

Abstract
A novel synthetic strategy towards the asymmetric synthesis of vicinal diols bearing a tertiary center is presented. The method encompasses the dinuclear Mg-catalyzed asymmetric addition of ethyl diazoacetate into several aldehydes, oxidation of the diazo functionality, and diastereoselective alkyl transfer of various organometallics into the resulting chiral β-hydroxy-α-ketoesters to afford a diverse range of 1,2-diols in high yield, diastereoselectivity, and chirality transfer.
1. INTRODUCTION
Vicinal diols bearing a tertiary carbinol have played a vital role in the synthesis of natural products.1 Several strategies towards the enantioselective synthesis of these structural motifs exist.2 Asymmetric dihydroxylation and epoxidation/ring opening reactions have found broad utility.3 In these processes, product diastereoselectivity is highly dependent on the preparation of stereochemically defined tri-substituted olefins. Chemoselective control in these reactions may be attenuated by pendant olefins often present in complex molecules.4a Our work on the total synthesis of alternaric acid reveals the challenges of such a motif wherein a seven step sequence was required to assemble the vicinal diol fragment based upon an asymmetric dihydroxylation strategy.4b To address these limitations, Misaki, Sugimura, and Lu have recently reported an aldol based approach towards vicinal diols via C–C bond formation by the development of direct asymmetric catalytic aldol reactions using α-oxycarbonyls.5
As a complementary strategy, we envisioned accessing a broad range of 1,2-diols bearing a tertiary carbinol employing the direct catalytic asymmetric addition of ethyl diazoacetate to aldehydes and manipulation of the diazo group to access a diverse range of vicinal diols via sequential oxidation and stereoselective alkyl transfer as illustrated in Scheme 1.6 At the outset, we recognized the conversion of β-hydroxy-α-diazo esters 4 to β-hydroxy-α-ketoester electrophiles A and the subsequent addition of carbon nucleophiles to such reactive intermediates with high chirality transfer could prove to be challenging due to the high propensity of these electrophiles to undergo competitive enolization or an α-ketol rearrangement.7 Furthermore, accessibility of the resulting 1,2-diols without recourse to protecting group manipulation would rely upon the identification of carbon nucleophiles that are compatible with a free alcohol. Despite these challenges, the flexibility of this approach to afford 1,2-diol motifs contained in numerous natural products led us to investigate the viability of such a protocol. Herein we provide a full account of our studies that led to the development of a highly enantioselective magnesium-catalyzed addition of ethyl diazoacetate to aldehydes. In this paper, we also report the ability of the resulting β-hydroxy-α-diazo esters to undergo the chemo- and diastereoselective synthesis of diverse 1,2-diols bearing a tertiary alcohol with high chirality transfer (see Scheme 1).5a,8
Scheme 1.
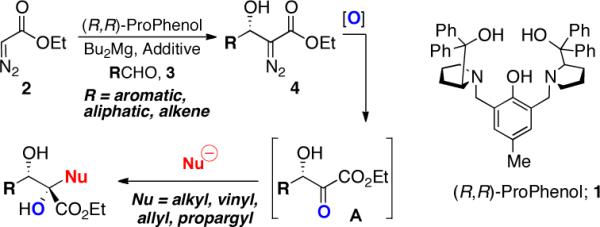
Strategy for the Asymmetric Synthesis of α,β- Dihydroxy -α-alkyl Esters.
2. RESULTS AND DISCUSSION
2.1 The Asymmetric Addition of Diazoesters to Aldehydes
The catalytic asymmetric direct aldol reaction constitutes a powerful strategy toward the atom economical synthesis of β-hydroxy carbonyl compounds.5a Although much progress has been made in the development of a direct ketone aldol, significantly fewer methods exist for the equivalent ester aldol reaction. The chemical reactivity of esters precludes their use in enamine catalysis, the higher pKa values of α-protons of esters relative to aldehydes or ketones also makes them less reactive substrates in direct aldol reactions. Furthermore, the thermal instability of simple ester enolates has limited the development of asymmetric aldol addition reaction of simple esters.
A tactic that has been employed in accessing β-hydroxyester containing scaffolds involves the asymmetric addition of diazoesters to aldehydes. A summary of methods utilizing asymmetric catalysis are presented in Scheme 2. Wang and coworkers first demonstrated the utility of diazoesters as nucleophiles in an asymmetric aldol reaction.9 A BINOL derived zirconium catalyst afforded the desired products in 53%–83% ee, where aromatic aldehydes resulted in higher levels of enantioinduction than aliphatic aldehydes. Arai and coworkers employed a chiral phase transfer cinchonidium derived catalyst that afforded the desired products in 0%–81% ee and demonstrated the utility of the resulting products in accessing α-amino acids.10 We became interested in obtaining a related product in high enantiocontrol to access a natural product fragment. This necessitated the development of a new method aimed at affording products with higher enantiocontrol. Herein we describe our efforts towards developing such a process, a portion of which has appeared in a preliminary communication, and the elaboration of such adducts for the asymmetric synthesis of vicinal diol motifs.6
Scheme 2.
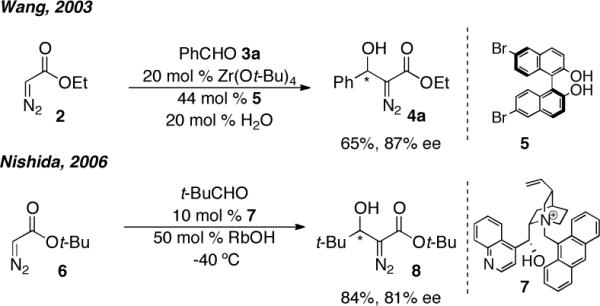
The Asymmetric Addition of Diazoesters to Aldehydes.
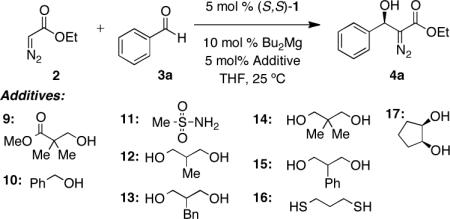
In our work, initial evaluation of the catalyst derived from the treatment of 5 mol % ProPhenol 1 with 10 mol % diethylzinc using ethyl diazoacetate 2 as the nucleophile and benzaldehyde 3a as the electrophile afforded the desired product 4a in high yields but in low enantioinduction. Further optimization (Table 1) revealed that the replacement of diethylzinc with di-n-butylmagnesium afforded the desired product in up to 27% ee (entries 1–5). Previous work on the use of the catalyst derived from ProPhenol 1 and diethylzinc wherein the remaining coordinative unsaturation had revealed that addition of complexing agents (additives) presumably to bind to the catalyst, provided a considerably more selective reaction pathway.11 Exploration of this concept with a dinuclear magnesium complex derived from the treatment of 10 mol % di-n-butylmagnesium, 5 mol % ProPhenol 1 and 5 mol % of β-hydroxy ester 9, the preferred additive in a previous Zn catalyzed process, enhanced the enantioselectivity to 37% ee (entry 6).
Table 1.
Selected Optimization Studies
| Entrya | Solvent | Additive | eeb (%) |
|---|---|---|---|
| 1 | PhCH3 | - | 9 |
| 2 | Et2O | - | 8 |
| 3 | Dioxane | - | −11 |
| 4 | DME | - | −16 |
| 5 | THF | - | 27 |
| 6 | THF | 9 | 37 |
| 7 | THF | 10 | 35 |
| 8 | THF | 11 | 27 |
| 9 | THF | 12 | 44 |
| 10 | THF | 13 | 38 |
| 11 | THF | 14 | 47 |
| 12 | THF | 15 | 51 |
| 13 | THF | 16 | 48 |
| 14 | THF | 17 | 65 |
All reactions run on a 0.47 mmol scale at 1M concentration in both donor 2 and acceptor 3.
Determined by chiral HPLC.
Coordinating groups possessing a mono alcohol such as 9 and 10 enhanced the enantiomeric excess to 37% ee (entries 6 and 7). The addition of methane sulfonamide 11 had no influence on the enantioinduction (entry 8). Replacement of mono-alcohols with bis-alcohols such as 2-substitutued-1,3 propane diols 12–15 further enhanced the enantioselectivity up to 51% ee (entries 9–12). Although the use of dithiol 16 increased the enantioselectivity, markedly low conversion was observed (entry 13). Finally the use of meso cis-1,2-cyclopentanediol 17 resulted in the greatest increase in the enantiocontrol affording 4a in 65% ee (entry 14).
Having identified that the addition of complexing groups can markedly influence the enantioselectivity in the formation of diazoester 4a, we reinvestigated the effect of various solvents employing additive 17 (Table 2). These studies revealed the superiority of THF over other coordinating and non-coordinating solvents such as dimethoxyethane (DME), 2-methyltetrahydrofuran, toluene, and heptane. Comparison of the results presented in Tables 1 and 2 also reinforce the role of the additive 17 in achieving higher optical purity of product 4a (compare experiments employing toluene, DME, and THF in Tables 1 and 2). Additionally, as a direct consequence of conducting the reaction on a larger scale, we observed that higher selectivity was achieved when ethyl diazoacetate 2 was added over a period of two minutes.
Table 2.
Influence of Solvent with the Incorporation of Additive 17.
| Entrya | mmol EDA | Solvent | Yield (%) | eeb (%) |
|---|---|---|---|---|
| 1 | 0.47 mmol | THF | NDc | 65 |
| 2d | 1.76 mmol | THF | 72 | −77e |
| 3d | 1.76 mmol | DME | 25 | −47e |
| 4d | 1.76 mmol | 2-MeTHF | 67 | −65e |
| 5d | 1.76 mmol | PhCH3 | 47 | −19e |
| 6d | 1.76 mmol | Heptane | 52 | −51e |
All reactions run on a 0.47 mmol scale at 1M concentration in both donor 2 and acceptor 3.
Determined by chiral HPLC.
ND = Not determined.
Addition over 2 min.
(R,R)-ProPhenol 1 employed.
Next, we investigated the influence of varying the stoichiometric ratio between (R,R)-ProPhenol 1, di-n-butylmagnesium, and meso cis-1,2-cyclopentanediol 17 (Table 3). Equalizing the molar ratio of magnesium and ligand 1 afforded only trace amounts of the desired product 4a (entry 2). Increasing the molar ratio of magnesium and (R,R)-ProPhenol 1 to 4:1 resulted in the formation of unidentified byproducts and lowered the optical purity of diazoester 4a (compare entries 1 and 3). Presumably, the addition of adventitious base promotes the formation of product 4a via the direct deprotonation of ethyl diazoaceate 2 with excess di-n-butylmagnesium. Furthermore, studies revealed that increasing the stoichometric ratio between cis-1,2-cyclopentanediol 17 and (R,R)-ProPhenol 1 also had a negative influence on the enantiocontrol (entries 4 and 5). Interestingly, a significant drop in optical purity was observed when the quantity of additive was increased from 5 mol % to 10 mol %. Greater enantiocontrol could be regained using 30 mol % meso cis-1,2-cyclopentanediol 17.
Table 3.
Investigating the Ratio of (R,R)-1, Magnesium, and Additive 17.
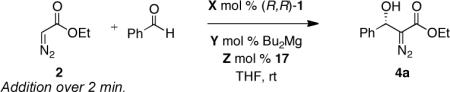
| Entrya | Solvent | X | Y | Z | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|---|
| 1 | THF | 5 | 10 | 5 | 20 | 72 | 77 |
| 2 | THF | 5 | 5 | 5 | 20 | Trace | NDd |
| 3 | THF | 5 | 20 | 5 | 20 | 52 | 47 |
| 4 | THF | 5 | 10 | 10 | 20 | 69 | 31 |
| 5 | THF | 5 | 10 | 30 | 20 | 66 | 47 |
All reactions were conducted using 1.76 mmol both ethyl diazoacetate 2 and benzaldehyde 3a at a concentration of 1.0 M in THF unless otherwise noted.
Isolated yields.
Determined by chiral HPLC.
ND = Not determined.
The influence of temperature on the enantioinduction was investigated employing either 1,3- or 1,2-diol additives 15 and 17 respectively (Table 4). These studies reinforced the superiority of 1,2-diols over 1,3-diols. Presumably, the conformational bias present in diol 17 enhances its coordination ability to the dinuclear magnesium catalyst. Additionally, it was observed that the highest level of enantioinduction was achieved at a reaction temperature of −20 °C employing either additive 15 or 17. The small decrease in selectivity by lowering the temperature to −40 °C could potentially be due to lowered reactivity leading to an increase in overall concentration of the diazoester during the slow addition that may result in the formation of a catalyst involving the coordination of multiple diazoesters. Another explanation invokes a shift of the relative energies of the diasteromeric transition states due to their differential temperature dependence. Experimental results support that the decrease in enantioselectivity is likely not due to a background reaction. The influence in the ratio of ligand to Bu2Mg (Table 3, entries 1–3) suggests that the presence of a background reaction only when a ratio of ProPhenol:magnesium is 1:4 is employed. Subsequent control experiments (vide infra) further support that the temperature effect is not due to a background reaction. Regardless of the exact explanation the effect is small.
Table 4.
Influence of Temperature on the Enantioinduction Employing diols 15 or 17.
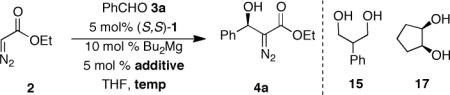
| Entrya | Additive | Temp (°C) | eeb (%) |
|---|---|---|---|
| 1 | 15 | −40 | 74 |
| 2 | 15 | −20 | 78 |
| 3 | 15 | 4 | 68 |
| 4 | 15 | 25 | 51 |
|
| |||
| 5 | 17 | −40 | 72 |
| 6 | 17 | −20 | 82 |
| 7 | 17 | 4 | 78 |
| 8 | 17 | 25 | 65 |
All reactions conducted on a 0.47 mmol scale at 1M concentration in both donor 2 and benzaldehyde acceptor 3a.
Determined by chiral HPLC.
Additionally, the evaluation of various ring sizes of commercially available 1,2-diols 17–19 revealed little influence of the ring size on enantioinduction (Table 5, entries 2, 4, and 5). The use of diol 17 offered a practical advantage as it can be added to the catalyst via syringe without further dilution and was selected as the additive of choice for further evaluation of this reaction. Further, we wondered whether the diazoester coordinated so strongly to the catalyst that multiple such units would be simultaneously involved which, in turn, would decrease the enantioselectivity. Interestingly, slow addition of ethyl diazoacetate 2 to the reaction mixture employing diol additive 17 afforded the desired product in highest enantiomeric excess of 95% ee (Table 5, entry 3). Presumably by maintaining a low concentration of ethyl diazoacetate 2 during the reaction, competition between the additive 17 and the nucleophile 2 is minimized thereby leading to greater enantiocontrol.
Table 5.
Influence of Ring Size of Additive on Enantioinduction.
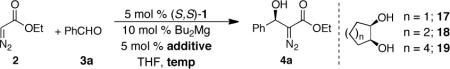
| Entrya | n | Temp. (°C) | Additive | ee (%)b |
|---|---|---|---|---|
| 1 | 1 | rt | 17 | 65 |
| 2 | 1 | −20 | 17 | 82 |
| 3 | 1 | −20 | 17 | 95c |
| 4 | 2 | −20 | 18 | 79 |
| 5 | 4 | −20 | 19 | 80 |
All reactions run on a 0.47 mmol scale at 1M concentration in both donor and acceptor.
Determined by chiral HPLC.
Diazoester 2 added via slow addition using a syringe pump.
2.2 Scope
Evaluation of the reaction scope revealed that high levels of enantioinduction could be achieved for a broad range of aldehydes. Carbocyclic aromatic aldehydes possessing both electron donating and withdrawing groups (Table 6, entries 1–9) all afforded products 4a–f with high levels of enantioinduction. The use of 2-furaldehyde (furfural) also afforded the aldol addition product 4g in 83% yield and 96% ee (entry 10).12 Aliphatic aldehydes such as those presented in entries 11–16 can be challenging substrates in metal-catalyzed direct aldol reactions due to the presence of enolizable protons. Previous studies employing ProPhenol 1 for direct aldol reactions with unbranched aliphatic aldehydes required a 10-fold excess of the donor and resulted in the isolation of products in modest yields and enantioselectivity.13 Employing aliphatic aldehydes in this process affords β-hydroxy diazoester products in yields ranging from 50–77% and in 90–98% ee using ethyl diazoacetate 2 and only up to two molar equivalents of the respective aldehyde. With cyclopropylcarboxaldehyde 3n the yield increased from 52% to 77% by increasing from one to two equivalents of the aldehyde (Table 6, entry 15 vs 16). For cyclohexanecarboxaldehyde higher yields were obtained by increasing the catalyst loading to 10 mol % (Table 6, entry 13). Lastly, of the two α,β-unsaturated aldehydes evaluated, cinnamyl aldehyde 3p (Table 6, entry 18) afforded the desired products in high enantioinduction. Further experiments revealed that the yield for this substrate was variable. Greater success was achieved with aldehyde 3o (Table 6, entry 17).
Table 6.
Expanded Reaction Scope for Asymmetric Catalytic Diazoester Aldol.
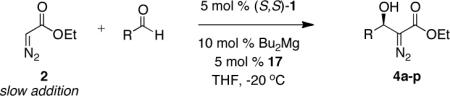
| Entrya | RCHO | Product | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | Ph 3a | 4a | 18 | 92 | 95 |
| 2d | Ph 3a | 4a | 24 | 95 | −95 |
| 3 | m-CH3OC6H4 3b | 4b | 18 | 83 | 90 |
| 4 | p-CH3OC6H4 3c | 4c | 18 | 70 | 87 |
| 5 | o-CIC6H4 3d | 4d | 18 | 91 | 89 |
| 6 | m-FC6H4 3e | 4e | 24 | 78 | 91 |
| 7e | p-FC6H4 3f | 4f | 24 | 87 | 94 |
| 8 | m-CIC6H4 3g | 4g | 18 | 88 | 98 |
| 9f | p-CIC6H4 3h | 4h | 18 | 85 | 96 |
| 10f | 2-Furyl 3i | 4i | 18 | 83 | 96 |
| 11f | i-Pr 3j | 4j | 24 | 56 | 97 |
| 12 | n-Bu 3k | 4k | 24 | 50 | 97 |
| 13g | Cyclohexyl 3l | 4l | 24 | 49 | 91 |
| 14h | Et 3m | 4m | 24 | 52 | 96 |
| 15 | cyclopropane 3n | 4n | 24 | 52 | >99 |
| 16h | cyclopropane 3n | 4n | 24 | 77 | >99 |
| 17 | E-BDMSCH=CH 3o | 4o | 24 | 86 | 90 |
| 18i | PhCHCH 3p | 4p | 18 | 50 | 94 |
All reactions were conducted using 0.88 mmol both ethyl dlazoacetate 2 and the respective aldehyde 3a–p at a concentration of 1.0 M in THF unless otherwise noted.
Isolated yields.
Determined by chiral HPLC.
Reaction conducted on 1.76 mmol scale of benzaldehyde 3a using (R,R)-1.
Ethyl diazoacetate 2 added at ~12.5 uL/hr.
Reactions conducted at 0.5 M.
Reaction conducted using 10 mol % (R,R)-1, 20 mol % Bu2Mg, 10 mol % 17 and 2 equiv. of diazoester 2.
Reactions conducted using 2 equiv aldehyde with respect to ethyl diazoacetate 2.
See discussion.
2.3 Mechanistic Discussion
A catalytic cycle for the transformation supported by experimental evidence is illustrated in Figure 1a. Treatment of ProPhenol 1 with two equivalents of di-n-butylmagnesium presumably affords species II. Although direct structural evidence for this complex II could not be ascertained by electrospray mass spectral analysis or X-ray crystallography, previous work by Ding and coworkers suggests the formation of a dinuclear complex (Figure 1b).14 Experimental results underscore the importance of the 1:2 ratio between (R,R)-ProPhenol 1 and magnesium and reveal that minimal background reaction takes place in the presence of free di-n-butylmagnesium alone or free ProPhenol 1 alone (Table 7). In the presence of an equivalent ratio of ProPhenol 1 and magnesium only trace amounts of the desired product are formed (Table 7, entry 1). A significant increase in yield is observed when the ratio between ProPhenol:magnesium is adjusted to 1:2. When the ratio is further increased to 1:4 (ProPhenol:magnesium) lowered yields and enantioselectivity are obtained. Initially we hypothesized that the lowered enantioselectivity was due to a competitive background reaction catalyzed either directly by free di-n-butylmagnesium, a magnesium alkoxide dissociated from the chiral ligand (such as the magnesium alkoxide of the product), or from free ProPhenol serving as a tertiary amine base. Further experimentation revealed that, in the absence of ProPhenol or di-n-butylmagnesium, no reaction at room temperature was observed (Table 7, entry 4). The presence of 5 mol % ProPhenol but no di-n-butylmagnesium afforded only trace amounts of product. Presumably the tertiary amine present in ProPhenol is promoting the formation of the trace product (Table 7, entry 5). Whether, di-n-butylmagnesium itself could catalyze the reaction led us to evaluate the reaction in the absence of ProPhenol. Interestingly, with 10 mol % Bu2Mg the desired product was obtained in 10% yield after 24h at room temperature (Table 7, entry 6). Replacing benzaldehyde with valeraldehyde 3k led to the formation of a complex mixture of products (Table 7, entry 7). In contrast, conducting the reaction in the presence of a catalyst derived from Bu2Mg, ProPhenol 1, and diol 17 at −20 °C with valeraldehyde 3k as the electrophile and with the slow addition of diazoester 2 (optimized results presented in Table 6) afforded the desired product in 50% yield (Table 7, entry 8).
Figure 1.

Proposed Catalytic Cycle for Asymmetric Addition of Diazoester 2 to Aldehyde 3a.
Table 7.
Evaluation of a Background Reaction.
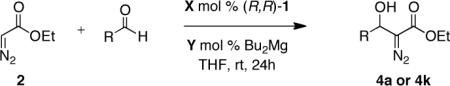
| Entrya | R | X | Y | Yield (%)b |
|---|---|---|---|---|
| 1 | Ph | 5 | 5 | Trace |
| 2c | Ph | 5 | 10 | 72 |
| 3 | Ph | 5 | 20 | 52 |
| 4 | Ph | 0 | 0 | 0 |
| 5 | Ph | 5 | 0 | Trace |
| 6d | Ph | 0 | 10 | 10 |
| 7d | n-Bu | 0 | 10 | complex mixture |
| 8c,e | n-Bu | 5 | 10 | 50 |
All reactions were conducted using 1.76 mmol both ethyl dlazoacetate 2 and benzaldehyde 3a at a concentration of 1.0 M In THF unless otherwise noted.
Isolated yields.
Reaction conducted in the presence of 5 mol % diol 17.
10 mol% Bu2Mg added to a solution of ethyl dlazoacetate prior to the addition of the respective aldehyde.
Reaction conducted under optimized conditions at −20 °C with slow addition of dlazoester 2.
These results suggest that free Bu2Mg promotes a detectable background reaction but only stoichiometrically to the amount of Bu2Mg in the case of benzaldehyde. In the case of an enolizable aldehyde valeraldehyde 3k a plethora of products is formed where only a minor amount of the desired product is detected. Furthermore, the studies support that the dinuclear metal complex illustrated in Figure 1 is indeed the reactive form. The influence of the slow addition of ethyl diazoacetate in enhancing the optical purity of the desired product is less due to non-ProPhenol catalyzed processes, but potentially because a higher concentration of ethyl diazoacetate leads to multiple coordination of diazoesters to the dinuclear complex leading to ambiguity of which one adds to the carbonyl group. Presumably, one role of the addition of the bidentate additive (diol 17) is that minimizes the number of available coordination sites where nucleophile 2 can complex.
Based on these results we postulate that upon initial addition of di-n-butylmagnesium three of the alkyl groups are consumed by the three protons on the ligand. The fourth alkyl group serves as the base to remove a proton from the 1,2-diol 17 to afford the desired complex III. Previous results on the incubation of diazoester 2 with D2O lead to significant H/D exchange at ambient temperature without the incorporation of an extraneous base thereby suggesting that the pKa of ethyl diazoacetate is suitably low for it to be deprotonated by the putative magnesium alkoxide in III (Figure 1c). The deprotonation would be expected to dramatically increase the rate of addition of ethyl diazoacetate to the aldehyde substrate. Thus, the deprotonation of ethyl diazoacetate (EDA) 2 employing magnesium alkoxide III affords complex IV. Coordination of benzaldehyde 3a then affords species V that undergoes an intramolecular aldol reaction affording the magnesium alkoxide complex VI. Protonation of this alkoxide in complex VI with ethyl diazoacetate 2 reveals the desired product and recycles to the catalytic species IV.
3. SYNTHESIS OF 1,2-DIOLS
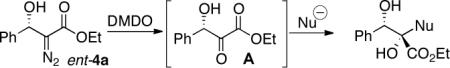
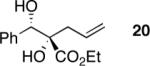
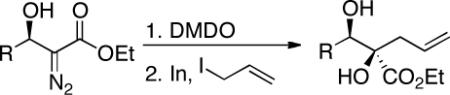
Treatment of readily accessible (S)-4a (95% ee) with dimethyldioxirane,15 concentration, followed by the addition of alkyl Grignard reagents at low temperatures resulted in complete conversion of the starting material, however, in disappointingly low yields and diastereocontrol of the tertiary alcohol products. We then turned our attention to less basic organoindium and organozinc reagents. Allylindium reagents readily undergo 1,2-addition with both aldehydes and ketones.16 These reagents, generated in situ, are efficacious in protic media. Paquette and coworkers have demonstrated the addition of allylindium reagents to achiral α-hydroxy carbonyl compounds.17 The transfer in chirality upon the addition of such nucleophiles to enantiomerically enriched β-hydroxy-α-ketoesters remained unknown. Gratifyingly, the addition of In(0) and allyl iodide in DMF to the crude reaction mixture afforded 1,2-diol 20 in 99% yield, 91% ee, and >20:1 dr over two steps (Table 8, entry 1).
Table 8.
Diastereoselective Addition of Carbon Nucleophiles to β-Hydroxy-α-ketoesters A.
| Entrya | Conditions | Product | % yieldb | drc | ct (%)d |
|---|---|---|---|---|---|
| 1 |
|
|
99 | >20:1e | 96 |
| 2 |
|
|
72 | >20:1 (1:1.3f) | 99,95 |
| 3 |
|
|
62 | >20:1 | >99 |
| 4 |
|
|
85 | >20:1e (5:1f) | 97,95 |
| 5 |
|
|
60g | >20:1e | >99 |
| 6 | Me2Zn |
|
71 | >20:1h | 99 |
| 7 | Et2Zn |
|
80 | >20:1 | >99 |
| 8 |
|
|
69 | 8:1 | 88 |
Conditions: (i) DMDO, concentrate; (ii) In (1.1 equiv), allyl halide (2.0 equiv) (entries 1–5); Me2Zn/Et2Zn/vinylzinc bromide (entries 6–8).
Combined isolated yield (over two steps).
Determined by crude NMR.
ct (%) (chirality transfer)=ee (%) product/ee (%) (S)-4a (HPLC).
Assigned by nOe on acetonide.
Diastereoselectivity at allylic position.
Isolated as a 5:1 mixture of homopropargyl alcohol and allenyl alcohol.
See supporting information for assignment.
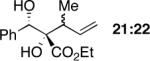
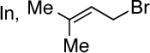
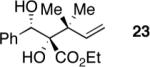
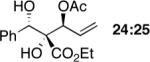
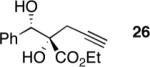
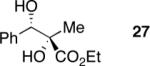
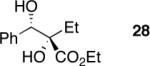
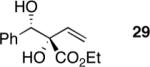
Exploration of the nucleophiles that can be employed is presented in Table 8. Using crotyl bromide, high levels of diastereocontrol at the carbinol centers was accomplished, however, a 1:1.3 ratio of products 21 and 22 epimeric at the methyl-bearing stereocenter was obtained potentially due to the rapid E/Z isomerization of the crotyl indium intermediate as has been previously observed by Paquette and coworkers (entry 2).18 Reverse prenylation of the ketoester derived from 4a led to the exclusive formation of 23 in 95% ee (entry 3). Stereotriad 24 was accessed from In(0) and 3-bromo-1-acetoxy-propene to generate a formal 1-hydroxyallyl anion equivalent.19 The desired product was obtained with a 5:1 dr with respect to the acetoxy-bearing stereocenter and 92% ee (entry 4). Using 3-bromoprop-1-yne led to propargyl alcohol 26 in 95% ee (entry 5). Organozinc reagents are also effective nucleophiles for the transfer of non-allyl groups. Commercially available Et2Zn and Me2Zn afforded 1,2-diols 27 and 28 both in >20:1 dr and 95% ee (entries 6 and 7).20 The addition of vinylzinc chloride, an sp2 hybridized nucleophile, afforded the desired diol 29 in 8:1 dr and 85% ee (entry 8). In contrast to these results, ethylmagnesium bromide afforded diol 28 in 24% and 4:1 dr and vinylmagnesium bromide afforded diol 29 in 24% and 1.9:1 dr in the absence of zinc chloride.
Next, we turned our attention to the diastereo- and enantio-selective allyl transfer to β-hydroxy-α-ketoesters where R ≠ Ph. Replacement of the phenyl group in diazoester 4a with other aromatic, aliphatic, and olefinic groups also affords vicinal diols 26–30 with high dr and chirality transfer (Table 9).
Table 9.
Reaction Scope with In-mediated Allyl Transfer
| Entrya | R | Product | % yieldb | drc | ct (%)d |
|---|---|---|---|---|---|
| 1 | p-MeOC6H4 | 30 | 89 | >20:1 | 98 |
| 2 | p-CIC6H4 | 31 | 90 | >20:1 | >99 |
| 3 | cyclopropane | 32 | 86 | 13:1 | >99 |
| 4 | n-Bu | 33 | 91 | >20:1 | >99 |
| 5 | PhCHCH | 34 | 33 | 6:1 | 94 |
Substrates prepared from (S,S)-ProPhenol.
Isolated yield (over two steps).
Determined by crude NMR.
ct (%) (chirality transfer) = ee (%) product/ee (%) starting material (HPLC).
To further demonstrate the synthetic utility of this method towards the streamlined synthesis of biologically active targets we rapidly assembled the western fragment of several macrolide antibiotics, including azithromycin which is pivotal in the containment of human bacterial infections. Magnesium-ProPhenol-catalyzed diazoester aldol with propionaldehyde affords 3m in 96% ee. Oxidation followed by the addition of Me2Zn initially afforded a complex mixture with trace quantities of the desired product. Interestingly, we found that the addition of racemic ProPhenol 1 promoted the alkyl transfer, thereby affording the requisite 1,2-diol. The ability of ProPhenol to facilitate alkyl transfer to such easily enolizeable substrates may prove to be a useful additional application of this catalytic zinc complex. Chemoselective acylation afforded western fragment 35 in 95% ee (Scheme 3).
Scheme 3.
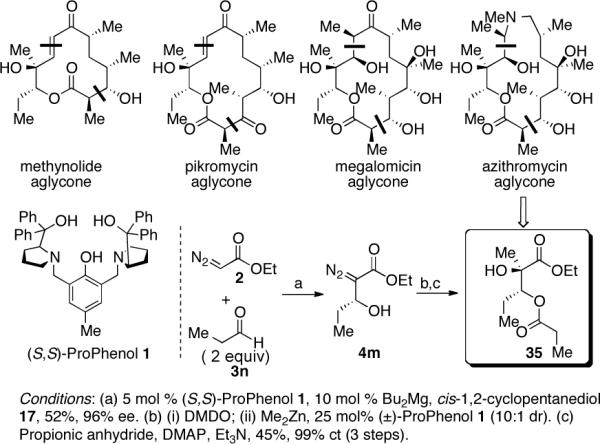
Synthesis of Polyketide Fragment
4. DETERMINATION OF RELATIVE AND ABSOLUTE CONFIGURATION
The relative and absolute stereochemistry were established by a combination of 1H NMR and X-ray techniques. In order to determine the absolute configuration of the stereogenic center created in the initial addition, we initially turned to the O-methyl-mandelate method2 which we applied to the more elaborated diol 31 derived from adduct (−)-4a (Scheme 4).
Scheme 4.
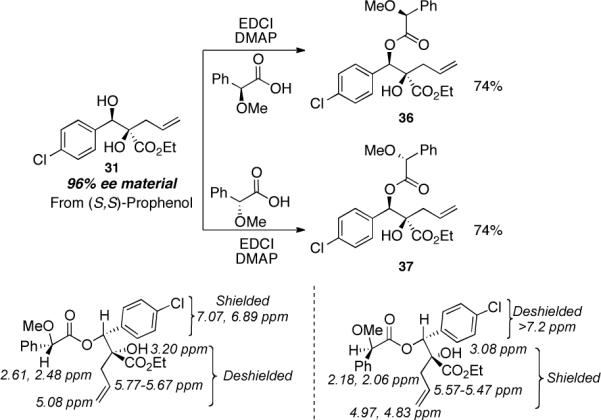
Synthesis of Mandelate Esters 36 and 37.
While this method establishes the absolute stereochemistry of the benzylic alcohol stereocenter, it does not establish the relative stereochemistry of the adjacent tertiary stereocenter. To address this deficiency, we turned to x-ray crystallographic analysis of acetonide 38 derived from initial adduct (S)-4a. As shown in Scheme 5a, conversion of diazoester 4a to diol 24, protection as the acetonide and acylation of the allylic alcohol with p-bromobenzoyl chloride afforded crystalline product 38. To determine whether the relative configuration was influenced by the functional nature of the allylating agent leading to adduct 24, the relative stereochemistry of two more diols 20 and 26 was determined via 1H NMR analysis of their corresponding acetonides 39 and 40 (Scheme 5b). Thus, the relative stereochemistry for all three acetonides, 38–40, is the same. The rationale for the diastereocontrol is based upon a chelation-controlled Zimmerman-Traxler transition state18 with preferential si-face attack (Scheme 5c).
Scheme 5.

Determination of Relative and Absolute Configuration.
5. CONCLUSIONS
We have developed a highly general catalyst system for the addition of commercially available ethyl diazoacetate to several aliphatic and aromatic aldehydes to access diverse β-hydroxy-α-diazoesters. Initial results employing a catalyst derived from the treatment of 5 mol % ProPhenol and 10 mol % Et2Zn or Bu2Mg furnished a catalyst capable of promoting the addition reaction in up to 37% ee. The incorporation of 5 mol % of several additives that putatively complex with the metal afforded the desired products in higher enantiocontrol. Further development of the reaction led to the identification of a catalyst system that afforded the desired products in up to 98% ee employing cis-1,2-cyclopentanediol as the additive and at −20 °C with the slow addition on ethyl diazoacetate. We hypothesize that the combination of the additive and the slow addition minimizes the coordination of ethyl diazoacetate to the catalyst thereby enhancing the enantioselectivity in the formation of β-hydroxydiazoesters.
Given our long-standing interest in accessing natural products containing vicinal diols, we envisioned the use of these enantiomerically enriched products to access 1,2-diols bearing a teritiary alcohol in high enantio- and diastereomeric purities using atom economical means. We envision that these motifs could be accessed via a sequence involving the oxidation of β-hydroxy-α-diazoesters to β-hydroxy-α-ketoesters followed by an alkyl transfer using a carbanion equivalent. The validity of this concept hinged upon the ability to identify carbon nucleophiles that would maintain the configurational stability of highly enolizable β-hydroxy-α-ketoesters without promoting α-ketol rearrangment and at the same time afford products in high diastereocontrol without recourse to protecting groups. Although initial efforts employing alkyl magnesium reagents led to limited yields and diastereoselectivity it was determined that less reactive allyl and propargyl indium species could be employed to access several diols and a triol in high optical purity. Furthermore, the addition of alkyl and vinyl nucleophiles was accomplished using vinyl and alkyl zinc reagents also affording products in high diastereoselectivity and chirality transfer. This strategy complements traditional asymmetric oxidations when chemoselectivity difficulties would preclude their use for the formation of products 20–26 and 29. Further, ability to access numerous α-hydroxy-β-diazoesters via a direct diazoester aldol coupled with the range of nucleophiles that can be employed in the sequential alkyl transfer to the corresponding β-hydroxy-α-ketoester reinforces the utility of the method in the rapid generation of molecular complexity.
6. EXPERIMENTAL SECTION
General Information
All magnesium-ProPhenol-catalyzed asymmetric additions of ethyl diazoacetate to aldehydes were performed in flame-dried glassware with magnetic stirring under an atmosphere of nitrogen in a −20 °C cryostat. All indium-mediated reactions were performed using “open- flask” conditions. All organozinc-mediated couplings were performed under an atmosphere of nitrogen.
Anhydrous DMF, dichloromethane, chloroform, tetrahydrofuran, acetone, acetonitrile, and toluene were obtained from a Seca solvent purification system by Glass Contour. Solvents and reagents were transferred via a syringe, which had been oven dried and cooled in a dessicator. Microliter syringes were dried under high vacuum for 1 h prior to use. Aldehydes for the asymmetric addition were freshly distilled prior to use. All other reagents were purchased from Aldrich Chemical Company and were used without further purification.
Analytical thin-layer chromatography was preformed on precoated 250μm layer thickness silica gel 60 F254 plates (EMD Chemicals Inc.). Visualization was performed by ultraviolet light and staining with ceric ammonium molybdate, potassium permanganate, or p-anisaldehyde. Organic solutions were concentrated by rotary evaporation at ambient temperature (essential for all diazo compounds). Flash column chromatography was performed using 40–63 μm silica gel (Silicycle silica gel) using compressed air. The eluents employed for flash chromatography are reported as volume: volume percentages.
Proton nuclear magnetic resonance (1H NMR) spectra were acquired using Varian Inova 400 MHz, 500 MHz, 600 MHz, and Mercury 400 MHz spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) and are calibrated to residual solvent peaks: proton (CDCl3 7.26 ppm). Coupling constants (J) are reported in Hz. Multiplicities are reported using the following abbreviations: s = singlet; br s = broad singlet; d = doublet; t = triplet; q = quartet; m = multiplet. 13C NMR spectra were recorded using a Varian Unity INOVA spectrometer at 125 MHz or a Varian Mercury at 100 MHz. Chemical shifts (δ) are reported in parts per million (ppm) and are calibrated to residual solvent peaks: carbon (CDCl3 77.0 ppm).
Infrared spectroscopic data was recorded on NaCl plates as thin films on a Thermo Scientific Nicolet IR100 FT-IR spectrometer. The absorbance frequencies are recorded in wavenumbers (cm−1). High resolution mass spectra (HRMS) were acquired by the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (http://mass-spec.stanford.edu) on a Micromass Q-Tof API-US mass spectrometer (Waters Corporation, Milford, MA). Chiral HPLC analysis was performed on Daicel Chiralpack columns (AD, AS, OB-H, OC, OD, or OJ) using heptane/2-propanol mixtures. The respective ratio of the eluent mixture, flow rate, detection wavelength, and column are indicated within the experimental details. Retention times (Tr) are reported in minutes (min). Optical rotations were measured using a JASCO DIP-1000 digital polarimeter in 50-mm cells and the sodium D line (589 nm) at the temperature, solvent, and concentration indicated.
Representative Procedure for the asymmetric addition of ethyl diazoacetate to aldehydes
(S)-ethyl 2-diazo-3-hydroxy-3-phenylpropanoate (4a)
To a solution of (R,R)-ProPhenol 1 (56 mg, 0.088 mmol, 0.05 equiv) in anhydrous THF (1.65 mL) was added a solution of di-n-butylmagnesium (176 μl of a 1 M solution in heptane, 0.10 equiv). After stirring for 30 minutes, cis-1,2-cyclopentanediol (9 μL, 0.088 mmol, 0.05 equiv) was added and the reaction was stirred for an additional 45 minutes. To the reaction was added benzaldehyde (177 μL, 1.76 mmol, 1.0 equiv). After stirring for 5 minutes at room temperature, the reaction was cooled to −20 °C and ethyl diazoacetate (201 mg, 1.76 mmol, 1.0 equiv) was added over 8 hours (25 μL/h). Upon completion, the reaction was quenched with pH 7 buffer and extracted with Et2O (5 × 5 mL). The organic layers were combined, dried (Na2SO4), and concentrated. Silica gel chromatography using a gradient of 15%–20% EtOAc/petroleum ether afforded 370 mg (95%, 95% ee) of the desired product as a yellow oil. This oil can be stored for several weeks in the freezer, however, transforms into the corresponding β-keto ester when stored at room temperature. TLC Rf = 0.50 (20% EtOAc/petroleum ether); [α]D25 –31.8 (95% ee, c. 11.5 mg/mL, CHCl3); 1H NMR (400 MHz, CDCl3): % 7.44–7.31 (m, 5H), 5.91 (d, J = 3.0 Hz, 1H), 4.26 (q, J = 7.2 Hz, 2H), 2.97 (br s, 1H), 1.29 (t, J = 7.2 Hz, 3H); HPLC Tr = 26.5 (minor) and 27.9 min (major) (Chiracel® OJ, λ = 254 nm, isocratic elution: heptane:iPrOH = 97:3, flow rate = 0.8 mL/min). The spectroscopic data was consistent with literature reported values.6
Representative Procedure for the conversion of β-hydroxy-α-diazoesters to vicinal diols
Indium Based Method: (S)-ethyl 2-hydroxy-2-((S)-hydroxy(phenyl)methyl)pent-4-enoate (20)
To a solution of (S)-ethyl 2-diazo-3-hydroxy-3-phenylpropanoate (40 mg, 0.18 mmol, 1.0 eq) in acetone (2 mL) was added DMDO (~0.1 M solution in acetone, 3.7 mL, 0.37 mmol, 1.7 eq) at −35 °C. Upon completion of the reaction (determined by TLC, ~ 1h) the reaction mixture was warmed to room temperature and concentrated under reduced pressure. Methylene chloride was added, the solution was dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in DMF (1.6 mL) and indium powder (23 mg, 0.2 mmol, 1.1 eq.) and allyl iodide (33 μL, 0.36 mmol, 2.0 eq) were added sequentially. The reaction mixture was stirred 12h and quenched with saturated aqueous NaHCO3. After extraction with CH2Cl2 (4 × 10 mL) the organic layers were combined, washed with H2O (50 mL), dried (Na2SO4), and concentrated under reduced pressure. Silica gel chromatography using a 20%–33% EtOAc/petroleum ether gradient afforded 45 mg (>99%, 91% ee) of the desired product as a white solid. Rf = 0.25 (20% EtOAc/Petroleum ether – KMnO4 stain); mp = 49–51 °C; [α] 25–34.2 (91% ee, c. 9.3 mg/mL, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.32–7.28 (m, 5H), 5.78 (dddd, J = 17.0, 10.3, 8.2, 6.5 Hz, 1H), 5.13 (m, 1H), 4.77 (d, J = 8.0 Hz, 1H), 4.10 (dq, J = 10.8, 7.2 Hz, 1H), 3.99 (dq, J = 10.8, 7.2 Hz, 1H), 3.36 (s, 1H), 3.06 (d, J = 8.0 Hz, 1H), 2.86 (ddt, J = 14.0, 6.9, 1.2, 0.6 Hz, 1H), 2.63 (ddd, J = 14.0, 7.4, 0.6 Hz, 1H), 1.17 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 173.6, 139.4, 132.5, 128.5, 128.3, 127.4, 119.4, 80.7, 77.7, 62.3, 40.7, 14.3 ppm; IR (neat) νmax 3649, 3503, 1733, 1717, 1699, 1558, 1541, 1507, 1456 1223, 1150 cm−1; HPLC Tr = 13.1 (minor) and 14.6 (major) (Chiracel® AD Chiral HPLC, λ = 220 nm, heptane:iPrOH = 90:10, 1.0 mL/min); HRMS (ES+) calcd for C14H18NaO4 [M+Na]+ 273.1103 found 273.1100.
(S)-ethyl 2-hydroxy-2-((S)-hydroxy(phenyl)methyl)-3,3-dimethylpent-4-enoate (23)
To a solution of (S)-ethyl 2-diazo-3-hydroxy-3-phenylpropanoate (40 mg, 0.18 mmol, 1.0 eq) in acetone (2 mL) was added DMDO (~0.1 M solution in acetone, 6 mL, 0.6 mmol, 3.3 eq) at −35 °C. Upon completion of the reaction (checked by TLC, approx. 1 h) the reaction mixture was warmed to RT and concentrated. CH2Cl2 was added, the solution dried over Na2SO4 and concentrated. The residue was dissolved in DMF (1.6 mL) and indium powder (23 mg, 0.2 mmol, 1.1 eq) and 3,3-dimethylallyl bromide (90% pure, 46.6 μL, 0.36 mmol, 2.0 eq) were added. After 16 h additional 3,3-dimethylallyl bromide (90% pure, 53.4 μL, 0.44 mmol, 2.0 eq) was added and the reaction mixture stirred for 2 h at 35 °C. The reaction was quenched with saturated aqueous NaHCO3 solution. After extraction with Et2O (4 × 10 mL) the organic layers were combined, washed with H2O (50 mL), dried over Na2SO4 and concentrated. The crude product was purified by silica gel chromatography (gradient 20%–33% EtOAc/petroleum ether) and afforded the title compound (20.7 mg, 62%, 95% ee) as a white solid. Rf = 0.40 (20% EtOAc/petroleum ether); mp = 97–99 °C; [α]D25 +24.0 (95% ee, c. 3.3 mg/mL, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.31–7.24 (m, 5H), 6.29 (dd, J = 17.6, 10.8 Hz, 1H), 5.19–5.09 (m, 3H), 4.00 (dq, J = 10.8, 7.2 Hz, 1H), 3.81 (dq, J = 10.8, 7.2 Hz, 1H), 3.55 (s, 1H), 2.90 (d, J = 8.2 Hz, 1H), 1.31 (s, 3H), 1.27 (s, 3H), 1.16 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 173.6, 144.7, 140.2, 128.1, 127.9, 127.6, 113.1, 83.1, 74.9, 62.0, 43.7, 23.5, 23.2, 13.8 ppm; IR (neat): νmax 3465, 2979, 1715, 1261, 1105 cm−1; HPLC Tr = 7.3 (minor) and 9.1 (major) (Chiracel® IA Chiral HPLC, l = 220 nm, heptane:iPrOH = 90:10, 1.0 mL/min); ee = 95%; HRMS (ES+) calcd for C16H22NaO4 [M+Na]+ 301.1416 found 301.1408.
Zinc Based Method: (2S,3S)-ethyl 2,3-dihydroxy-2-methyl-3-phenylpropanoate (27)
To a solution of (S)-ethyl 2-diazo-3-hydroxy-3-phenylpropanoate (50 mg, 0.22 mmol, 1.0 eq) in acetone (2 mL) was added DMDO (~0.1M solution in acetone, 6 mL, 0.6 mmol, 2.7 eq) at −35 °C. Upon completion of the reaction (checked by TLC, approx. 1 h) the reaction mixture was warmed to RT and concentrated. CH2Cl2 was added, the solution dried over Na2SO4 and concentrated. The residue was dissolved in THF (1 mL) and cooled to −78 °C. Dimethylzinc (1.2M solution in toluene, 0.6 mL, 0.66 mmol, 3.0 eq) was added dropwise and the reaction was allowed to warm gently to room temperature. After 40 h the reaction was quenched with pH 7 buffer and extracted with diethyl ether (4 × 10 mL). The organic layers were combined, dried over Na2SO4 and concentrated. The crude product was purified by silica gel chromatography using a gradient of 15%–33% EtOAc/petroleum ether to afford 35 mg (71%, 94% ee) of the desired product as a light yellow oil. Rf = 0.15 (20% EtOAc/petroleum ether); [α]D25 +10.0 (94% ee, c. 10.0 mg/mL, CHCl3);1H NMR (400 MHz, CDCl3): δ 7.36–7.27 (m, 5H), 4.74 (d, J = 7.4 Hz, 1H), 4.11 (dq, J = 10.4, 7.0 Hz, 1H), 4.03 (dq, J = 10.4, 7.0 Hz, 1H), 3.34 (s, 1H), 3.01 (d, J = 7.4 Hz, 1H), 1.56 (s, 3H), 1.18 (dd, J = 7.0, 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3): δ 174.5, 139.1, 128.2, 128.0, 127.0, 77.9, 775, 62.0, 22.6, 13.9 ppm; IR (neat): νmax 3473, 2984, 2939, 1730, 1453, 1243, 1160, 1049, 1026, 702 cm−1; HPLC Tr = 16.5 (minor) and 18.0 (major) (Chiracel® AD Chiral HPLC, l = 220 nm, heptane:iPrOH = 90:10, 0.8 mL/min). Submitted for MS.
Supplementary Material
Acknowledgements
This work has been supported by the National Institutes of Health (GM33049) and the National Science Foundation. S.M. acknowledges Stanford University for a graduate fellowship. P.K. acknowledges Landesstiftung BW for the Baden-Württemberg-Stipendium undergraduate scholarship. P. E. acknowledges DAAD and the Bayer Fellowship Program.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Selected examples: Forbes JE, Pattenden GJ. Chem. Soc., Perkin. Trans. 1991;1:1959.. Clive DLJ, Minaruzzaman Org. Lett. 2007;9:5315. doi: 10.1021/ol702509a.. Xie W, Ding D, Zi W, Li G, Ma D. Angew. Chem. Int. Ed. 2008;47:2844. doi: 10.1002/anie.200705557.. Boger DL, Ichikawa S, Zhong W. J. Am. Chem. Soc. 2001;123:4161. doi: 10.1021/ja010195q.. Trost BM, Friedreksen MU, Papillon JP, Harrington PE, Shin S, Shireman BT. J. Am. Chem. Soc. 2005;127:3666. doi: 10.1021/ja042435i.. Maki K, Motoki R, Fujii K, Kanai M, Kobayashi T, Tamura S, Shibasaki M. J. Am. Chem. Soc. 2005;127:17111. doi: 10.1021/ja0562043.. Trost BM, Probst GD, Schoop S. J. Am. Chem. Soc. 1998;120:9228.
- 2.Selected examples: Hatkeyama S, Matsui Y, Suzuki M, Sakurai K, Takano S. Tetrahedron Lett. 1985;26:6485.. Shao H, Rueter JK, Goodman M. J. Org. Chem. 1998;63:5240.. Claudel S, Olszewski TK, Mutzenardt P, Aroulanda C, Coutrot P, Grison C. Tetrahedron. 2006;62:1787.
- 3.Recent examples: Lim SM, Hill N, Myers AG. J. Am. Chem. Soc. 2009;131:5763. doi: 10.1021/ja901283q.. Kim HC, Kang SH. Angew. Chem. Int. Ed. 2009;48:1827. doi: 10.1002/anie.200805334.
- 4.a) Robles O, McDonald F. Org. Lett. 2009;11:5498. doi: 10.1021/ol902365n. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Probst G, Schoop A. J. Am. Chem. Soc. 1998;120:9228. [Google Scholar]
- 5.(a) Misaki T, Takimoto G, Sugimura T. J. Am. Chem. Soc. 2010;132:6286. doi: 10.1021/ja101216x. [DOI] [PubMed] [Google Scholar]; (b) Luo J, Wang H, Han X, Xu L-W, Kwiatkowski J, Huang K-W, Lu Y. Angew. Chem., Int. Ed. 2011;50:1861. doi: 10.1002/anie.201006316. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Dou X, Lu Y. Org. Lett. 2011;13:5248. doi: 10.1021/ol2021274. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Brindle CS. Chem. Soc. Rev. 2010;39:1600. doi: 10.1039/b923537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trost BM, Malhotra S, Fried BA. J. Am. Chem. Soc. 2009;131:1674. doi: 10.1021/ja809181m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For the addition of hydride nucleophiles a β-hydroxy-α-ketoesters see Liao M, Yao W, Wang J. Synthesis. 2004;16:2633.. Yao W, Wang J. Org. Lett. 2003;5:1527. doi: 10.1021/ol0343257.. For precedence for α-ketol rearrangement: Zhu Y, Tu Y, Yu H, Shi Y. Tetrahedron Lett. 1998;39:7819.; Paquette LA, Lobben PC. J. Org. Chem. 1998;63:5604. doi: 10.1021/jo980825f.; Steward KM, Johnson JS. Org. Lett. 2010;12:2864. doi: 10.1021/ol100996w.
- 8.Recent methods for the asymmetric synthesis of 1,2-diols bearing a tertiary carbinol: Giampietro NC, Kampf JW, Wolfe JP. J. Am. Chem. Soc. 2009;131:12556. doi: 10.1021/ja905930s.. Jiao P, Kawasaki M, Yamamoto H. Angew. Chem. Int. Ed. 2009;48:3333. doi: 10.1002/anie.200900682.. Nicewicz DA, Johnson JS. J. Am. Chem. Soc. 2005;127:6170. doi: 10.1021/ja043884l.
- 9.Yao W, Wang J. Org. Lett. 2003;5:1527. doi: 10.1021/ol0343257. [DOI] [PubMed] [Google Scholar]
- 10.(a) Arai S, Hasegawa K, Nishida A. Tetrahedron Lett. 2004;45:1023. [Google Scholar]; (b) Hasegawa K, Arai S, Nishida A. Tetrahedron. 2006;62:1390. [Google Scholar]
- 11.Trost BM, Fettes A, Shireman BT. J. Am. Chem. Soc. 2004;126:2660. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- 12.Ester 4g was light sensitive
- 13.For representative examples see Trost BM, Ito H. J. Am. Chem. Soc. 2000;122:12003.. Trost BM, Shin S, Sclafani JA. J. Am. Chem. Soc. 2005;127:8602. doi: 10.1021/ja051526s.
- 14.Xiao Y, Wag Z, Ding K. Macromolecules. 2006;39:128. [Google Scholar]
- 15.Adam W, Bialas J, Hadjiarapoglou L. Chem. Ber. 1991;124:2377. [Google Scholar]
- 16.For In(0)-mediated allylation of simple a-keto esters: Lee PH, Lee K, Chang S. Synth. Commun. 2001;31:3189.
- 17.(a) Paquette LA, Mitzel TM. J. Am. Chem. Soc. 1996;118:1931. [Google Scholar]; (b) Paquette LA, Mitzel TM. Tetrahedron Lett. 1995;36:6863. [Google Scholar]; (c) Paquete LA, Lobben PC. J. Org. Chem. 63:5604. doi: 10.1021/jo980825f. [DOI] [PubMed] [Google Scholar]
- 18.Paquette LA, Mitzel TM. J. Org. Chem. 1996;61:8799. doi: 10.1021/jo9613489. [DOI] [PubMed] [Google Scholar]
- 19.Lombardo M, Girotti R, Morganti S, Trombini C. Org. Lett. 2001;3:2981. doi: 10.1021/ol016315g. [DOI] [PubMed] [Google Scholar]
- 20.Diastereoselectivity for Me2Zn addition based on literature: Green JE, Bender DM, Jackson S, O'Donnell MJ, McCarthy JR. Org. Lett. 2009;11:807. doi: 10.1021/ol802325h.. Ethyl and vinyl addition products assigned by analogy
- 21.a) Dale JA, Mosher HS. J. Am. Chem. Soc. 1973;95:512. [Google Scholar]; b) Trost BM, Belletire JL, Godleski S, McDougal PG, Balkovec JM, Baldwin JJ, Christy ME, Ponticello GS, Varga SL, Springer JP. J. Org. Chem. 1986;51:2370. [Google Scholar]
- 18.Chen X, Hortelano ER, Eliel EL, Frye SV. J. Am. Chem. Soc. 1992;114:1778. [Google Scholar]; and references therein. Stanton GR, Johnson CN, Walsh PJ. J. Am. Chem. Soc. 2010;132:4399. doi: 10.1021/ja910717p. and references therein. Chelation control was also observed with the TBS protected (S)-1 consistent with reference 18a (supporting information)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.