application of PET biodistribution studies thus far has been to the development of drugs targeting the central nervous system (CNS), they also can play an important role in other areas, e.g. in optimizing anti-cancer drugs [7], as up-regulation of pumps that exclude drugs from tumours is well described [8]. PET biodistribution studies can be integrated with conventional stable isotope DMPK studies [9] or with pharmacodynamic measures [10].
It is important to recognize, however, that it is the distribution of the radionuclide, not the molecule, that is directly measured in the PET experiment. A creative extension of the traditional biodistribution experiment that provided information on drug metabolism directly from the PET study illustrates this well [11]. Temozolomide, an alkylating agent used in cancer chemotherapy, undergoes decarboxylation and ring opening in the 3–4 position to produce the highly reactive methyldiazonium ion (which then can alkylate DNA for pharmacological action of the molecule). To evaluate this directly in vivo in humans, a dual radiolabelling strategy was employed in which [11C]-temozolomide was radiolabelled separately both in the 3-N-methyl and 4-carbonyl positions (Figure 2A,B, respectively). 11C in the C-4 position of [4-11C-carbonyl]-temozolomide was converted to [11C]-CO2 and an inactive metabolite. Paired studies were performed with the two labelled forms of [11C]-temozolomide in a small number of patients with gliomas. A third PET scan was performed with 11C-radiolabelled bicarbonate to provide data allowing quantitative modelling of the labelled CO2 release. Data were obtained on activities of [11C]-temozolomide and [11C]-metabolites in plasma collected during scanning and [11C]-CO2 was measured in the expired air. Greater amounts of [11C]-CO2 in the plasma and exhaled air and lower tumour [11C]-temozolomide signal with the [4-11C-carbonyl]-temozolomide relative to that labelled in the 3-N-methyl position confirmed ring-opening as a mechanism for metabolic activation of temozolomide.
Figure 2.
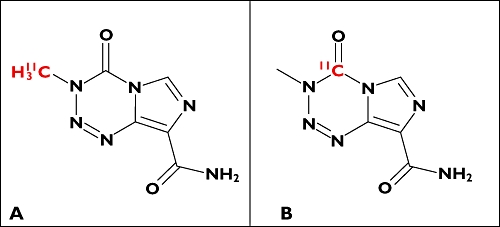
Chemical structures of 3-N-[11C-methyl]-temozolomide (A) and [4-11C-carbonyl]-temozolomide (B)
A new extension of the above techniques is being pioneered with first efforts to characterize the biodistribution behaviours of monoclonal antibodies and other biopharmaceuticals. A range of methods are available for labelling such large molecules [12]. Because of the much slower approach to an equilibrium biodistribution (typically expected to be days to weeks) for these large molecules, long-lived positron emitters such as 89Zr, 64Cu or 124I have been used. Considerable information is potentially available from such experiments, but confounds arising from the slow approach to steady-state, the need to account for physical barriers to free diffusion and different kinds of non-specific interactions (e.g. with molecule uptake into the reticuloendothelial system) make these studies technically more challenging than those with small molecules. Although promising, this area is still in an early stage of development.
Assessing target interactions with PET
Demonstration of interaction of a drug molecule with its target in a tissue also provides direct evidence of biodistribution into that tissue. If possible, it should be considered the approach of choice for defining drug–target pharmacokinetics.
Target interaction studies are most informative if there is a strong hypothesis regarding the extent of target interaction needed for a pharmacological effect. In such cases, data relating plasma concentration to target occupancy can guide dose selection directly. For example, for inhibitors of G-protein coupled receptors, preclinical (and clinical) studies typically suggest that free concentrations sufficient to provide above 70% receptor occupancy are needed (see e.g. [13, 14]). If information concerning the relationship between plasma concentration and target interactions is available before dose ranging studies, the range of doses that need to be explored in early phase studies can be deduced.
Target interaction studies require a radioligand that binds selectivity to the target of interest with a high enough affinity to provide useful signal-to-noise in a PET study. The usual outcome measure of interest from a radioligand study is the ‘binding potential’ (BP), which is proportional to the specific binding divided by the free concentration of the radioligand [15]. If the PET study is performed after administration of an unlabelled (drug) molecule that binds to the same target, the measured radioligand BP will vary with the local free drug concentration. Conducting such studies over a range of doses, allows binding affinity of the unlabelled molecule to be estimated the variation in radioligand BP (Figure 3). Characterization of the relationship between plasma concentration and target interaction for alternative candidate molecules can be important particularly if there are dose limiting toxicities (Figure 4).
Figure 3.
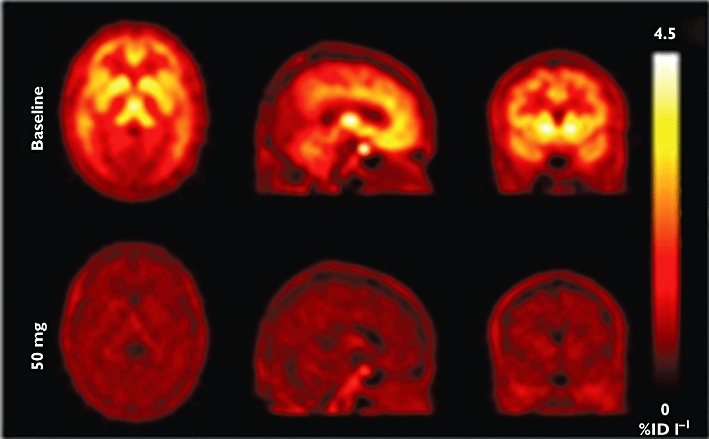
One image set from an illustrative drug occupancy study illustrating the radioligand signal before (upper) and after (lower) administration of cold drug competing for the same binding site. Brighter regions define increased radioisotope concentration. With repetition of a similar image pair over a range of doses of cold drug (or by varying timing of radioligand injection after cold drug administration), varying plasma concentrations at the time of scanning allow estimation of an in vivo IC50 for the cold drug based on measures of relative displacement of the radiotracer
Figure 4.
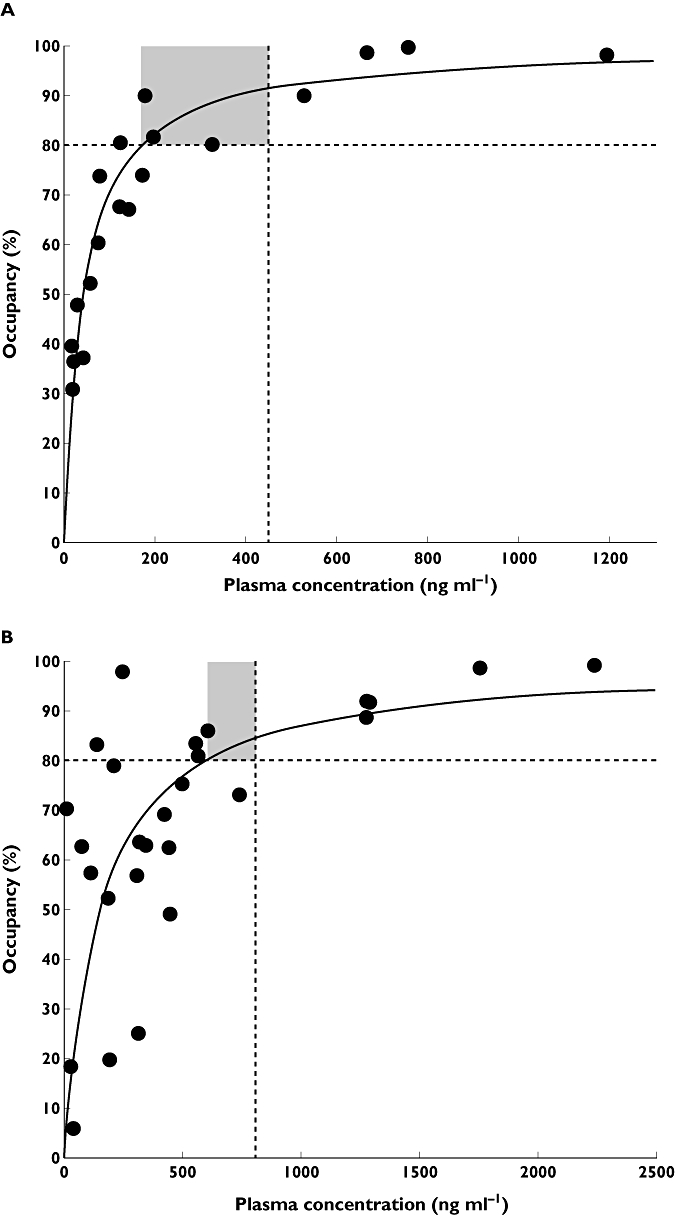
An example of data from target occupancy studies with two molecules in candidate selection. The molecules were antagonists and previous work suggested that occupancy by approximately 80% or more would be needed for the desired pharmacological effects (horizontal broken line). However, both molecules had recognized potential toxicities at plasma concentration shown by the vertical solid line. The in vivo human target occupancy-plasma concentrations defined in separate sets of PET experiments (see panels A and B) established a range of plasma concentrations (and thus doses) over which pharmacological effects were likely to be seen. In doing so, they also estimated the therapeutic index for the molecules (grey areas). The molecule used in the study for panel A, which has the higher therapeutic index, was selected for further development
PET studies are expensive and there is an ethical imperative to minimize exposure of volunteers to even the low additional radiation exposure of PET studies. Adaptive designs that use information gained from each single observation to improve the selection of the subsequent dose can optimize the efficiency of a study [16]. The less prior knowledge concerning dosing that is available, the greater the potential efficiency gains with adaptive designs [16].
Human in vivo target interaction studies can reduce sources of substantial uncertainty in drug development. For example, in some cases the affinity of a molecule in humans in vivo is very different from that measured in isolated tissue in ex vitro or in preclinical models. A histamine H3-receptor antagonist we have studied, for example, was shown to have an in vivo human affinity a full order of magnitude higher from that measured in preclinical studies [17]. This observation had a substantial impact on the drug development programme, as it gave rationale and confidence for a major reduction in dosing into a range that was well-tolerated by patients. The study also defined unexpectedly slow receptor ‘off’ rates for the molecule, leading to a re-estimation of the optimal dosing frequency. This example thus also highlights that, in general, the time course for target interaction does not reflect plasma pharmacokinetics except for the limiting case of molecules with fast equilibrium binding properties that diffuse passively between compartments. An interesting variant of this application of target occupancy studies is to the ‘reverse engineering’ of empirically established treatments to define better those interactions that may be driving therapeutic efficacy [18].
Target interaction studies have typically been conducted as single dose studies for experimental convenience. Repeat dose occupancy studies may induce changes in target expression, in which case application of single dose occupancy measures will be inaccurate. However, if the pharmacokinetic model appropriate to the drug can be estimated, repeat dose brain target occupancy can be estimated based on the basis of the combined occupancy data obtained after administration of a single dose and plasma pharmacokinetic data [19]. Theoretical arguments show that the models used in these analyses can predict repeat dose occupancy even when the relationship following single dose is not described by a simple direct model dependent on the instantaneous plasma concentration.
Applications of PET to studies of pharmacodynamics
Some radiotracers (e.g. [18F]-fluorodeoxyglucose (FDG), [18F]-6-fluoro-L-3,4-dihydroxyphenylalanine (FDOPA), [18F]-3′-fluorothymidine (FLT)) can be used to assess specific metabolic or synthetic rates, allowing inferences concerning the functional state or integrity of a tissue. Radioligands can be used to measure the concentration of specific receptor or transporter sites, allowing for the assessment of the integrity or distribution of a specific target that may correspond to its expression. Quantitative compartmental analysis methods can be used to take account of the potential confounds from differences in blood flow (and, thus, availability of the radioligand) between tissues.
Two applications illustrate the complementary ways in which radiotracers and radioligands can be used for pharmacodynamic studies. FDG has been used as a PET radiotracer for defining brain metabolic activity in Alzheimer's disease (AD) and its pharmacological modulation. For example, effects of treatments expected to enhance metabolism or slow rates of its impairment with the progression of neurodegeneration can be assessed with serial FDG PET scanning [20]. By contrast, [11C]-PIB has been used as a radioligand to provide specific information concerning the deposition of amyloid, which is thought to be related directly to mechanisms of neurodegeneration and is a current target for AD therapy. A number of pharmaceutical companies are developing anti-amyloid antibodies intended to provide a ‘peripheral sink’ which binds blood amyloid and thus reduces brain amyloid concentrations. The radioligand [11C]-PIB, which has a high affinity for the beta sheet structure of the deposits, can be used to localize and estimate changes in relative concentrations of amyloid, as demonstrated in a recent phase IIa study of bapineuzumab [21].
Similar considerations hold for applications of pharmacodynamic PET to development of therapeutics in oncology. For example, use of FDG PET as a radiotracer provides an index of the enhanced glucose transport and phosphorylation in many tumours (the ‘Warburg’ effect). Qualitative assessment of the FDG PET signal is used routinely in the clinic as a diagnostic marker for tumours. Quantitative measurements before and after treatment can define pharmacodynamic effects expressed as changes in glucose transport, glycolytic enzyme activity or cell viability [22] (Figure 5). An atypical but illustrative example of this for drug development came with the demonstration of dramatic FDG PET responses to imatinib within 24 h of dosing for gastrointestinal stromal tumours in some patients [23]. Complementary information comes with use of specific radioligands. An 18F-tagged peptide dimer of arginine-glycine-aspartate (E[c(RGDyK)]2) that binds to the αvβ2 integrin that is up-regulated with tumour angiogenesis defines integrin-positive tumours more specifically, for example [24]. A growing ‘toolkit’ of radiotracers able to assess the activity of biological processes commonly altered by many therapies and specifically-targeted radioligands is available (Table 2).
Figure 5.
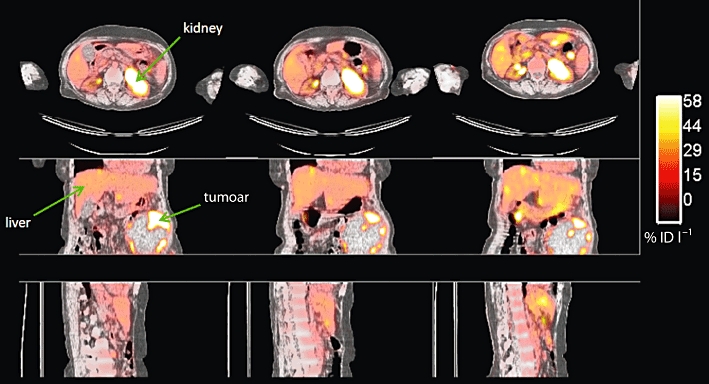
[18F]-FDG PET study of a patient with an abdominal ovarian tumour (arrow). A significant decrease in [18F]-FDG uptake (SUVmax) together with volumetric tumour reduction was observed at the second visit. Images courtesy of Dr A. Saleem, GSK Clinical Imaging Centre
Table 2.
Selected positron emission tomography radiotracers well characterized for pharmacodynamic studies (adapted from [43], but see also http://www.ncbi.nlm.nih.gov/books/NBK5330/)
| PET radiotracer | Clinical application |
|---|---|
| [13N]-ammonia | Myocardial perfusion |
| [18F]-fluorodeoxyglucose FDG) | Glucose uptake and phosphorylation |
| [11C]-methionine | Protein synthesis |
| [18F]-fluoromizonidazole | Tumour hypoxia |
| [11C]-acetate | Oxidative metabolism |
| [18F]-DOPA | Presynaptic dopaminergic function |
| [18F]-fluoride | Bone scintigraphy |
| [82Rb]-rubidium | Myocardial perfusion |
| [18F]-fluorotyrosine | Amino acid uptake, protein synthesis |
| [11C]-thymidine | DNA synthesis |
| [18F]-fluorothymidine (FLT) | Tumour cell proliferation |
| [64Cu]-ATSM or [18F]MISO | Hypoxia |
While the major applications of PET in drug development thus far have been to CNS or oncology therapeutics, there is potential for much wider applications of PET in drug development. One of the most promising new general areas of application is to inflammatory diseases [25]. The best characterized radioligand target has been the 18 kDa translocator protein (TSPO, previously known as the peripheral benzodiazepine receptor), expression of which is increased with macrophage or microglial activation, to provide a molecular marker of innate immune responses [26]. The most widely used radioligand thus far has been [11C]-PK11195 [18], but interpretation of studies is limited by its relatively poor signal-to-noise ratio [23]. While several alternative radioligand candidates have been evaluated in humans, differences in their binding affinity between subjects raised concerns about whether studies with them can be interpreted quantitatively [27]. However, identification of a genetic polymorphism in the TSPO gene that is responsible for this behaviour now promises to make quantitative studies possible after simple genetic testing [28].
Integration of data from PET target occupancy studies and functional MRI (fMRI) methods provides a novel strategy for directly relating information on drug–target interactions directly with a measure of functional effects in the brain. Recent studies illustrating this approach have related the extent of binding of an antagonist to its µ-opioid target with modulation of fMRI reward responses to the administration of a palatable food stimulus [29] or a dopamine receptor occupancy to reward responses in a gambling task (Figure 6). The study simultaneously provided direct evidence validating the target as a modulator of satiety responses in humans and suggested a pharmacological dose range based both on the measures of target interaction and pharmacodynamic effect. New potential for extension of this kind of work has come with advances in detector technology that have made possible a first generation of fully integrated human PET/MRI systems [30, 31]. Integrated acquisition of data will increase the precision of registration of the MRI and PET data particularly for applications outside of the head.
Figure 6.
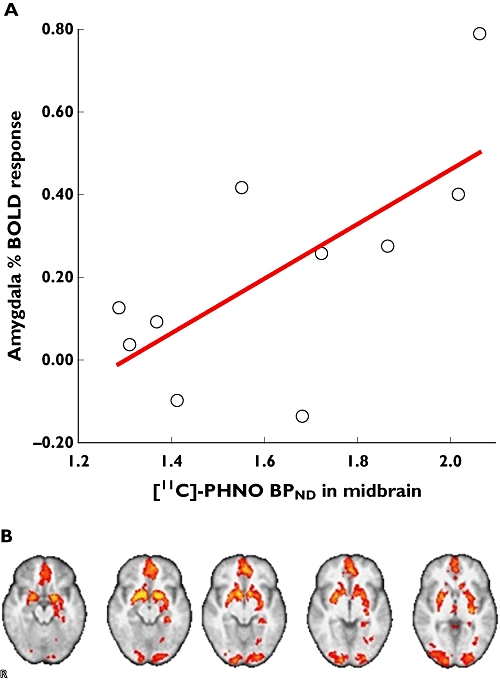
(A) Group statistical maps (overlaid on structural MRI image) showing baseline fMRI BOLD response to receipt of monetary rewards. (B) Relationship between study population variability in midbrain dopamine receptor subtype 3 (D3R) availability and amygdala response to reward. Correlation between midbrain [11C]-(+)-PHNO BPND and amygdala activation to monetary rewards for individual subjects studied, demonstrating that subjects with higher midbrain D3R availability have greater amygdala BOLD response to rewards
Development of target-specific radioligands for PET
Availability of appropriate radioligands is a major challenge for PET molecular imaging in applications to drug development. Target interaction studies demand availability of a radioligand that has good affinity for the target, and binds with high specificity and selectivity [32]. There are many examples of such molecules, particularly for targets in the CNS [33], but novel drug targets will demand novel radioligands and the discovery of radioligands for new targets is a complex and resource-intensive undertaking requiring highly specialized and skilled staff.
Typically, the first steps in the discovery of a novel radioligand involve screening relevant compounds (e.g. from a library of molecules with some selectivity for binding to the target of interest) for feasibility of introduction of a positron-emitting radioisotope label (e.g. 11C or 18F) and for chemical parameters such as lipophilicity (e.g. log P or log [solubility in octanol]/[solubility in aqueous solution at pH 7.4]) and affinity and selectivity for the target (e.g. through measures of Ki, IC50, EC50). Additional pharmacological criteria include plasma clearance, metabolic fate, plasma protein binding and the potential to access the target tissue. For applications in the brain, assessment of the potential for crossing the blood–brain barrier is critical. Compounds need to be sufficiently lipophilic to allow passive diffusion across cell membranes while not being so lipophilic that a substantial fraction interacts non-specifically with membranes. As a ‘rule of thumb’, initial consideration is given to compounds with a measured log P of 1–3.
The affinity of a radioligand for its target and the amount of target available are fundamental parameters that will determine the observed signal. The desired affinity range for radioligand candidates therefore depends on the expected target density. The signal-to-noise ratio can be approximated by the BP (BP = (Bmax– B) × (kon/koff), where Bmax is the concentration of the target of interest and B is the concentration of the target occupied by the ligand). The ratio of first order kinetic rate constants for radioligand binding and release from the target, kon/koff, can be expressed as 1/KD. Under true tracer conditions (B < < Bmax), the equations can be simplified as BP = Bmax/KD. A practical range for the binding potential, when allowing for non-specific binding in the tissues of interest, is between 0.5 −15. Values less than 0.5 or greater than 15 suggest that a candidate radioligand may suffer from either undesirably high variability or low precision, respectively.
Target selectivity is governed by the relative affinity, density and tissue distribution of potentially competing interactions. Under usual circumstances, adequate target specificity is expressed with a similar density in the same tissue demands at least an order of magnitude difference in affinity. However, for applications in which receptors have known distributions and are not anatomically co-localized, similar affinities for receptors can be allowed.
The potential for a compound to access the tissue of interest also should be carefully considered. Our personal experience in discovery efforts for radioligands targeting the brain has been that the brain : blood ratio observed in preclinical rodent or porcine biodistribution studies should be >1 for practical utility. Invasive, equilibrium dialysis measurements of the fraction of ligand free in plasma (fP) and tissue proteins (fND) can provide even in vitro data that predicts brain penetration; the ratio fP : fND correlates well with the non-specific volume of distribution (VND) [3, 15].
Care should be taken to ensure the ligand is not subject to fast active transport from the target tissue back into the blood. Examples of such active transport systems are P-gp, the organic anion transporters (OATP), lung cancer resistant protein (LRP), brain cancer resistant protein (BCRP) and multidrug resistant proteins (MRP) [8]. In brain the most prominent of these is P-gp. In silico or in vitro methods can be used to screen out compounds that may be substrates for this transporter [2, 34]. However, weak to moderate substrates can still be useful radioligands, e.g. [11C]-carfentanil and [18F]-4-(2′-methoxyphenyl)-1-[2′-(N-2-pyridinyl)-p-fluorobenzamido]-ethyl-piperazine ([18F]-p-MPPF) [35].
In order to optimize signal-to-noise, encourage rapid tracer kinetics and facilitate equilibrium between plasma and tissue concentrations within the period of the scan (typically 90–120 min), clearance of the ligand from plasma should be relatively fast. This is usually challenging with chemical structures derived directly from molecules developed as drugs, because most therapeutics are designed for dosing no more frequently than once or twice daily. The compounds with high plasma clearance that are preferred for radioligands are typically not seen as viable drug candidates, so the drug development process actively screens against them.
Radioisotope labelling is perhaps the most flexible part of initial candidate screening, as there is a well-developed arsenal of chemistry methodology for both 11C and 18F chemistries. Work with 18F has focused primarily on nucleophilic substitution reactions (both aliphatic and aromatic). Nonetheless, feasibility of the chemistry can be challenging for 11C syntheses. As a rule of thumb, a PET radiopharmaceutical should be available for clinical use within three half-lives of receiving the radioactivity from the cyclotron. For 11C, this means that the entire process, including quality control release testing to specification, should ideally take less than 1 h. As a result, most chemistry with 11C involves introduction of the label as a single, final step to limit loss of product as a result of radioactive decay [36].
Finally, the position of labelling should be carefully considered with regard to the known or probable metabolic fate of the PET radioligand. Where possible, the ligand should be labelled in a position which, upon oxidation or hydrolysis, leads to a labelled hydrophilic fragment, as these are less likely to enter tissues. An example illustrating this is provided by the 5-HT1A receptor radioligand, [11C]-WAY 100635. Initially, this compound was labelled in the O-methyl position (Figure 7A) to achieve similar criteria based on the reported primary route of metabolism in rat. Rodent preclinical studies supported the potential suitability of this radioligand with demonstration of a high signal in the hippocampus and a very low signal in the cerebellum, tissues known to have high and very low 5-HT1A receptor expression, respectively [37]. However, translation to humans gave a surprising result: the observed medial temporal cortex (MTC) : cerebellum signal ratio was more than five-fold lower than expected [38]. Contrary to the experience with rats, it was found that in man the primary route of metabolism in humans is through hydrolysis of the amide bond (Figure 7B). The hydrolysis product ([11C]-WAY 100634) readily enters the brain and has a high affinity for α1-adrenergic receptors, reducing the specific signal-to-noise for the 5-HT1A receptor. Subsequent labelling in the carbonyl position generated a radioligand (11C-carbonyl]-WAY 100635) that gave a much more specific signal (as supported by the much improved MTC/cerebellum signal ratio) [39].
Figure 7.
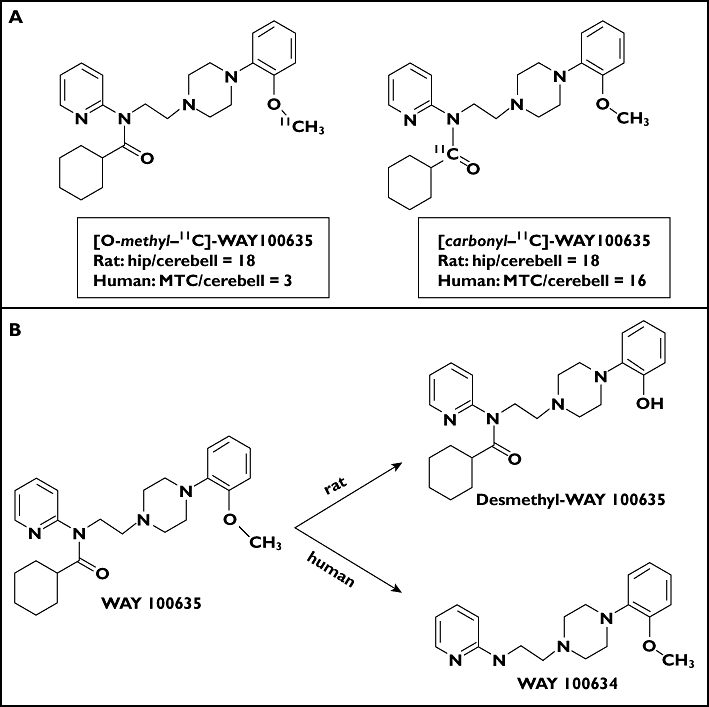
(A) Structures for alternately radiolabelled forms of the radioligand WAY100635 which show different relative radioisotope accumulation in hippocampus (hip) and medial temporal cortex (MTC) relative to the cerebellum (cerebell) in rat and human because of different routes of metabolism in the two species (B)
Despite an apparent thorough knowledge of critical physicochemical parameters that define the boundaries the overall rate of discovery of new PET tool compounds compared with the effort invested by the field is low. Recent advances in design-based biomathematical modelling [40], better understanding of factors predicting non-specific binding [41] and new approaches to medicinal and PET chemistry promise opportunities for more efficient development in the future.
Conclusions
PET allows a new ‘precision pharmacology’ that can have an important role in drug development. While imaging experimental medicine can add complexity to planning clinical development and increase the cost per patient studied, well-designed studies can answer key questions earlier and with smaller numbers of subjects for more confident decision-making. In the future, applications of molecular imaging to the development of drugs can add further value with their translation to clinical use as a companion diagnostic for patient stratification enabling higher efficacy and value. More responsive patient populations can be identified not just to enable smaller, more informative clinical trials, but also to direct medicines to patients who will experience the greatest benefit [42]. We urge that the potential value of a translational molecular imaging strategy be considered routinely and at the earliest stages of planning for the development of new drugs.
Acknowledgments
PMM is supported by the Medical Research Council for work that has contributed to this review. The authors also want to acknowledge the support of their colleagues at the GSK Clinical Imaging Centre for work described in this review and particularly to Professor Marc Laruelle, whose leadership and insight helped all of them to appreciate better how best to deliver value from molecular imaging for drug development. The authors additionally thank Jasper van der Aart for help with the figures and Rachel Green for general editorial support.
Competing Interests
The authors all are full-time employees of GlaxoSmithKline and hold stocks and options in the company.
REFERENCES
- 1.Bergstrom M, Grahnen A, Langstrom B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur J Clin Pharmacol. 2003;59:357–66. doi: 10.1007/s00228-003-0643-x. [DOI] [PubMed] [Google Scholar]
- 2.Summerfield SG, Stevens AJ, Cutler L, del Carmen Osuna M, Hammond B, Tang SP, Hersey A, Spalding DJ, Jeffrey P. Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J Pharmacol Exp Ther. 2006;316:1282–90. doi: 10.1124/jpet.105.092916. [DOI] [PubMed] [Google Scholar]
- 3.Gunn RN, Summerfield SS, Salinas C, Read KR, Searle G, Ruffo AD, Parker C, Stevens AJ, Bonasera T, Jeffrey PM, Laruelle MA. Combining PET and equilibrium dialysis to assess blood-brain barrier transport. J Cereb Blood Flow Metab. 2007;27(Suppl. 1) [Google Scholar]
- 4.Slifstein M, Laruelle M. Models and methods for derivation of in vivoneuroreceptor parameters with PET and SPECT reversible radiotracers. Nucl Med Biol. 2001;28:595–608. doi: 10.1016/s0969-8051(01)00214-1. [DOI] [PubMed] [Google Scholar]
- 5.Passchier J, Lawrie KWM, Bender D, Fellows I, Gee AD. [11C] Loperamide as highly sensitive PET probe for measuring changes in P-glycoprotein functionality. J Labelled Comp Radiopharm. 2003;46:S94. [Google Scholar]
- 6.Loscher W, Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol. 2005;76:22–76. doi: 10.1016/j.pneurobio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Rosso L, Brock CS, Gallo JM, Saleem A, Price PM, Turkheimer FE, Aboagye EO. A new model for prediction of drug distribution in tumor and normal tissues: pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009;69:120–7. doi: 10.1158/0008-5472.CAN-08-2356. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–76. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 9.Wagner CC, Simpson M, Zeitlinger M, Bauer M, Karch R, Abrahim A, Feurstein T, Schutz M, Kletter K, Muller M, Lappin G, Langer O. A combined accelerator mass spectrometry-positron emission tomography human microdose study with 14C- and 11C-labelled verapamil. Clin Pharmacokinet. 2011;50:111–20. doi: 10.2165/11537250-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talbot DC, Ranson M, Davies J, Lahn M, Callies S, Andre V, Kadam S, Burgess M, Slapak C, Olsen AL, McHugh PJ, de Bono JS, Matthews J, Saleem A, Price P. Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clin Cancer Res. 2010;16:6150–8. doi: 10.1158/1078-0432.CCR-10-1932. [DOI] [PubMed] [Google Scholar]
- 11.Saleem A, Brown GD, Brady F, Aboagye EO, Osman S, Luthra SK, Ranicar AS, Brock CS, Stevens MF, Newlands E, Jones T, Price P. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003;63:2409–15. [PubMed] [Google Scholar]
- 12.van Dongen GA, Vosjan MJ. Immuno-positron emission tomography: shedding light on clinical antibody therapy. Cancer Biother Radiopharm. 2010;25:375–85. doi: 10.1089/cbr.2010.0812. [DOI] [PubMed] [Google Scholar]
- 13.Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–20. doi: 10.1176/appi.ajp.157.4.514. [DOI] [PubMed] [Google Scholar]
- 14.Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des. 2009;15:2550–9. doi: 10.2174/138161209788957528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, Holden J, Houle S, Huang SC, Ichise M, Iida H, Ito H, Kimura Y, Koeppe RA, Knudsen GM, Knuuti J, Lammertsma AA, Laruelle M, Logan J, Maguire RP, Mintun MA, Morris ED, Parsey R, Price JC, Slifstein M, Sossi V, Suhara T, Votaw JR, Wong DF, Carson RE. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab. 2007;27:1533–9. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 16.Zamuner S, Di Iorio VL, Nyberg J, Gunn RN, Cunningham VJ, Gomeni R, Hooker AC. Adaptive-optimal design in PET occupancy studies. Clin Pharmacol Ther. 2010;87:563–71. doi: 10.1038/clpt.2010.9. [DOI] [PubMed] [Google Scholar]
- 17.Ashworth S, Rabiner EA, Gunn RN, Plisson C, Wilson AA, Comley RA, Lai RY, Gee AD, Laruelle M, Cunningham VJ. Evaluation of 11C-GSK189254 as a novel radioligand for the H3 receptor in humans using PET. J Nucl Med. 2010;51:1021–9. doi: 10.2967/jnumed.109.071753. [DOI] [PubMed] [Google Scholar]
- 18.Girgis RR, Xu X, Miyake N, Easwaramoorthy B, Gunn RN, Rabiner EA, Abi-Dargham A, Slifstein M. In vivo binding of antipsychotics to D(3) and D(2) receptors: a PET study in baboons with [(11)C]-(+)-PHNO. Neuropsychopharmacology. 2011;36:887–95. doi: 10.1038/npp.2010.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abanades S, van der Aart J, Barletta JA, Marzano C, Searle GE, Salinas CA, Ahmad JJ, Reiley RR, Pampols-Maso S, Zamuner S, Cunningham VJ, Rabiner EA, Laruelle MA, Gunn RN. Prediction of repeat-dose occupancy from single-dose data: characterisation of the relationship between plasma pharmacokinetics and brain target occupancy. J Cereb Blood Flow Metab. 2010;31:949–52. doi: 10.1038/jcbfm.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzimopoulou S, Cunningham VJ, Nichols TE, Searle G, Bird NP, Mistry P, Dixon IJ, Hallett WA, Whitcher B, Brown AP, Zvartau-Hind M, Lotay N, Lai RY, Castiglia M, Jeter B, Matthews JC, Chen K, Bandy D, Reiman EM, Gold M, Rabiner EA, Matthews PM. A multi-center randomized proof-of-concept clinical trial applying [(1)F]FDG-PET for evaluation of metabolic therapy with rosiglitazone XR in mild to moderate Alzheimer's disease. J Alzheimers Dis. 2010;22:1241–56. doi: 10.3233/JAD-2010-100939. [DOI] [PubMed] [Google Scholar]
- 21.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 22.Wahl RL, Zasadny K, Helvie M, Hutchins GD, Weber B, Cody R. Metabolic monitoring of breast cancer chemohormonotherapy using positron emission tomography: initial evaluation. J Clin Oncol. 1993;11:2101–11. doi: 10.1200/JCO.1993.11.11.2101. [DOI] [PubMed] [Google Scholar]
- 23.Van den Abbeele AD, Badawi RD. Use of positron emission tomography in oncology and its potential role to assess response to imatinib mesylate therapy in gastrointestinal stromal tumors (GISTs) Eur J Cancer. 2002;38(Suppl. 5):S60–5. doi: 10.1016/s0959-8049(02)80604-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Xiong Z, Wu Y, Cai W, Tseng JR, Gambhir SS, Chen X. Quantitative PET imaging of tumor integrin alphavbeta3 expression with 18F-FRGD2. J Nucl Med. 2006;47:113–21. [PMC free article] [PubMed] [Google Scholar]
- 25.Matthews PM, Comley R. Advances in the molecular imaging of multiple sclerosis. Expert Rev Clin Immunol. 2009;5:765–77. doi: 10.1586/eci.09.66. [DOI] [PubMed] [Google Scholar]
- 26.Owen DR, Piccini P, Matthews PM. Towards molecular imaging of multiple sclerosis. Mult Scler. 2010;17:262–72. doi: 10.1177/1352458510390070. [DOI] [PubMed] [Google Scholar]
- 27.Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, Innis RB, Pike VW, Reynolds R, Matthews PM, Parker CA. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2010;52:24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, Rhodes C, Pulford DJ, Bennacef I, Parker CA, StJean PL, Cardon LR, Mooser VE, Matthews PM, Rabiner EA, Rubio J. An 18kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2011 doi: 10.1038/jcbfm.2011.147. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabiner EA, Beaver J, Makwana A, Searle G, Long C, Nathan PJ, Newbould RD, Howard J, Miller SR, Bush MA, Hill S, Reiley R, Passchier J, Gunn RN, Matthews PM, Bullmore ET. Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry. 2011;16:826–35. doi: 10.1038/mp.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heiss WD. The potential of PET/MR for brain imaging. Eur J Nucl Med Mol Imaging. 2009;36(Suppl. 1):S105–12. doi: 10.1007/s00259-008-0962-3. [DOI] [PubMed] [Google Scholar]
- 31.Pichler BJ, Kolb A, Nagele T, Schlemmer HP. PET/MRI: paving the way for the next generation of clinical multimodality imaging applications. J Nucl Med. 2010;51:333–6. doi: 10.2967/jnumed.109.061853. [DOI] [PubMed] [Google Scholar]
- 32.Cunningham VJP, Parker CA. PET studies in drug development: methodological considerations. Drug Discov Today Technologies. 2005;2:311–15. doi: 10.1016/j.ddtec.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Iwata R, Pascali C, Bogni A, Horvath G, Kovacs Z, Yanai K, Ido T. A new, convenient method for the preparation of 4-[18F]fluorobenzyl halides. Appl Radiat Isot. 2000;52:87–92. doi: 10.1016/s0969-8043(99)00117-7. [DOI] [PubMed] [Google Scholar]
- 34.Gombar VK, Polli JW, Humphreys JE, Wring SA, Serabjit-Singh CS. Predicting P-glycoprotein substrates by a quantitative structure-activity relationship model. J Pharm Sci. 2004;93:957–68. doi: 10.1002/jps.20035. [DOI] [PubMed] [Google Scholar]
- 35.Elsinga PH, Hendrikse NH, Bart J, Vaalburg W, van Waarde A. PET Studies on P-glycoprotein function in the blood-brain barrier: how it affects uptake and binding of drugs within the CNS. Curr Pharm Des. 2004;10:1493–503. doi: 10.2174/1381612043384736. [DOI] [PubMed] [Google Scholar]
- 36.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem Int Ed Engl. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 37.Laporte AM, Lima L, Gozlan H, Hamon M. Selective in vivo labelling of brain 5-HT1A receptors by [3H]WAY 100635 in the mouse. Eur J Pharmacol. 1994;271:505–14. doi: 10.1016/0014-2999(94)90812-5. [DOI] [PubMed] [Google Scholar]
- 38.Pike VW, McCarron JA, Lammerstma AA, Hume SP, Poole K, Grasby PM, Malizia A, Cliffe IA, Fletcher A, Bench CJ. First delineation of 5-HT1A receptors in human brain with PET and [11C]WAY-100635. Eur J Pharmacol. 1995;283:R1–3. doi: 10.1016/0014-2999(95)00438-q. [DOI] [PubMed] [Google Scholar]
- 39.Pike VW, McCarron JA, Lammertsma AA, Osman S, Hume SP, Sargent PA, Bench CJ, Cliffe IA, Fletcher A, Grasby PM. Exquisite delineation of 5-HT1A receptors in human brain with PET and [carbonyl-11 C]WAY-100635. Eur J Pharmacol. 1996;301:R5–7. doi: 10.1016/0014-2999(96)00079-9. [DOI] [PubMed] [Google Scholar]
- 40.Guo Q, Brady M, Gunn RN. A biomathematical modeling approach to central nervous system radioligand discovery and development. J Nucl Med. 2009;50:1715–23. doi: 10.2967/jnumed.109.063800. [DOI] [PubMed] [Google Scholar]
- 41.Baciu M, Sebai SC, Ces O, Mulet X, Clarke JA, Shearman GC, Law RV, Templer RH, Plisson C, Parker CA, Gee A. Degradative transport of cationic amphiphilic drugs across phospholipid bilayers. Philos Transact A Math Phys Eng Sci. 2006;364:2597–614. doi: 10.1098/rsta.2006.1842. [DOI] [PubMed] [Google Scholar]
- 42.Scheinin Noora M, Scheinin M, Rinne JO. Amyloid imaging as a surrogate marker in clinical trials in Alzheimer's disease. Q J Nucl Med Mol Imaging. 2011;55:265–79. [PubMed] [Google Scholar]
- 43.Kitson S, Cuccurullo V, Ciarmello A, Salvo D, Mansi L. Clinical applications of positron emission tomography (PET) imaging in medicine, oncology, brain diseases and cardiology. Curr Radiopharm. 2009;2:224–53. [Google Scholar]