

Abstract
To economically produce recombinant human α-galactosidase A (GLA) with a cell culture system that does not require bovine serum, we chose methylotrophic yeast cells with the OCH1 gene, which encodes α-1,6-mannosyltransferase, deleted and over-expressing the Mnn4p (MNN4) gene, which encodes a positive regulator of mannosylphosphate transferase, as a host cell line. The enzyme (yr-hGLA) produced with the gene-manipulated yeast cells has almost the same enzymological parameters as those of the recombinant human GLA produced with cultured human fibroblasts (agalsidase alfa), which is currently used for enzyme replacement therapy for Fabry disease. However, the basic structures of their sugar chains are quite different. yr-hGLA has a high content of phosphorylated N-glycans and is well incorporated into the kidneys, the main target organ in Fabry disease, where it cleaves the accumulated glycosphingolipids. A glycoprotein production system involving this gene-manipulated yeast cell line will be useful for the development of a new enzyme replacement therapy for Fabry disease.
INTRODUCTION
Fabry disease (MIM 301500) is an X-linked inborn error of metabolism with a high incidence of 1 in 1,250–4,000 male live births (1–3). In Fabry disease, a deficiency of α-galactosidase A (GLA) (MIM 300644; EC 3.2.1.22) activity causes the systemic accumulation of glycosphingolipids including globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3), and patients with the disease exhibit progressive renal, cardiac and vascular disorders (4–7).
So far, two different recombinant human GLAs, agalsidase alfa (Replagal®; Shire HGT, Cambridge, MA, USA) (8), produced with cultured human fibroblasts, and agalsidase beta (Fabrazyme®; Genzyme, Cambridge, MA, USA) (9,10), generated by Chinese hamster ovary (CHO) cells, have been developed, and they are used for enzyme replacement therapy (ERT) for Fabry disease. These recombinant GLAs are glycoproteins having both complex type and high-mannose type sugar chains produced by cultured mammalian cells, and it is thought that they are incorporated into cells mainly via mannose 6-phosphate (M6P) receptors in many organs (11) except for the liver, in which uptake by hepatocytes and Kupffer cells occurs mainly through asialoglycoprotein receptors and mannose ones, respectively (12). The incorporated GLAs cleave the glycosphingolipids deposited in Fabry organs, and there have been many clinical reports describing the efficacy of these recombinant enzymes (13–16). However, there are some disadvantages of producing recombinant enzymes using mammalian cells. Because mammalian cell cultures usually require bovine serum or a serum-free synthetic medium, the production of recombinant enzymes is expensive and/or careful monitoring for contamination by microbes is required to exclude infectious diseases such as bovine spongiform encephalopathy (BSE). Furthermore, the efficacy of uptake of the currently available recombinant GLAs by the kidneys is not so high (17,18). As renal insufficiency is the most significant disorder determining the prognosis of Fabry disease, the economical production of a safe recombinant enzyme that can be highly incorporated into the kidneys is urgently needed.
Previously, we produced a recombinant human GLA using budding yeast Saccharomyces cerevisiae and revealed that the enzyme cleaved the Gb3 accumulated in cultured Fabry fibroblasts and organs from Fabry mice. However, the productivity of the enzyme with the S. cerevisiae strain was very low (0.1 mg per 1-liter culture) (19,20).
In this study, we used a methylotrophic yeast, Ogataea minuta, in which the OCH1 gene is disrupted (21) and the Mnn4p (MNN4) gene is overexpressed (22,23), as host cells to produce a recombinant GLA having mammalianlike, phosphorylated N-glycans, and examined its effects on the organs of Fabry mice.
MATERIALS AND METHODS
Plasmid Construction and Transformation of a Methylotrophic Yeast
To prepare a human GLA expression vector for a methylotrophic yeast, O. minuta, an open reading frame encoding human GLA, was amplified using primers Xb-GLA-F (5′-CCCTCTAGAA AAATGCAGCTGAGGAACC-3′) and Ba-GLA-R (5′-GGGGGGATCCTTAAA GTAAGTCTT-3′), with pCXN2Gal (24) including the human GLA cDNA sequence as a template. The polymerase chain reaction fragment obtained was digested with XbaI and BamHI, and the resultant DNA fragment was inserted between the XbaI and BamHI sites of the pOMEU1 vector (21). On the other hand, plasmid pOMEG-ScMNN4 (25) was used for overproduction of ScMnn4p in O. minuta. These vectors were cut with NotI and used for transformation of the O. minuta TK-3-A strain (Δoch1) (21), which was provided by Kyowa Hakko Kirin (Tokyo, Japan). Transformation of O. minuta was performed as described previously (21,25). The correct integration was confirmed by polymerase chain reaction.
Expression and Purification of a Yeast Recombinant Human GLA (yr-hGLA)
The transformed O. minuta was precultured in 100 mL broth (1% yeast extract, 2% peptone, 0.2 mg/mL adenine, 2% glucose [YPAD]) and then transferred to 6 L broth (6% peptone, 1% yeast extract, 1.34% yeast nitrogen base without amino acids, 1% glycerol and 0.1 mmol/L potassium phosphate, pH 6.0 [BMGY]) in a jar fermentor. When the glycerol was completely consumed, methanol was added as a carbon source and inducer. Methanol induction was performed at 28°C and continued until the GLA activity in the culture broth reached saturation. The temperature and dissolved oxygen concentration were monitored and controlled by a computer during fermentation. After induction, the supernatant of the cultured medium was concentrated by ultrafiltration (Microza UF; Asahi Kasei Chemicals, Tokyo, Japan) and used as the crude enzyme.
Purification of GLA was performed at 4°C, with all of the column materials being purchased from GE Healthcare Bio-Sciences (Piscataway, NJ, USA). The crude enzyme was precipitated by 55% ammonium sulfate saturation, and the precipitate was dissolved in 25 mmol/L 2-(N-morpholino) ethanesulfonic acid (MES) buffer, pH 6.0. A sample was dialyzed against the same buffer and then applied to a HiLoad 16/10 Q Sepharose HP column equilibrated with 25 mmol/L MES buffer, pH 6.0. After the column was washed, the GLA protein was eluted with a linear gradient of NaCl, from 0 to 1 mol/L, in the same buffer. The fractions containing the enzyme activity were recovered, and then ammonium sulfate was added to the solution to a final concentration of 0.3 mol/L. The resultant solution was applied to a HiLoad 16/10 Phenyl Sepharose HP column equilibrated with 25 mmol/L MES buffer, pH 6.0, containing 0.3 mol/L ammonium sulfate. After the column was washed, the GLA protein was eluted with 25 mmol/L MES buffer, pH 6.0. The fractions containing the enzyme activity were pooled and concentrated by ultrafiltration. This solution was applied to a HiLoad 16/60 Superdex 200-pg column using 0.2 mol/L NaCl in the same buffer. The fractions containing the enzyme activity were pooled. Treatment of the fractions with a culture supernatant of the Cellulomonas sp. SO-5 strain for exposure of M6P residues on glycans was performed as described previously (19,25). After the treatment, a sample was further purified with a Q Sepharose HP column followed by a Superdex one under the same conditions as those given above, and the active fractions were collected.
Biochemical Analyses of yr-hGLA
To determine the purity and molecular mass of yr-hGLA, sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) followed by staining with Coomassie brilliant blue R was performed using yr-hGLA treated or not treated with glycopeptidase F (PNGase F). Deglycosylation of the enzyme was performed with an enzymatic deglycosylation kit (Prozyme, San Lendodro, CA, USA). Briefly, the enzyme proteins were electrophoresed on a Tris-glycine polyacrylamide gel and then stained with Coomassie brilliant blue R. Immunoblotting analysis with anti-GLA antibodies was performed as described previously (24). As a control, agalsidase alfa was used.
GLA activity was fluorometrically measured with 4-methylumbelliferyl (MU)-α-d-galactopyranoside (Calbiochem, La Jolla, CA, USA) as a substrate in the presence of N-acetyl-d-galactosamine (Sigma-Aldrich, St. Louis, MO, USA), a specific inhibitor of N-acetylgalactosaminidase, in 0.1 mol/L citrate-phosphate buffer, pH 4.6, as described previously (26). The fluorescence of 4-methylumbelliferone released on the enzyme reaction was measured with a Wallac 1420 ARVO MX Multilabel Counter (PerkinElmer, Waltham, MA, USA) at excitation and emission wavelengths of 355 nm and 460 nm, respectively. Protein determination was performed with a Micro BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA), using bovine serum albumin as a standard. A kinetic experiment involving yr-hGLA and various concentrations of the substrate was performed to determine the Michaelis constant (Km) value.
Pyridylamine (PA)-labeled N-glycans derived from yr-hGLA were separated by high-performance liquid chromatography (HPLC), and the percentage of phosphorylated N-glycans was calculated based on the peak areas, as described previously (25,27). Briefly, after PNGase F digestion, the yr-hGLA N-glycans were PA-labeled and fractionated on a normal-phase column, Shodex Asahipak NH2P-50 4E (Showa Denko, Tokyo, Japan), to roughly separate phosphorylated and nonphosphorylated glycans. Then, each fraction was applied to a TSK-gel Amide-80 column (Tosoh, Tokyo, Japan), and the amount of each fraction was calculated as the percentage of the total peak area.
The percentage of phosphorylated N-glycans was also confirmed by bacterial alkaline phosphatase digestion. PA-labeled N-glycans derived from 10 μg yr-hGLA and agalsidase alfa were treated with 2 units alkaline phosphatase from Escherichia coli C75 (Takara Bio, Shiga, Japan) in 50 mmol/L Tris-HCl, pH 9.5, at 37°C for 15 h. After boiling, the samples were filtered and analyzed on a COSMOSIL 5C18-AR-II column (4.6 × 250 mm; Nacalai Tesque, Kyoto, Japan). Isocratic separation was performed with 10 mmol/L sodium phosphate, pH 3.8, containing 0.075% 1-butanol for 30 min at a flow rate of 1 mL/min at 55°C. Samples before alkaline phosphatase treatment were used as controls. When the retention times shifted on the treatment, the fractions were judged to be phosphorylated N-glycans, and the amount of phosphorylated N-glycans was calculated as the percentage of the total peak area. Three independent experiments involving three independent preparations were performed, and average values with ± standard deviation (SD) are calculated.
Examination of the Effects of yr-hGLA on Organs of Fabry Mice
Mice were bred and maintained at the Animal Center of our university. Both male and female Fabry mice (GLA knock-out mice denoted by T Ohshima, et al. [28,29]) and wild-type C57BL/6 mice (14 months old) were used in this experiment, which was approved by the animal ethics committee of our university.
To determine the biodistribution of the enzyme, a single dose (1.0 mg/kg body weight) of yr-hGLA was injected into a tail vein of three Fabry mice. As a control, agalsidase alfa was used. The mice were sacrificed 1 h after administration of the enzymes, and then the GLA activity in the kidneys, heart and liver was measured. The mice were perfused with phosphate-buffered saline (pH 7.4) before removal of the organs.
To examine cleavage of the glycosphingolipids accumulated in the organs, two groups of Fabry mice, each consisting of three mice, were injected with yr-hGLA or agalsidase alfa (1.0 mg/kg body weight) separately every day for 4 d and were then sacrificed 24 h after the last injection.
For determination of the Gb3 levels, tissues including the kidneys, heart and liver were analyzed by means of high performance thin-layer chromatography-immunostaining with an anti-Gb3 monoclonal antibody (30), followed by densitometry, as described previously (31).
For measurement of lyso-Gb3 in mouse tissues, lyso-Gb3 was extracted and then derivatized with o-phthalaldehyde (OPA) reagent. The OPA-derivatized lyso-Gb3 was separated by HPLC and then quantitated by fluorescence detection. A calibration curve for lyso-Gb3 was prepared by the addition of authentic lyso-Gb3 to a normal tissue homogenate (7). For morphological analysis, mouse tissues were analyzed electron microscopically, as described previously (20).
Statistical Analyses
Data are expressed as mean ± SD. We performed statistical analyses with a Student t test. Values were considered statistically significant at P < 0.05.
RESULTS
Production and Purification of yr-hGLA
To produce a recombinant human GLA having mammalianlike, phosphomannosylated sugar chains, we expressed human GLA in a gene-manipulated O. minuta strain with the OCH1 gene, which encodes α-1,6-mannosyltransferase catalyzing the initial reaction of the outer sugar chain synthesis specific to yeast, deleted and overexpressing the S. cerevisiae MNN4 gene, which encodes a positive regulator of mannosylphosphate transferase. The productivity of the yr-hGLA was estimated to be 12 mg per 1-liter culture.
Then, the enzyme was purified from the culture medium by means of column chromatography. The purification degree was 5,230-fold, and the recovery of the enzyme was 35%.
Characterization of yr-hGLA
yr-hGLA was detected as two or more broad bands on SDS-PAGE, but it only gave one band after deglycosylation. This result suggested that yr-hGLA has heterogeneous sugar chains (Figure 1).
Figure 1.

Coomassie brilliant blue R staining and immunoblotting of yr-hGLA. The purified yr-hGLA and agalsidase alfa were separated on a Tris-glycine polyacrylamide gel and then stained with Coomassie brilliant blue R (left). Before the electrophoresis, the enzyme was treated (PNGase F+) or not treated (PNGase F–) with glycopeptidase F. Immunoblotting with anti-GLA antibodies was performed (right).
The enzymological parameters of yr-hGLA were determined using MU-α-d-galactopyranoside, an artificial substrate for GLA. The specific activity of yr-hGLA was 2.0 mmol/h/mg protein, and its Km value was 3.0 mmol/L, with these values being almost the same as those of agalsidase alfa (specific activity, 2.1 mmol/h/mg protein; Km, 2.8 mmol/L).
The monosaccharide composition of yr-hGLA was determined and then compared with that of agalsidase alfa, which had been reported previously (32). yr-hGLA has mannose residues, but does not contain any fucose, galactose or sialic acid residues (data not shown). To determine the content of phosphorylated N-glycans, N-glycans derived from yr-hGLA and agalsidase alfa were PA- labeled and then separated by HPLC. For agalsidase alfa, 15.3 ± 2.6% of the total N-glycans was found to be phosphorylated; on the other hand, for yr-hGLA, the phosphorylated N-glycan content was 28.7 ± 2.7%.
Effects of yr-hGLA in Fabry Mice
We injected a single dose of yr-hGLA into Fabry mice, and 1 h after the administration, the GLA activity in the kidneys, heart and liver was measured. As a control, agalsidase alfa was used. The results are shown in Figure 2. An apparent increase in enzyme activity was observed in the organs of Fabry mice after the yr-hGLA administration. The increase in enzyme activity on the administration of yr-hGLA was greater in the kidneys than in the case of agalsidase alfa, although it was apparently lower in the liver.
Figure 2.
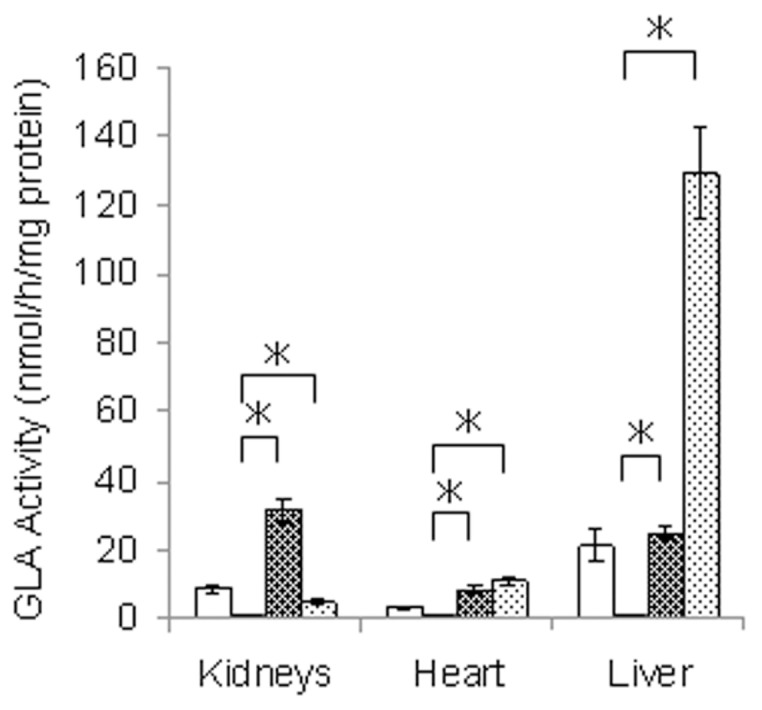
GLA activity in organs from Fabry mice treated with yr-hGLA or agalsidase alfa. Fabry mice were injected with a single dose (1.0 mg/kg body weight) of yr-hGLA or agalsidase alfa separately and sacrificed 1 h after the administration. Then, the GLA activity in the organs was measured. □, Wild-type mice; ■, Fabry mice;
 , Fabry mice treated with yr-hGLA;
, Fabry mice treated with yr-hGLA;
 , Fabry mice treated with agalsidase alfa. Error bars represent mean ± SD (n = 3). *P < 0.05.
, Fabry mice treated with agalsidase alfa. Error bars represent mean ± SD (n = 3). *P < 0.05.
The effects of yr-hGLA and agalsidase alfa on the degradation of Gb3 and lyso-Gb3 that had been accumulated in organs were examined separately after repeated administration. The results are summarized in Figure 3. Both the enzymes decreased the Gb3 and lyso-Gb3 accumulated in the kidneys, heart and liver. There were no differences in the decreases in the glycosphingolipid levels between yr-hGLA and agalsidase alfa under the experimental conditions. The lyso-Gb3 level in the kidneys decreased to approximately 30% of that in untreated Fabry mice, but the Gb3 level remained at about 80% of that in untreated Fabry mice.
Figure 3.

Levels of Gb3 and lyso-Gb3 in Fabry mouse organs. Fabry mice were repeatedly injected with yr-hGLA or agalsidase alfa (1.0 mg/kg body weight) separately every day for 4 d and sacrificed 24 h after the last injection. Then, the levels of Gb3 (left) and lyso-Gb3 (right) in the organs were determined. ■, Wild-type mice; □, Fabry mice;
, Fabry mice treated with yr-hGLA;
, Fabry mice treated with agalsidase alfa. Error bars represent mean ± SD (n = 3). *P < 0.05. ND, not detected.
Morphological analysis revealed that many lamellar inclusion bodies resulted from the accumulation of glycolipids in the kidneys, hearts and livers of Fabry mice and that the number of the inclusion bodies markedly decreased after the repeated administration of yr-hGLA, as in the case of agalsidase alfa (Figure 4).
Figure 4.

Morphological effects of repeated administration of yr-hGLA and agalsidase alfa. yr-hGLA or agalasidase alfa was repeatedly injected into Fabry mice separately, and then the kidneys, heart and liver were examined electron microscopically. The circles indicate pathological changes resulting from Fabry disease (inclusion bodies). Wild-type, wild-type mice; Fabry, Fabry mice; Fabry + yr-hGLA, Fabry mice treated with yr-hGLA; Fabry + Agalsidase alfa, Fabry mice treated with agalsidase alfa. The scale bar represents 1 μm.
DISCUSSION
ERT with recombinant human GLAs produced by mammalian cells has been reported to have beneficial effects in Fabry patients (alleviation of neuropathic pain, improvement of sweat function, regression of hypertrophic cardiomyopathy and also stabilization of kidney function), although it is not effective in the advanced stages of the disease (8–10,13–16). However, there are various disadvantages of the production of enzymes using mammalian cells (that is, the high cost, the complicated procedure and difficulty in performing a large scale-up and the risk of infections through cultivation in the presence of bovine serum). Although there is no experimental evidence that the BSE infectious agent is present in bovine serum, both the Center for Biologics Evaluation and Research of the U.S. Food and Drug Administration and the European Medicines Agency have issued guidelines for the controlled use of materials of animal origin to reduce the risk of BSE infection (33,34). To overcome these problems, we chose O. minuta cells as candidate host cells for the production of a recombinant GLA for ERT, because a methylotrophic yeast is suitable for large-scale and economic production of glycoproteins and does not require bovine serum for culture.
In this study, we used an OCH1- disrupted strain to produce a recombinant GLA having mammalianlike N-glycans. Because humans do not have any β-linked mannose, α-1,3–linked mannose or mannose residue with phosphodiester linkage (Manα1-PO4-), they could exhibit strong immunogenicity in humans (35–37). However, glycoproteins produced by O. minuta do not contain any β-mannose or α-1,3-mannose residues (21), and mannose attached to phosphate residues would be removed by the α-mannosidase treatment in the procedure for exposure of M6P residues on N-glycans. There have been reports that there is no difference in antibody production on ERT between agalsidase alfa produced by human fibroblasts and agalsidase beta produced by CHO cells when they are administrated at the same dose (38,39). Considering this, it is expected that the mammalianlike sugar chains of yr-hGLA produced by the gene-manipulated O. minuta do not exhibit high antigenicity in humans. We obtained a productivity level of >10 mg/L, which will be increased by further improvement and scaling up of the culture in the future.
We examined the enzymological characteristics of the recombinant human GLA produced by the gene-manipulated O. minuta cells. There is no large difference in enzyme activity or substrate affinity between yr-hGLA and agalsidase alfa. However, results of monosaccharide composition analysis suggested that yr-hGLA has high-mannose type sugar chains but not complex type ones and that its content of M6P residues is very high. These features will be advantageous for incorporation through M6P receptors into the cells of many organs, including the kidneys.
Lee et al. (40) examined the biodistribution of agalsidase alfa and agalsidase beta injected into tail veins of Fabry mice. The results suggested that the enzymes are predominantly incorporated into the liver. Recombinant lysosomal enzymes produced by mammalian cells usually contain both complex type and high-mannose type sugar chains, and most of the infused enzymes are rapidly incorporated into hepatocytes and Kupffer cells via asialoglycoprotein and mannose receptors, respectively. So, effective delivery of lysosomal enzymes to various organs except for the liver through M6P receptors should be compromised on rapid clearance of administered enzymes by asialoglycoprotein and mannose receptors in the liver. Sly et al. (41) reported that genetical elimination of mannose receptors slowed the plasma clearance of recombinant β-glucuronidase, resulting in enhanced degradation of the accumulated substrates in the kidneys. They also reported that saturation of the mannose receptor clearance system with high doses of enzyme improved the targeting to M6P receptor–containing tissues, including the kidneys. Thus, inhibition of uptake of lysosomal enzymes by the liver should facilitate their incorporation into the kidneys via M6P receptors. Our present animal study also revealed that the administration of agalsidase alfa greatly increased the GLA activity in the livers of Fabry mice. On the other hand, the increase in enzyme activity in the liver was moderate when yr-hGLA, which does not have complex type sugar chains, was injected. As with the kidneys, yr-hGLA exhibited markedly increased enzyme activity compared with in the case of agalsidase alfa. Because there should be no difference in protein sequence between yr-hGLA and agalsidase alfa, the difference in their biodistributions after injection should be due to a difference in their sugar chains. The mechanism underlying the high incorporation of yr-hGLA into the kidneys is unknown. But low clearance of the enzyme by the liver and/or a high content of phosphorylated N-glycans of yr-hGLA may play a role in the mechanism. Recently, Prabakaran et al. revealed that at least three endocytic receptors including M6P, megalin and sortilin were related to GLA delivery in human podocytes (Prabakaran et al. Receptor-mediated uptake of α-galactosidase A in human podocytes in Fabry disease. The 11th European Round Table on Fabry Disease, October 15, 2010, Istanbul). Considering the results, many receptors other than M6P are involved in the uptake of GLA in the kidneys, and they may play an important role in the mechanism.
Takamatsu et al. (42) reported that a recombinant fibroblast growth factor produced by gene-manipulated yeast cells was predominantly incorporated into the kidneys after intravenous injection into mice. Glycoproteins with a specific sugar chain structure produced by gene-manipulated yeast cells may be applicable to kidney-targeting enzyme incorporation.
As to the cleavage of glycosphingolipids accumulated in the kidneys, heart and liver, there is no apparent difference between yr-hGLA and agalsidase alfa on repeated injection every day for 4 d. Both the enzymes decreased the Gb3 and lyso-Gb3 levels in these organs. In the kidneys, the levels of Gb3 and lyso-Gb3 were reduced to approximately 80% and 30% that of the basal levels, respectively. The reason why the lyso-Gb3 level decreased more impressively on administration of the enzymes has not yet been clarified, but the difference in their hydrophobicity might underlie the effect of the hydrophilic enzyme. Morphological analysis also showed pathological improvement of the organs including the kidneys on the administration of yr-hGLA, as in the case of agalsidase alfa. There have been many reports that highly phosphorylated enzymes used for ERT are incorporated into the target organs and degrade the accumulated substrates more highly than normal enzymes (43–45). An animal study involving low doses of enzymes and long-term investigation is required to determine whether or not there is a difference in the cleavage of glycosphingolipids accumulated in the organs of Fabry mice between yr-hGLA and agalsidase alfa.
CONCLUSION
In conclusion, we produced a recombinant human GLA with a high content of phosphorylated N-glycans using a methylotrophic yeast. The enzyme was well incorporated into the kidneys and cleaved the glycosphingolipids accumulated in them. Because an enzyme production system involving a methylotrophic yeast is economic and does not require bovine serum for the procedure, it should be useful for the development of a new ERT for Fabry disease.
ACKNOWLEDGMENTS
We wish to thank Ashok B Kulkarni (National Institutes of Health) and Toshio Oshima (Waseda University) for providing the Fabry mice. We also thank Yoshiko Tanabe for typing the manuscript. This work was supported by the Program for Research on Intractable Diseases of Health and Labor Science Research Grants (H Sakuraba); the Program for the Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (ID: 09-15, H Sakuraba); the JAPS Asia/Africa Scientific Platform Program (H Sakuraba); the Japan Society for the Promotion of Science (JSPS ID: 21390314, H Sakuraba); and the High-Tech Research Center Project of the Ministry of Education, Culture, Sports, Science and Technology of Japan (S0801043, H Sakuraba).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Spada M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Gene. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwu WL, et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A) Hum Mutat. 2009;30:1397–405. doi: 10.1002/humu.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin HY, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Gene. 2009;2:450–6. doi: 10.1161/CIRCGENETICS.109.862920. [DOI] [PubMed] [Google Scholar]
- 4.Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3733–74. [Google Scholar]
- 5.Aerts JM, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105:2812–7. doi: 10.1073/pnas.0712309105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Togawa T, et al. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab. 2010;100:257–61. doi: 10.1016/j.ymgme.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Togawa T, et al. Tissue and plasma globotriaosylsphingosine could be a biomarker for assessing enzyme replacement therapy for Fabry disease. Biochem Biophys Res Commun. 2010;399:716–20. doi: 10.1016/j.bbrc.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Schiffmann R, et al. Infusion of α-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A. 2000;97:365–70. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eng CM, et al. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. 2001;68:711–22. doi: 10.1086/318809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eng CM, et al. Safety and efficacy of recombinant human α-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 11.Kornfeld S, Sly WS. I-cell disease and pseudo-Hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York: 2001. pp. 3469–82. [Google Scholar]
- 12.Rosenfeld EL, Belenky DM, Bystrova NK. Interaction of hepatic asialoglycoprotein receptor with asialoorosomucoid and galactolyzed lysosomal alpha-glucosidase. Biochim Biophys Acta. 1986;883:306–12. doi: 10.1016/0304-4165(86)90323-5. [DOI] [PubMed] [Google Scholar]
- 13.Mehta A, et al. Fabry Outcome Survey investigators: enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986–96. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- 14.Fervenza FC, Torra R, Warnock DG. Safety and efficacy of enzyme replacement therapy in the nephropathy of Fabry disease. Biologics. 2008;2:823–43. doi: 10.2147/btt.s3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thurberg BL, et al. Cardiac microvascular pathology in Fabry disease: evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation. 2009;119:2561–7. doi: 10.1161/CIRCULATIONAHA.108.841494. [DOI] [PubMed] [Google Scholar]
- 16.Salviati A, Burlina AP, Borsini W. Nervous system and Fabry disease, from symptoms to diagnosis: damage evaluation and follow-up in adult patients, enzyme replacement, and support therapy. Neurol Sci. 2010;31:299–306. doi: 10.1007/s10072-009-0211-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakuraba H, et al. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J Hum Genet. 2006;51:180–8. doi: 10.1007/s10038-005-0342-9. [DOI] [PubMed] [Google Scholar]
- 18.Ioannou YA, Zeidner KM, Gordon RE, Desnick RJ. Fabry disease: preclinical studies demonstrate the effectiveness of α-galactosidase A replacement in enzyme-deficient mice. Am J Hum Genet. 2001;68:14–25. doi: 10.1086/316953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiba Y, et al. Production in yeast of α-galactosidase A, a lysosomal enzyme applicable to enzyme replacement therapy for Fabry disease. Glycobiology. 2002;12:821–8. doi: 10.1093/glycob/cwf096. [DOI] [PubMed] [Google Scholar]
- 20.Sakuraba H, et al. Corrective effect on Fabry mice of yeast recombinant human α-galactosidase with N-linked sugar chains suitable for lysosomal delivery. Hum Genet. 2006;51:341–52. doi: 10.1007/s10038-006-0369-6. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda K, et al. Production of Man5GlcNAc2-type sugar chain by the methylotrophic yeast Ogataea minuta. FEMS Yeast Res. 2006;6:1052–62. doi: 10.1111/j.1567-1364.2006.00116.x. [DOI] [PubMed] [Google Scholar]
- 22.Odani T, Shimma Y, Tanaka A, Jigami Y. Cloning and analysis of the MNN4 gene required for phosphorylation of N-linked oligosaccharides in Saccharomyces cerevisiae. Glycobiology. 1996;6:805–10. doi: 10.1093/glycob/6.8.805. [DOI] [PubMed] [Google Scholar]
- 23.Odani T, Shimma Y, Wang XH, Jigami Y. Mannosylphosphate transfer to cell wall mannan is regulated by the transcriptional level of the MNN4 gene in Saccharomyces cerevisiae. FEBS Lett. 1997;420:186–90. doi: 10.1016/s0014-5793(97)01513-5. [DOI] [PubMed] [Google Scholar]
- 24.Ishii S, Kase R, Sakuraba H, Suzuki Y. Characterization of a mutant α-galactosidase gene product for the late-onset cardiac form of Fabry disease. Biochem Biophys Res Commun. 1993;197:1585–9. doi: 10.1006/bbrc.1993.2659. [DOI] [PubMed] [Google Scholar]
- 25.Akeboshi H, et al. Production of recombinant β-hexosaminidase A, a potential enzyme for replacement therapy for Tay-Sachs and Sandhoff diseases, in the methylotrophic yeast Ogataea minuta. Appl. Environ. Microbiol. 2007;73:4805–12. doi: 10.1128/AEM.00463-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayes JS, Scheerer JB, Sifers RN, Donaldson ML. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin Chim Acta. 1981;112:247–51. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 27.Akeboshi H, et al. Production of human β-hexosaminidase A with highly phosphorylated N-glycans by the overexpression of the Ogatae minuta MNN4 gene. Glycobiology. 2009;19:1002–9. doi: 10.1093/glycob/cwp080. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima T, et al. α-Galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci U S A. 1997;94:2540–4. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohshima T, et al. Aging accentuates and bone marrow transplantation ameliorates metabolic defects in Fabry disease mice. Proc Natl Acad Sci U S A. 1999;96:6423–7. doi: 10.1073/pnas.96.11.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kotani M, et al. Generation of one set of murine monoclonal antibodies specific for globo-series glycolipids: evidence for differential distribution of the glycolipids in rat small intestine. Arch Biochem Biophys. 1994;310:89–96. doi: 10.1006/abbi.1994.1144. [DOI] [PubMed] [Google Scholar]
- 31.Kawashima I, et al. Phospholipid storage in the myocardium of a unique Japanese case of idiopathic cardiomyopathy. Clin Chim Acta. 2006;372:154–7. doi: 10.1016/j.cca.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Tajima Y, et al. Use of a modified α-N-acetylgalactosaminidase in the development of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2009;85:569–80. doi: 10.1016/j.ajhg.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoon K. To manufacturers of biological products (Letter) Rockville, MD: U.S. Department of Health and Human Services, Center for Biologics Evaluation and Research; 2003. [Google Scholar]
- 34.European Medicines Agency (EMEA) Note for guidance on minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products (EMEA/410/01). Revised. London, UK: EMEA; 2003. [Google Scholar]
- 35.Shibata N, Suzuki A, Kobayashi H, Okawa Y. Chemical structure of the cell-wall mannan of Candida albicans serotype A and its difference in yeast and hyphal forms. Biochem J. 2007;404:365–72. doi: 10.1042/BJ20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Faille C, et al. Immunoreactivity of neoglycolipids constructed from oligomannosidic residues of the Candida albicans cell wall. Infect Immun. 1990;58:3537–44. doi: 10.1128/iai.58.11.3537-3544.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ballou CE. Isolation, characterization, and properties of Saccharomyces cerevisiae mnn mutants with nonconditional protein glycosylation defects. Methods Enzymol. 1990;185:440–70. doi: 10.1016/0076-6879(90)85038-p. [DOI] [PubMed] [Google Scholar]
- 38.Vedder AC, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. 2007;2:e598. doi: 10.1371/journal.pone.0000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vedder AC, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab. 2008;94:319–25. doi: 10.1016/j.ymgme.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Lee K, et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003;13:305–13. doi: 10.1093/glycob/cwg034. [DOI] [PubMed] [Google Scholar]
- 41.Sly WS, et al. Enzyme therapy in mannose receptor-null mucopolysaccharidosis VII mice defines roles for the mannose 6-phosphate and mannose receptors. Proc Natl Acad Sci U S A. 2006;103:15172–7. doi: 10.1073/pnas.0607053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takamatsu S, et al. Monitoring of the tissue distribution of fibroblast growth factor containing a high mannose-type sugar chain produced in mutant yeast. Glycoconj J. 2004;20:385–97. doi: 10.1023/B:GLYC.0000033995.95412.1f. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Y, et al. Glycoengineered acid α-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther. 2009;17:954–63. doi: 10.1038/mt.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Q, et al. Strategies for neoglycan conjugation to human acid α-glucosidase. Bioconjug Chem. 2011;22:741–51. doi: 10.1021/bc1005416. [DOI] [PubMed] [Google Scholar]
- 45.Tsuji D, et al. Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis. Ann Neurol. 2011;69:691–701. doi: 10.1002/ana.22262. [DOI] [PubMed] [Google Scholar]