

Abstract
Peritoneal dissemination is a common and fatal clinical manifestation of gastric cancer with few effective therapies available. Natural Newcastle disease virus (NDV) has been shown to be an effective oncolytic agent, and recent advances now allow genetic manipulation of this virus to improve cancer killing and safety. This study was designed to investigate the effectiveness of a genetically engineered NDV in the treatment of peritoneally disseminated gastric carcinoma. NDV mutant virus containing a modified F cleavage site and insertion of enhanced green fluorescent protein (GFP), NDV(F3aa)-GFP, was tested in vitro against human gastric cancer cells by standard cytotoxicity at different multiplicities of infection. To test NDV(F3aa)-GFP in vivo in a peritoneal carcinomatosis gastric tumor model, MKN-74 human gastric cancer cells were injected intraperitoneally (IP) in severe combined immunodeficient mice. Mice were treated with NDV (F3aa)-GFP either once or multiple times after tumor challenge. Effective killing of MKN-74 cells by NDV (F3aa)-GFP was found in vitro. This cancer killing was dose-related and correlated with viral replication. GFP expression was a good marker of infection. The virus was also effective as an antitumor therapy in a peritoneal cancer model that simulates clinical disease. Half the animals treated with virus had no evidence of disease. Genetically engineered NDV [NDV(F3aa)-GFP] administered IP is an effective antitumor therapy against peritoneal carcinomatosis from human gastric cancer in a xenograft model, without significant toxicity. These data provide further rationale for clinical trials involving NDV for peritoneal carcinomatosis from gastric cancer.
Keywords: Newcastle disease virus, Oncolysis, Peritoneal carcinomatosis, Stomach cancer
Introduction
Gastric cancer remains one of the most common malignant tumors worldwide [1]. In the last few decades, patient survival has improved as a result of early detection, development of surgical technique, and use of more effective anticancer agents [2, 3]. However, the prognosis of patients with advanced gastric cancer remains poor. Peritoneal carcinomatosis (PC) from gastric cancer occurs frequently, with estimates that 15–50% of patients have widespread peritoneal disease at the time of initial diagnosis [4, 5]. PC is the most common reason for failure after treatment for curative intent and, once established, is associated with a 5-year survival of less than 20% [6]. The most widely accepted therapies for PC are systemic or intraperitoneal chemotherapy, which are largely ineffective. Therefore, there has been a dire need for novel effective therapies for PC.
Oncolytic therapy using viruses is a promising form of cancer therapy, and it has become particularly attractive with improvements in techniques for manipulating RNA viruses to genetically modify these viruses into highly potent and tumor-specific vectors [7]. Among these is Newcastle disease virus (NDV), a member of the family Paramyxoviridae, which contains a 15,186 base-long, single-stranded, nonsegmented negative-sense RNA genome coding for seven viral proteins [8]. NDV causes serious infections in almost all birds but does not cause disease in humans. Only mild flu-like symptoms and conjunctivitis have been described in humans working with live NDV vaccines [9]. NDV was first noted to replicate in and destroy tumor cells in 1955, and the interest in using this virus as an antineoplastic agent has increased steadily in recent years [10, 11]. The inherent viral oncolytic properties are believed to derive from defective interferon (IFN) signaling pathways in tumor cells [12]. Normal cells, with an effective antiviral response, inhibit viral replication before significant damage is instigated, thereby allowing use of NDV as a safe and effective anticancer therapeutic agent. The known sensitivity of NDV to IFN and its inability to replicate in normal mammalian cells are likely to contribute to its safety in humans. In addition, NDV can be used against a variety of tumors, as it is known to enter cells by binding to sialic acid residues, which are present on a wide distribution of human and murine cancer cell lines [13].
Although naturally occurring strains of NDV used as oncolytic agents have shown some promising results and have been used in clinical trials, genetic modification of NDV affords the opportunity to improve antitumor efficacy. A cleavage of the F protein is known to be required for initiation of infection and is considered to be a major determinant of NDV virulence [14]. The reverse-genetics system has been used to modify the F protein to contain a multibasic cleavage and activation site. Mice in a metastatic colon tumor model (CT26) treated with an intravenous injection of this genetically engineered NDV, so-called “NDV(F3aa),” exhibited prolonged survival compared with wild-type control virus [15]. In this study, we evaluated the efficacy of NDV(F3aa)-GFP therapy against PC from human gastric cancer. We show that this therapeutic approach has the potential to improve current treatment of disseminated gastric cancer.
Materials and methods
Cells
Human gastric adenocarcinoma (AGS) cell lines were used in this study. These cells were maintained in Ham’s F-12 media, and MKN-74 cells were maintained in Roswell Park Memorial Institute 1640 media. All media contained 10% fetal calf serum, 100 μg/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained in a 5% CO2 humidified incubator at 37°C.
Virus
Virus was grown in eggs or cell culture per methods used by Park et al. [15]. Recombinant virus was amplified in 10-day-old embryonated chicken eggs and grown to a titer of 8.0 log10 egg 50% infectious dose per milliliter. In Madin–Darby canine kidney cell culture, the virus grew to a titer of 5×108 plaque-forming units (pfu) per milliliter and produced plaques approximately two thirds of the size of WSN.
The NDV mutant viruses with modified F cleavage site [NDV(F3aa)] were previously described. Briefly, the attenuated NDV Hitchner B1 strain (NDV-B1) was modified with the reverse-genetics system, and the cleavage site of the F protein modified with three amino acid changes, making it more fusogenic [15].
To generate NDV(F3aa) expressing GFP, a DNA fragment GFP flanked by the appropriate NDV-specific RNA transcriptional signals was inserted into the XbaI site created between the P and M genes of pT7NDV/F3aa. Viruses were rescued from complementary cDNA using methods described previously and sequenced by reverse transcription-polymerase chain reaction for insertion fidelity.
After NDV(F3aa)-GFP was generated as described above, it was produced, tittered, and stored. For virus preparation, 10-day-old embryonated hen eggs were inoculated (1,000 pfu) with the virus of interest and incubated (37°C, 48 h). At 48 h, the eggs were refrigerated and the allantoic fluid was harvested. After the cellular debris were cleared by centrifugation (15×g), the viruses were aliquoted and directly frozen (−80°C) until use. Virus titers were determined by inoculation of serial virus dilutions into Vero cells and counting by immunofluorescence. For each viral treatment, stored aliquots were taken from the −80°C freezer, thawed on ice, and reconstituted to the appropriate volume with cold phosphate buffered saline (PBS).
In vitro cytotoxicity assay
The human gastric cancer cell lines MKN-74 and AGS were evaluated for sensitivity to NDV(F3aa)-GFP in vitro. Cells were plated at 5×104 cells/well in 12-well plates (Costar, Corning, Corning, NY, USA) and infected 6 h later at multiplicities of infection (MOIs, virus particles per tumor cell) of 0, 0.01, 0.1, and 1. Cells were infected in 12-well plates for 1, 3, 5, and 7 days. At each time point, the media was aspirated and the cells were washed with 1 mL of PBS. The cells were subsequently incubated with 1.35% Triton X-100 at 37°C for 30 min to lyse the cells and release intracellular lactate dehydrogenase (LDH). LDH was then quantified with a Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Results are expressed as the surviving fraction of cells as determined by the measured absorbance of each sample relative to control, untreated cell lysates. All samples were analyzed in triplicate.
GFP expression
Cells were plated at 5×104 per well in 12-well plates in 1 mL of medium per well. Following 6 h of incubation, NDV(F3aa)-GFP in 100 μL of PBS was added to each well at MOIs of 0.01, 0.1, and 1.0. At time intervals of 1, 3, 5, and 7 days post-infection, cells were examined with an inverted fluorescence microscope (Nikon Eclipse TE300, Nikon, Tokyo, Japan) containing a GFP emission filter for GFP expression, and photographs were taken. All samples were analyzed in triplicate.
In vitro viral proliferation
Viral titering was performed to determine the ability of NDV(F3aa)-GFP to replicate in the MKN-74 human gastric cancer cell line. Cells were plated at 5×104 cells/well in 12-well plates and infected 6 h later with an MOI of 1.0. At 1, 3, 5, and 7 days post-infection, the supernatants were collected and the virus titers determined by serial dilution and immunofluorescence in Vero cells. Briefly, the day before infection, Vero cells were plated to confluence in a 96-well plate. Vero cells were infected with serial supernatant dilutions in a total volume of 20 μL. Twelve hours post-infection, Vero cells were fixed with 100 μL of 4% formaldehyde in PBS and permeabilized with 100 μL of 1% triton X-100 in PBS. Primary and secondary antibodies were anti-NDV polyclonal rabbit serum 1:500 and Alexa fluor 488 (green) 2° antirabbit 1:1,000, respectively. All samples were analyzed in triplicate.
Animal studies
All animal procedures were performed under approved protocols and in accordance with ethical guidelines by the Institutional Animal Care and Use Committee (IACUC) at Memorial Sloan-Kettering Cancer Center. Female severe combined immunodeficient (SCID) mice (National Cancer Institute, Bethesda, MD, USA) were used for all in vivo experiments and were housed in a temperature- and light-controlled animal facility. Food and water were permitted ad libitum. Anesthesia of animals was induced with a mixture of isoflurane (2 l/min) and oxygen (2 l/min) in an induction chamber and maintained with a nasal cone. All animals were sacrificed by CO2 inhalation at the termination of the experiment.
Toxicity study
Eight-to-10-week-old C57/Bl6 immune-competent mice underwent intraperitoneal (IP) injection of either 100 μL PBS or 2×106, 5×106, or 1×107 pfu of NDV(F3aa)-GFP. The mice were monitored for behavioral changes including failure to eat, drink, or groom normally. Toxicity studies were performed to evaluate the extent of inflammatory response. On day 1 and day 3 after injections, mice were sacrificed and whole blood was collected. After rapid centrifugation, serum was separated and analyzed by enzyme-linked immunosorbent assay for interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α; eBioscience, Inc., San Diego, CA, USA). Serum samples were also sent for alanine aminotransferase (ALT), aspartate aminotransferase (AST), and creatinine quantification.
Treatment of gastric carcinomatosis with intraperitoneal NDV(F3aa)-GFP
Twenty SCID mice were inoculated intraperitoneally with 5× 106 MKN-74 human gastric cancer cells suspended in 100 μL PBS. Viral treatment (n=15) groups included IP injection of 5×106 pfu of NDV(F3aa)-GFP in 100 μL PBS, either as a single treatment (day 1, n=7) or as multiple treatments (days 1, 4, and 7; n=8) after tumor challenge. The five control animals were treated with IP injection of PBS. For the duration of the study, all animals were weighed every other day, examined for cachexia, and monitored for signs of behavioral change, including abnormal eating, drinking, and grooming. Toxicity studies were performed to evaluate the extent of inflammatory response. Immune-deficient, virally treated mice were evaluated for behavioral changes, including changes in eating, drinking, or grooming habits. These mice experienced no deviation in behavior from their normal baseline. All animals were sacrificed 52 days after tumor challenge, or earlier because of large tumor size, in accordance with IACUC guidelines for ethical treatment of animals. At this time, all abdominal organs with associated peritoneum were removed en bloc. The mesentery, diaphragm, omentum, and all associated tumor were systematically stripped from the bowel and associated organs and weighed as peritoneal tumor specimens. After removing all organs and tumor, the carcasses were weighed. Pair-wise comparisons were performed using the exact Wilcoxon rank sum test and overall group comparisons were determined using the exact Kruskal-Wallis test.
Immunofluorescence staining of tumor
Peritoneal tumors were harvested and frozen in Tissue-Tek embedding medium (Sakura Finetek, Torrance, CA, USA), and immunofluorescence staining was performed to determine the presence of NDV(F3aa)-GFP in specimens. Primary and secondary antibodies were anti-NDV polyclonal rabbit serum 1:500 and Alexa fluor 488 (green) 2° antirabbit 1:1,000, respectively. Slides were counterstained with diamidino-2-phenylindole (SigmaAldrich Corp., St. Louis, MO, USA).
Results
In vitro cytotoxicity of NDV(F3aa)-GFP
The ability of NDV(F3aa)-GFP to kill two human gastric cancer cell lines was tested in vitro at MOIs of 0.01, 0.1, and 1 (Fig. 1). The virus effectively killed MKN-74 cells, and a dose response was observed. At an MOI of 1, more than 77% of the cells were killed at 7 days. Even at the lowest MOI of 0.01, over 30% of the cells had been killed by day 7. In contrast, the AGS cells were resistant to viral infection, with no significant cytotoxicity demonstrated 7 days after infection.
Fig. 1.
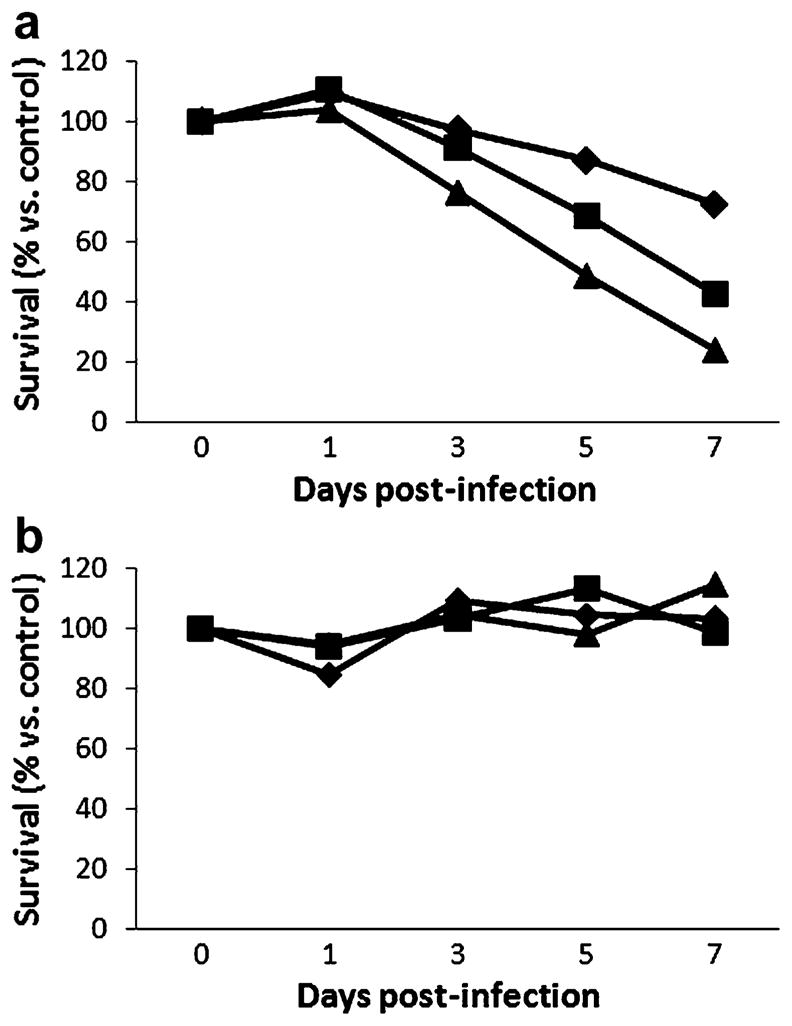
Killing of gastric cancer cells in vitro by NDV(F3aa)-GFP. The MKN-74 (a) cell line was highly sensitive to viral oncolysis at MOIs (multiplicities of infection, number of viral particles relative to cancer cells) of 1.0 (triangle), 0.1 (square), and 0.01 (diamond). The AGS (b) cell line showed minimal sensitivity to viral oncolysis
GFP expression in the infected cell lines
To monitor viral infection in the gastric cancer cell lines, GFP expression was assessed 1, 3, 5, and 7 days after viral infection at an MOI of 1.0 by fluorescence microscopy. MKN-74 cells demonstrated near 100% infectivity (all cells green) by day 5 (Fig. 2). AGS cells, on the contrary, demonstrated no GFP expression at all.
Fig. 2.
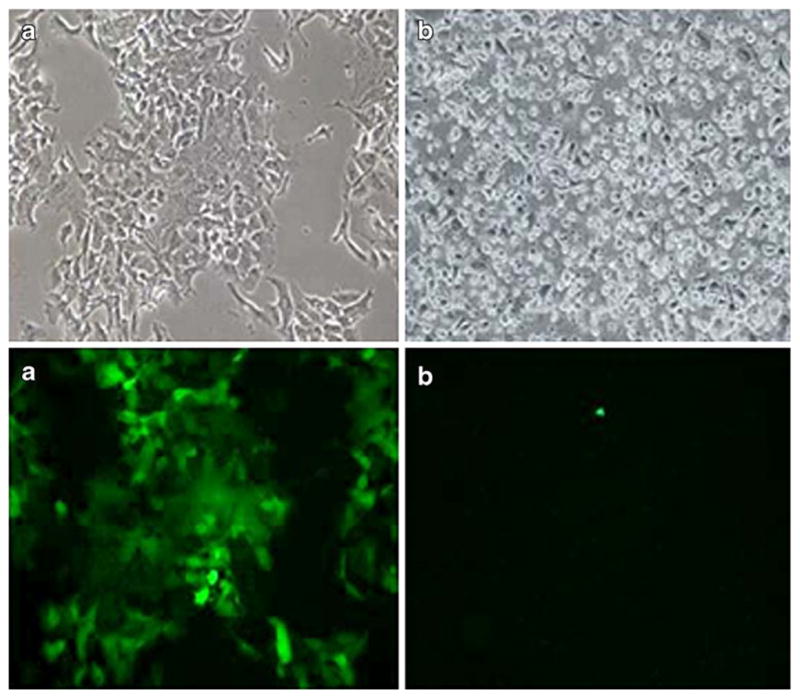
Green fluorescent protein (GFP) expression of MKN74 (a) and AGS (b) cell lines 5 days after infection with NDV(F3aa)-GFP. Abundant GFP expression was observed in the MKN74 (a) cell lines. AGS (b) displayed minimal GFP expression (×100 magnification)
In vitro viral proliferation
To assess for the efficiency of viral replication, supernatants were collected from the infected MKN-74 cells at different time points. MKN-74 strongly supported viral replication with a peak viral titer, measuring 1.7×104 pfu, being detected at 5 days after infection at an MOI of 1.0 (Fig. 3). No replication was detected for the resistant cell line AGS.
Fig. 3.

In vitro viral proliferation. NDV(F3aa)-GFP replication in MKN-74 gastric cancer cell line after in vitro infection with an MOI of 1.0. Supernatants were collected 1, 3, 5, and 7 days post-infection and titered by serial dilution and immunofluorescence on Vero cells. Peak viral titers measured 1.7×104 pfu at 5 days after infection
Toxicity study
None of the virally treated mice exhibited any behavioral changes that deviated from the eating, drinking, and grooming habits exhibited by mice treated with PBS. There were no deaths.
With respect to cytokine levels, the virally treated groups had serum IL-6 levels equivalent to or less than those obtained from the control mice on day 1, and at the higher doses (5×106 pfu group) had a mild 1.7-fold increased level, as compared with control levels, on day 3. The virally treated groups had a transient increase in TNF-α levels on day 1, with less than a threefold increased level as compared with mock-infected mice. These levels dropped to less than a 1.3-fold increase by day 3. All groups, including mock and virally treated groups, had serum IL-6 and TNF-α levels below 100 pg/mL at both day 1 and day 3 after IP injections. Certainly, all mice exhibited serum cytokine levels well below levels reported to be associated with toxicity of 40,000 pg/mL for IL-6 and 4,000 pg/mL for TNF-α [16, 17].
With respect to ALT, only the 1×107 pfu-treated group had an elevated serum level of 225 U/L, which normalized by day 3. AST levels were elevated in all groups on day 1, with the highest level in the 1×107 pfu-treated group at 369 U/L, compared with the control group level of 160.5 U/L. AST normalized in this group by day 3 as well. Creatinine levels remained normal in all groups at both day 1 and day 3 following IP injections.
In vivo treatment of gastric carcinomatosis with NDV(F3aa)-GFP
To assess the ability of a human gastric cancer cell line to develop peritoneal implantation, 5×106 MKN-74 cells in 100 μL in PBS were injected IP into SCID mice. No complications or toxicity from either IP injection of tumor or IP injection of NDV were observed. By 3 weeks after tumor inoculation, peritoneal nodules, including an omental mass, were observed; and by the fourth week, there was an increase in both size and number of tumor nodules. All collected nodules were confirmed to be poorly differentiated adenocarcinoma by hematoxylin and eosin staining. By the sixth week after tumor inoculation, control animals became lethargic, showed weight gain, and demonstrated progressively distended abdomens with overwhelming tumor, whereas the NDV(F3aa)-GFP-treated animals maintained baseline weight and continued to feed and groom normally (Fig. 4).
Fig. 4.

Body weight change of treatment groups. Four weeks after tumor injection, all control mice (dotted line) demonstrated weight gain due to tumor burden. Meanwhile, neither the single viral treatment (square) or multiple viral treatment (triangle) groups showed significant change of weight from baseline
In accordance with the IACUC guidelines for ethical treatment of animals, the control animals were sacrificed at 52 days or earlier after cancer cell injection, because of signs of lethargy, poor grooming, and distended, firm abdomens. The treated animals in the study were also sacrificed at this time so that assessment of tumor burden could be conducted in a consistent manner across the control and treatment groups.
All control animals had developed innumerable tumor nodules, ranging between several millimeters to 3.5 cm. However, there was no gross tumor in 6 (40%) NDV-treated mice (2 (29%) in the single treatment group; 4 (50%) in multiple treatment). Even in treated animals that had tumors, the nodules were significantly smaller, the largest being 1.5 cm (Fig. 5a) compared with untreated mice (Fig. 5b). The presence of NDV(F3aa)-GFP in tumor specimens in these treated animals was subsequently confirmed by immunofluorescence staining. Green cells representing virus were observed in a representative tumor section from a mouse treated with single virus injection (Fig. 5). Previous control analysis of the anti-NDV polyclonal rabbit antibody on VERO cells infected with NDV virus or mock infected with PBS confirmed that only cells infected with NDV virus demonstrated staining with the antibody. There was no background autofluorescence or non-specific antibody uptake detected (data not shown).
Fig. 5.

Efficacy of viral treatment for gastric carcinomatosis. A representative control mouse (a) had greater amounts of tumor burden (arrows) compared with a virally treated mouse (b). Below is a representative peritoneal tumor from a mouse treated with NDV(F3aa)-GFP. The tissue was fixed, permeabilized, stained with anti-NDV antibody, and counterstained with diamidino-2-phenylindole (DAPI; ×100 magnification)
Mean peritoneal tumor weight for the control group was significantly greater compared with the virally treated groups (7.22±1.59 g in control group; 1.46±2.06 g in single-treatment group; 1.36±2.01 g in multiple-treatment group, respectively; P=0.005). The control animals were also cachectic compared with the treated groups: carcass weight was significantly greater in the NDV-treated groups (P=0.005; Fig. 6). There was no significant difference of tumor weight between the single-treatment and multiple-treatment groups (P>0.05).
Fig. 6.

Peritoneal tumor burden (a) and carcass weight (b) of treatment groups. There was a significant difference in mean tumor weights (a) between control and virally treated groups (P=0.005), but no difference was noted between single and multiply treated groups. The weights of carcasses (b) in the control group were significantly lower compared with the virally treated groups (P=0.005). Error bars represent standard deviation
Discussion
NDV, a Paramyxoviridae family member, emerged as an anticancer therapeutic agent more than 50 years ago as a result of its potent oncolysis and limited toxicity to normal cells. NDV selectively replicates within tumor cells, making it an attractive anticancer therapeutic with a low toxicity profile [10]. This virus has been extensively studied as an oncolytic agent in different human tumor cell lines and tumor models. Lorence et al. demonstrated that a single local injection of NDV strain 73-T causes complete regression of human neuroblastoma [18] and fibrosarcoma [19] xenografts in athymic mice. This particular NDV strain also showed significant tumor growth inhibition in epidermoid, colon, breast, large cell lung, and prostate carcinoma models [20]. With these preclinical results, the naturally occurring strains of NDV have been used in clinical trials against advanced human cancers to confirm both safety and efficacy of the virus in human administration. More than 60% of patients with stage II melanoma treated with viral oncolysate after surgery were alive and had no detectable recurrent disease after 10 years [21]. In addition, patients with stage III malignant melanoma treated with an NDV oncolysate in 1975 as part of a phase II clinical study revealed a 55% overall 15-year survival [22].
Despite these successes, exploiting the immunostimulatory effects of NDV, the direct oncolytic effects of naturally occurring strains of NDV on human cancers was shown to be only marginal. To overcome this limitation, recombinant NDVs designed for enhanced cancer lysing therapeutic efficacy are being developed. A cleavage of the F protein is known to be required for initiation of infection and is considered to be a major determinant of NDV virulence [14]. Using reverse-genetics, the F protein of NDV-B1 strain was modified to contain a multibasic cleavage and activation site [15]. Modification of the cleavage site of the F protein allows the protein to be cleaved by intracellular proteases, making the virus more effective in entering cells and forming syncytia. Mice bearing implanted colon cancer CT26 tumors were treated with a recombinant NDV modified to contain a highly fusogenic F protein, NDV(F3aa). These treated mice exhibited significant reduction in tumor development and prolonged survival compared with wild-type control virus [16]. We demonstrate that experimental gastric cancer can be treated with this viral construct. Not only are tumor cells killed in vitro, significant anti-tumor efficacy in a gastric cancer PC model was also achieved.
PC is a frequent and eventually fatal complication of gastric carcinoma. Thus, the treatment and prevention of peritoneal dissemination may affect long-term survival. For primary tumors that breach the wall of the stomach (>T2), early postoperative peritoneal recurrence develops in more than half of patients. This finding implies that invisible micrometastases are already present in the peritoneal cavity at the time of operation. Unfortunately, there is no standard treatment for peritoneal dissemination. Surgery alone can only remove the bulky visible tumor nodules but is not suitable for control of invisible micrometastases. Systemic chemotherapy is the most widely accepted therapy for PC [23, 24], but, peritoneal metastases are known to be relatively resistant to systemic chemotherapy in gastric cancer due to poor blood supply and oxygenation of cancer cells in the peritoneum [25]. As peritoneal metastasis is considered to be caused by free cancer cells exfoliated from serosa-exposed tumor, some authors have advocated that it should be regarded as a locoregional extension of disease rather than another manifestation of systemic metastasis [26]. Therefore, many strategies utilizing instillation of therapeutic agents into the peritoneal cavity are being investigated. Bennett et al. demonstrated successful treatment of PC from human gastric cancer with a replication-competent herpes virus (G207) in an orthotopic cancer model [27]. G207 was highly effective in controlling intraperitoneal free cancer cells and eventually prolonged survival. Our data would suggest oncolytic NDV to be another potential agent that may be delivered in the peritoneal cavity to achieve cancer killing.
The NDV(F3aa) used in our study is well-tolerated. All animals receiving virus, including multiple injections, tolerated the treatment without any signs of behavioral changes and only mild, transient elevations in serum liver function and inflammatory markers. While control animals became cachectic from tumor, as evidenced by their significantly decreased carcass body mass, the treated animals maintained baseline weight throughout the observation period. This low toxicity, if verified in humans, would speak for this therapy as a potential adjuvant treatment after surgery to control micrometastases. While our study evaluated the therapeutic benefit of NDV(F3aa) against micrometastases and prevention of development of peritoneal disease, future studies evaluating its efficacy against established peritoneal tumor burden may also be important with significant clinical application.
This study demonstrated that NDV(F3aa)-GFP is effective in experimental treatment of a gastric tumor peritoneal model and, in some cases, completely cures the disease. Sensitivity of tumor to this virus can be correlated with evaluation of infectivity by the marker gene GFP and by in vitro viral replication assay. This virus can be safely administered and did not cause any signs of toxicity in the treatment groups. NDV(F3aa)-GFP, therefore, offers a novel therapeutic agent against gastric cancer, particularly against peritoneal dissemination, the most lethal form of this disease.
Footnotes
Disclosure of potential conflict of interests The authors declare no conflict of interests related to this study.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Nakajima T. Gastric cancer treatment guidelines in Japan. Gastric Cancer. 2002;5:1–5. doi: 10.1007/s101200200000. [DOI] [PubMed] [Google Scholar]
- 3.Park CH, Song KY, Kim SN. Treatment results for gastric cancer surgery: 12 years’ experience at a single institute in Korea. Eur J Surg Oncol. 2008;34:36–41. doi: 10.1016/j.ejso.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 4.Okajima K, Yamada S. Surgical treatment of far-advanced gastric cancer. Jpn J Cancer Clin. 1986;32:1203–1209. [PubMed] [Google Scholar]
- 5.Sugarbaker PH, Yonemura Y. Clinical pathway for the management of resectable gastric cancer with peritoneal seeding: best palliation with a ray of hope for cure. Oncology. 2000;58:96–107. doi: 10.1159/000012086. [DOI] [PubMed] [Google Scholar]
- 6.Ajani JA, Ota DM, Jackson DE. Current strategies in the management of locoregional and metastatic gastric carcinoma. Cancer. 1991;67:260–265. doi: 10.1002/1097-0142(19910101)67:1+<260::aid-cncr2820671309>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 7.Donahue JM, Mullen JT, Tanabe KK. Viral oncolysis. Surg Oncol Clin N Am. 2002;11:661–680. doi: 10.1016/s1055-3207(02)00025-x. [DOI] [PubMed] [Google Scholar]
- 8.Nemunaitis J. Live viruses in cancer treatment. Oncol Hunt. 2002;16:1483–1492. [PubMed] [Google Scholar]
- 9.Park MS, García-Sastre A, Cros JF, Basler CF, Palese P. Newcastle disease virus V protein is a determinant of host range restriction. J Virol. 2003;77:9522–9532. doi: 10.1128/JVI.77.17.9522-9532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson NJ. Scientific interest in Newcastle disease virus is reviving. J Natl Cancer Inst. 1999;91:1708–1710. doi: 10.1093/jnci/91.20.1708. [DOI] [PubMed] [Google Scholar]
- 11.Csatary LK. Viruses in the treatment of cancer. Lancet. 1971;2 (7728):825. doi: 10.1016/s0140-6736(71)92788-7. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamurthy S, Takimoto T, Scroggs RA, Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol. 2006;80:5145–5155. doi: 10.1128/JVI.02618-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reichard KW, Lorence RM, Cascino CJ, Peeples ME, Walter RJ, Fernando MB, Reyes HM, Greager JA. Newcastle disease virus selectively kills human tumor cells. J Surg Res. 1992;52:448–453. doi: 10.1016/0022-4804(92)90310-v. [DOI] [PubMed] [Google Scholar]
- 14.Sergel-Germano T, McQuain C, Morrison TG. Mutations in the fusion peptide and heptad repeat regions of the Newcastle disease virus fusion protein block fusion. J Virol. 1994;68:7654–7658. doi: 10.1128/jvi.68.11.7654-7658.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park MS, Steel J, García-Sastre A, Swayne D, Palese P. Engineered viral vaccine constructs with dual specificity: avian influenza and Newcastle disease. Proc Natl Acad Sci U S A. 2006;103:8203–8208. doi: 10.1073/pnas.0602566103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vigil A, Park MS, Martinez O, Chua MA, Xiao S, Cros JF, Martínez-Sobrido L, Woo SL, García-Sastre A. Use of reverse genetics to enhance the oncolytic properties of Newcastle disease virus. Cancer Res. 2007;67(17):8285–8292. doi: 10.1158/0008-5472.CAN-07-1025. [DOI] [PubMed] [Google Scholar]
- 17.Reid T, Galanis E, Abbruzzese J, Sze D, Wein LM, Andrews J, Randlev B, Heise C, Uprichard M, Hatfield M, Rome L, Rubin J, Kirn D. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): phase II viral, immunologic, and clinical endpoints. Cancer Res. 2002;62 (21):6070–6079. [PubMed] [Google Scholar]
- 18.Lorence RM, Katubig BB, Reichard KW, Reyes HM, Phuangsab A, Sassetti MD, Walter RJ, Peeples ME. Complete regression of human fibrosarcoma xenografts after local New-castle disease virus therapy. Cancer Res. 1994;54:6017–6021. [PubMed] [Google Scholar]
- 19.Lorence RM, Reichard KW, Katubig BB, Reyes HM, Phuangsab A, Mitchell BR, Cascino CJ, Walter RJ, Peeples ME. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J Natl Cancer Inst. 1994;86:1228–1233. doi: 10.1093/jnci/86.16.1228. [DOI] [PubMed] [Google Scholar]
- 20.Phuangsab A, Lorence RM, Reichard KW, Peeples ME, Walter RJ. Newcastle disease virus therapy of human tumor xenografts: antitumor effects of local or systemic administration. Cancer Lett. 2001;172:27–36. doi: 10.1016/s0304-3835(01)00617-6. [DOI] [PubMed] [Google Scholar]
- 21.Cassel WA, Murray DR. A ten year follow-up on stage II malignant melanoma patients treated postsurgically with New-castle disease virus oncolysate. Med Oncol Tumor Pharmacother. 1992;9:169–171. doi: 10.1007/BF02987752. [DOI] [PubMed] [Google Scholar]
- 22.Batliwalla FM, Bateman BA, Serrano D, Murray D, Macphail S, Maino VC, Ansel JC, Gregersen PK, Armstrong CA. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med. 1998;4:783–794. [PMC free article] [PubMed] [Google Scholar]
- 23.Hong YS, Song SY, Lee SI, Chung HC, Choi SH, Noh SH, Park JN, Han JY, Kang JH, Lee KS, Cho JY. A phase II trial of capecitabine in previously untreated patients with advanced and/or metastatic gastric cancer. Ann Oncol. 2004;15:1344–1347. doi: 10.1093/annonc/mdh343. [DOI] [PubMed] [Google Scholar]
- 24.Ohtsu A. Chemotherapy for metastatic gastric cancer: past, present, and future. J Gastroenterol. 2008;43:256–264. doi: 10.1007/s00535-008-2177-6. [DOI] [PubMed] [Google Scholar]
- 25.Ceelen WP, Påhlman L, Mahteme H. Pharmacodynamic aspects of intraperitoneal cytotoxic therapy. Cancer Treat Res. 2007;134:195–214. doi: 10.1007/978-0-387-48993-3_12. [DOI] [PubMed] [Google Scholar]
- 26.Bozzetti F, Yu W, Baratti D, Kusamura S, Deraco M. Locoregional treatment of peritoneal carcinomatosis from gastric cancer. J Surg Oncol. 2008;98:273–276. doi: 10.1002/jso.21052. [DOI] [PubMed] [Google Scholar]
- 27.Bennett JJ, Kooby DA, Delman K, McAuliffe P, Halterman MW, Federoff H, Fong Y. Antitumor efficacy of regional oncolytic viral therapy for peritoneally disseminated cancer. J Mol Med. 2000;78:166–174. doi: 10.1007/s001090000092. [DOI] [PubMed] [Google Scholar]