

Abstract
Wnt7a signals through its receptor Fzd7 to activate the planar-cell-polarity pathway and drive the symmetric expansion of satellite stem cells resulting in enhanced repair of skeletal muscle. In differentiated myofibres, we observed that Wnt7a binding to Fzd7 directly activates the Akt/mTOR growth pathway thereby inducing myofibre hypertrophy. Notably, the Fzd7 receptor complex was associated with Gαs and PI3kinase and these components were required for Wnt7a to activate the Akt/mTOR growth pathway in myotubes. Wnt7a/Fzd7 activation of this pathway was completely independent of IGF-receptor activation. Together, these experiments demonstrate that Wnt7a/Fzd7 activates distinct pathways at different developmental stages during myogenic lineage progression, and together identify a novel non-canonical anabolic signalling pathway for Wnt7a and its receptor Fzd7 in skeletal muscle.
Keywords: Wnt7a, Fzd7, Akt/mTOR, Hypertrophy, non-canonical Wnt signalling
The Wnt family of genes encodes for nineteen cystein-rich, secreted glycoproteins which bind to Frizzled (Fzd) receptors on target cells1. In canonical Wnt-signalling, binding of Wnt proteins to Fzd activates Disheveled (Dsh), leading to the inactivation of Glycogen Synthase Kinase-3β (GSK-3β), a cytoplasmic serine-threonine kinase leading to stabilization of β-catenin2. In non-canonical Wnt signalling, Wnt proteins bind Fzd and glypican-4, to activate Dsh at the cell membrane, leading to activation of Rho and JNK or stimulation of calcium influx and activation of NFAT, PKC and CamKII. An additional non-canonical Wnt pathway, the planar-cell-polarity pathway (PCP), plays a role in patterning by instituting polarity of cells within a tissue, such as in the cochlea3. Importantly, we recently demonstrated a role for Wnt-PCP signalling in driving symmetric cell divisions of satellite stem cells in adult skeletal muscle by enhancing the planar polarization of stem cells4.
During development, Wnts are essential for myogenic induction in the paraxial mesoderm5–7. Wnts are also involved in regulating differentiation during muscle fibre development8, and during muscle regeneration in adults9,10. In the adult, canonical Wnt/β-catenin signalling has been suggested to control myogenic lineage progression by limiting Notch signalling and thus promoting differentiation11.
Satellite stem cells represent a small subpopulation of satellite cells that are capable of self-renewal and long-term reconstitution of the satellite cell niche following transplantation12. Notably, Wnt7a exposure dramatically stimulates the symmetric expansion of satellite stem cells and this expansion requires Fzd7 and Vangl2, both components of the PCP signalling pathway4. Wnt7a deficiency results in impaired maintenance of the satellite cell compartment. Therefore, Wnt7a signalling through the PCP pathway controls the homeostatic level of satellite stem cells and hence regulates the regenerative potential of muscle4.
IGF/IGFR activation of Akt/mTOR represents the key signalling axis regulating skeletal muscle hypertrophy13,. Binding of IGF-1 to the IGF-receptor directly actives PI3K and subsequently Akt. Activation of Akt promotes protein synthesis by activating the mTOR complex and inhibiting GSK3β. The mTOR TORC1 complex stimulates phosphorylation of ribosomal protein S6 which results in enhanced protein translation initiation and elongation. Activation of this pathway has the dual ability to inhibit proteolytic degradation and to stimulate new protein synthesis13.
Over expression of Wnt7a during muscle regeneration results in a marked enhancement of the regeneration process, generating increased numbers of larger fibres, independent of an effect on myoblast proliferation or differentiation4. Here, we investigate the activity of Wnt7a on postmitotic muscle fibres and identify an undescribed non-canonical Wnt/Fzd7-signalling pathway that directly activates the anabolic AKT/mTOR hypertrophy pathway.
We hypothesized that Wnt7a was inducing hypertrophy of myofibres as tibialis anterior (TA) muscle electroporated with a CMV-Wnt7a plasmid displayed an increase in mass and myofibre calibre4. To investigate whether Wnt7a was in fact stimulating hypertrophic growth of myofibres, we first exposed differentiating cultures of satellite cell derived primary myoblasts with recombinant Wnt7a. After 5 days of differentiation, we observed a significant increase in myotube diameter (Figs.1a–c). Similarly, differentiated C2C12 myotubes stably transfected with a CMV-Wnt7a-HA plasmid displayed enhanced myotube diameters (Figs.1e–g). Only application of Wnt7a, but not Wnt5a or Wnt3a, resulted in myofibre hypertrophy (Figs.1h, S1e–h), underscoring the specificity of the response to Wnt7a. The Wnt7a exposed myotubes also displayed about a 3-fold increase in the numbers of myonulei (Fig. S1a).
Figure 1.

Wnt7a induces hypertrophy in differentiated myotubes and myofibres. (a, b) Primary myoblasts derived from satellite cells were differentiated for 5 days in medium containing 50 ng/ml Wnt7a recombinant protein or BSA as a control. Staining for myosin heavy chain (MyHC, in green) marks differentiated cells. (c) Quantification of the fibre diameter as in a, b. n=3 (d) Quantification of the fibre diameter of C2C12 cells treated with 50 ng/ml recombinant Wnt7a protein. The Wnt7a recombinant protein was applied at day 3 of differentiation when the majority of the cells are already differentiated. n=3 (e,f) C2C12 cells were stably transfected with a CMV-Wnt7a-HA expression plasmid and differentiated for 5 days. (g) Quantification of the fibre diameter as in e,f. n=3 (h) Application of different recombinant Wnt proteins (50 ng/ml each) revealed that induction of hypertrophy is a Wnt7a specific phenomenon. n=3 (i,j) Electroporation of the CMV-Wnt7a-HA expression plasmid (40 μg) into the tibialis anterior muscle of adult mice resulted in increased fibre diameters 2 weeks after electroporation compared to a control (CMV-lacZ) plasmid. (k) Quantification of the fibre diameter of TA muscles electroporated with expression plasmids for Wnt3a-HA, Wnt5a-HA, Wnt7a-HA or a lacZ control plasmid (40 ug plasmid each). n=4 (l) Quantification of a single injection of recombinant Wnt7a protein (2.5 ug) into the tibialis anterior muscle of adult mice, mice were sacrificed two weeks after injection. n=4 (m,n) representative images of immunostained sections of TA muscle showing Pax7- (red) and laminin-staining (green). Nuclei were counterstained with Dapi (blue) (o) Intramuscular injection of recombinant Wnt7a protein into the tibialis anterior (TA) muscle resulted in a significant increase in the muscle weight, without affecting the weight of the contralateral muscle three weeks after injection. Injection of long-IGF stimulated an equal increase in both injected and contralateral TA muscle. n=4 (p) Wnt7a stimulated an expansion in the satellite cell pool whereas IGF did not. n=4 (q) Wnt7a and IGF injection resulted in a significant increase in fibre calibre. Grey bars indicate the contralateral muscle.. n=4, *p<0.01, **p<0.001, *** p<0.0001. Error bars represent SEM.
To discriminate between induction of hypertrophy and enhanced fusion, recombinant Wnt7a was applied to myotubes after 3 days of differentiation. We observed an identical degree of hypertrophy (Figs.1d, S1b). Furthermore, myotubes were treated with Wnt7a after application of Cytosine arabinoside (AraC)14,15, an inhibitor of DNA replication, to eliminate mononuclear myoblasts. Notably, myotubes in AraC-treated cultures similarly displayed enhanced myofibre diameter (Fig. S1k–m). We next investigated the possibility that Wnt7a accelerates differentiation or enhances proliferation. Western blot and mRNA analyses revealed normal kinetics of various of myogenic markers (Fig. S1i,j). Lastly, the rate of proliferation of primary myoblasts4, or of C2C12 myoblasts was not affected (Fig. S1d). Therefore we conclude that Wnt7a acts on already established myotubes to induce hypertrophy and is not a consequence of accelerated kinetics of differentiation or enhanced myoblast proliferation.
Electroporation of plasmid CMV-Wnt7a into the TA muscle of adult muscle stimulates both satellite cell expansion and myofibre growth to induce productive hypertrophy4. Electroporation with CMV-Wnt3a and CMV-Wnt5a expression plasmids did not induce hypertrophy, providing further support for the specificity of the Wnt7a response (Fig. 1k). However, the electroporation conditions used also result in an injury to the muscle raising the question of whether active regeneration is required for the Wnt7a response.
To address whether Wnt7a is capable of stimulating productive hypertrophy with minimal induction of regeneration as compared with electroporation, recombinant Wnt7a protein was directly injected into the TA muscles of seven-week old mice (n=3). We observed that the maximum response occurred after injection of 2.5 μg of Wnt7a with the mass of the TA muscle significantly increased by over 40% (p<0.001) (Fig. 1o). Moreover, the numbers of satellite cells were also significantly increased by almost 2-fold per field (p<0.001) (Fig. 1p) as well as the fiber calibre (Fig. 1q,m,n). Interestingly, the entire muscle was affected suggesting that the injected Wnt7a protein was distributed throughout the muscle. While IGF injection enhanced muscle mass bilaterally, IGF had no effect on the number of satellite cells (Fig. 1p). Taken together, these data indicate that Wnt7a protein delivered by intramuscular injection results in an increased number of satellite cells together with sustained muscle hypertrophy which is independent of extensive regeneration.
Fzd7 is required for the induction of symmetric satellite stem cell divisions by Wnt7a4. Co-immunoprecipitation experiments confirmed the binding of Wnt7a to Fzd7 in cultured myocytes (Fig. S2a) and in COS cells (Fig. S2b). Wnt7a-HA coimmunoprecipitated specifically with Fzd7YFP, but not with Fzd3YFP or YFP alone. Therefore, we investigated whether Fzd7 was also required for the induction of hypertrophy by Wnt7a. Transfection of Fzd7 siRNA resulted in a complete abrogation of the ability of Wnt7a to induce myotube hypertrophy (Fig. 2a, S2e). Overexpression of Fzd7 has previously been demonstrated to activate downstream signalling16. Interestingly, we observed that overexpression of Fzd7 was sufficient to induce hypertrophy in myotubes derived from both C2C12 myoblasts (Fig. 2b,d,e) and primary myoblasts (Fig. 2c,f,g).
Figure 2.

Wnt7a induces muscle hypertrophy through its receptor Fzd7. (a) siRNA mediated knockdown of Fzd7 inhibited the induction of hypertrophy in Wnt7a-HA expressing myotubes. Expression of Fzd7YFP is sufficient to induce significant hypertrophy in (b) differentiated C2C12 cells and (c) differentiated primary myoblasts. (d,e) Representative images of C2C12 cells expressing Fzd7-YFP or a control plasmid after 5 days of differentiation. Staining for myosin heavy chain (MyHC, in green) marks differentiated cells. Nuclei are counterstained with Dapi (blue). (f,g) Representative images of differentiated primary myoblasts expressing Fzd7-YFP or a control plasmid. Differentiation was carried out for 5 days. Scale bar: 50 μm (n=3), *** p<0.0001. Error bars represent SEM
The planar cell polarity effector Vangl2 is required for Wnt7a induction of symmetric satellite stem cell divisions4. Knock down of Vangl2 did not interfere with Wnt7a induction of myotube hypertrophy (Fig. S2d, S2g). However, Vangl2 is strongly down-regulated through myogenic differentiation (not shown). Therefore, we conclude that PCP-signalling is not involved in the induction of myotube hypertrophy by Wnt7a/Fzd7.
Wnt7a has been implicated in both canonical and non-canonical Wnt signalling depending on the cell and tissue context4, 17. Previously, we demonstrated that Wnt7a stimulation of myogenic cells did not result in upregulation of Tcf7 or Axin2 or activation of activated-β-catenin6. Therefore, to confirm that Wnt7a was acting through the non-canonical pathway in differentiated muscle, we assessed the degree of GSK3β and β-catenin phosphorylation by immunoblot analyses. Wnt7a stimulation of differentiated myotubes resulted in no change in the levels or phosphorylation of these canonical Wnt-signalling components (Fig. S2h, S3a–d). Moreover, application of BIO (6-bromoindirubin-3′-oxime) or LiCl18 did not inhibit Wnt7a mediated hypertrophy (Fig. S2i–k). Together, these data unequivocally argue that Wnt7a is acting through a non-canonical signalling pathway to induce myofibre hypertrophy.
The central signalling pathway implicated in the regulation of myofibre hypertrophy in skeletal muscle is the Akt/mTOR pathway13. Therefore, we assessed the ability of Wnt7a to activate Akt/mTOR signalling. Strikingly, we observed that Wnt7a treatment resulted in markedly elevated levels of phosphorylated Akt and its downstream target S6 in cultured myotubes (Fig. 3a,c, S3e–j). Importantly, Wnt7a treatment of proliferating myoblasts did not activate the Akt/mTOR pathway (Fig. 3a). Neither Wnt3a nor Wnt5a activated Akt or S6 (Fig. S4d). A single injection of recombinant Wnt7a into an excised TA muscle resulted in robust activation of Akt and its downstream target S6 (Fig. 3b). Inhibition of mTOR by rapamycin completely blocked the induction of hypertrophy by Wnt7a without affecting myoblast fusion (Figs.3d, S4a–c). Foxo transcription factors are key effectors of the Akt/mTOR growth pathway in skeletal muscle13. Accordingly, we observed inhibitory phosphorylation of the Akt downstream targets Foxo1 and Foxo3a following Wnt7a stimulation (Fig. 3a).
Figure 3.

Wnt7a activates the Akt/mTOR pathway in differentiated myotubes and myofibres. (a) Immunoblot analyses revealed increased phosphorylation of members of the Akt/mTOR pathway in C2C12 expressing Wnt7a-HA compared to control cells. Members of other known non-canonical pathway as PKC, CamKII or c-jun, a downstream effector of JNK, are not activated in Wnt7a-HA expressing myotubes. Increased levels of phosphorylated Foxo1/3a were observed in Wnt7a-HA expressing cells as well as increased levels of total Foxo1 in myoblasts and myotubes and Foxo3a in myoblasts. n=3. (b) Injection of Wnt7a recombinant protein into tibialis anterior muscle of adult mice leads to an activation of the Akt/mTOR pathway as visualized by increased phosphorylation of S6 and Akt. n=3. (c) Application of 50 ng/ml Wnt7a recombinant protein to differentiated C2C12 cells (5 days differentiation) resulted in increased levels of phosphorylated Akt and phosphorylated S6 demonstrating the rapid activation of the Akt/mTOR pathway by Wnt7a. n=3. (d) Inhibition of mTOR by rapamycin abolished hypertrophy in Wnt7a-HA expressing C2C12 cells, the fibre diameter was measured 5 days after induction of differentiation (n=4), *** p<0.001. Error bars represent SEM. See S6 for uncropped images of blots.
To evaluate the involvement of other non-canonical Wnt-signalling pathways, we investigated the phosphorylation of PKC, CamKII and c-jun, the downstream effector of JNK (Fig. 3a). None of these components were significantly activated in Wnt7a-HA expressing C2C12 cells during differentiation or after application of Wnt7a to myotubes (Fig. S4e). Together, these data strongly support the assertion that Wnt7a acts on muscle fibres through activation of the anabolic Akt/mTOR signalling pathway.
In skeletal muscle, IGF-1 and its muscular isoform MGF are ligands of the IGF receptor which signals through IRS1 to activate the PI3K/Akt/mTOR growth pathway13. We therefore investigated the possibility that Wnt7a was indirectly activating the Akt/mTOR pathway by inducing an upregulation of IGF-1 expression. C2C12 myotubes treated with recombinant Wnt7a protein did not show any increase in phosphorylated IGF-receptor, while showing a robust phosphorylation of Akt and S6 (Fig. 3c). Western blot analyses using phospho-specific antibodies indicated that Wnt7a stimulation had no effect on the activation of IGFR or IRS1 (Fig. 4a, Fig. S4f,g). Specific inhibition of IGFR using AG102419, did not prevent stimulation of myotube hypertrophy by Wnt7a (Fig. 4b), although AG1024 treatment resulted in smaller myotubes in control cells as expected. Furthermore, examination of IGF-1 and MGF mRNA levels revealed that these genes were in fact down regulated following Wnt7a stimulation (Fig. 4c). Therefore, we conclude that Wnt7a induces hypertrophy independent of IGFR/IRS1 activity.
Figure 4.
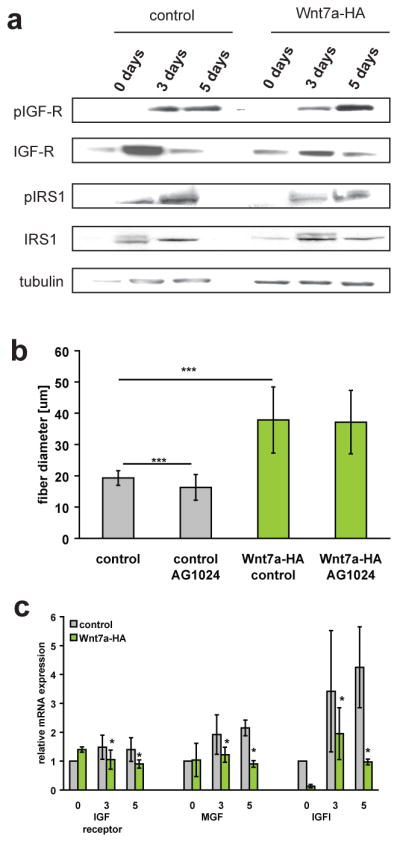
Wnt7a induces hypertrophy independent of IGF-receptor activity. (a) Immunoblot analyses revealed no changes in the phosphorylation of IGF receptor as well as IRS-1 in Wnt7a-HA expressing cells compared to control cells. Additionally the total amount of IGF-receptor and IRS1 were not significantly changed in Wnt7a-HA expressing cells compared to control cells. n=3. (b) Inhibition of IGF receptor activity by AG1024 did not result in changes in fibre diameter in Wnt7a-HA expressing cells suggesting that Wnt7a acts independently of IGF receptor activity. All analyses were performed in cells which were differentiated for 5 days. n=3. (c) Quantitative Real-time PCR of members of the IGF and IGF receptor family (n=3), *** p<0.0001. Error bars represent SEM. See S6 for uncropped images of blots.
In skeletal muscle, phosphorylation of PI3K directly activates the Akt/mTOR hypertrophic pathway20,21. Notably, we observed increased phosphorylation of PI3K following Wnt7a induction of cultured myotubes (Fig. 3a). Therefore, to investigate whether Fzd7 was directly activating the PI3K/Akt/mTOR pathway, we performed co-immunoprecipitation experiments to assess the potential association of Fzd7 and PI3K in a receptor complex. Strikingly, we observed that PI3K was detected by Western blot following immunoprecipitation of Fzd7, but not Fzd3, from extracts prepared from myotubes (Fig. 5a). Importantly, this interaction was not detectable in proliferating myoblasts (Fig. 5a). Treatment of the cells with the specific PI3K inhibitor LY294002 completely abrogated the ability of Wnt7a to induce myotube hypertrophy (Fig. 5b). Therefore, we conclude that a myotube-specific receptor complex, containing Fzd7 and PI3K, mediates the activation of the Akt/mTOR growth pathway following Wnt7a stimulation.
Figure 5.

The Fzd7 receptor complex contains PI3K and the G protein Gαs (a) Coimmunoprecipitation analyses demonstrated that Fzd7YFP binds the p85 subunit of PI3K in myotubes but not in myoblasts. (b) Blocking of PI3K activity with the specific inhibitor LY294002 abrogated the hypertrophy induced by Wnt7a. Fibre diameter was measured 5 days after induction of differentiation in C2C12 control cells as well as in C2C12 cells expressing Wnt7a-HA (n=4), *** p<0.001. (c) IP-Western coimmunoprecipitation analysis demonstrated that Fzd7 binds G protein Gαs. (d). Suramin, a specific inhibitor of the G protein Gαs23 completely blocked Wnt7a induced hypertrophy, n=3, *** p<0.001. (e) Suramin inhibits Wnt7a induced phosphorylation of Akt and S6. (f) Knockdown of GNAS1 abrogates Wnt7a induced hypertrophy in C2C12 cells, n=3, *** p<0.001. Error bars represent SEM. See S6 for uncropped images of blots.
We performed mass spectrometry to identify additional components of the Fzd7 receptor complex in myotubes. These experiments identified the G protein subunit alpha S (stimulatory G protein, Gαs) as an Fzd7 associated protein. Binding of Fzd7 to Gαs (encoded by the GNAS1 gene) was confirmed by co-immunoprecipitation analyses in myotubes of C2C12 cells. Gαs specifically bound Fzd7YFP but not YFP alone (Fig. 5c). These data led us to hypothesize that Gαs mediates the activation of PI3Kinase following Wnt7a binding to Fzd7. Therefore, we investigated whether differentiated myotubes treated with Suramin, a specific inhibitor of the association of Gαs and beta-gamma subunits22,23, was capable of abrogating Wnt7a induced hypertrophy. Strikingly, Suramin completely blocked Wnt7a evoked hypertrophy and phosphorylation of Akt and S6 (Fig. 5d,e), but did not inhibit IGF-1 induced hypertrophy (Fig. S4j). Furthermore knockdown of Gαs (Fig. S4k) resulted in abrogation of Wnt7a evoked hypertrophy (Fig. 5f). Taken together, these data unequivocally establish that the Wnt receptor Fzd7, and the associated components Gαs and PI3K, are required for the induction of myotube hypertrophy by Wnt7a.
We have described a role for Wnt signalling in the direct regulation of growth pathways in differentiated skeletal muscle. Binding of Wnt7a to Fzd7 directly activated PI3K and the Akt/mTOR pathway to induce myofibre hypertrophy both in vitro and in vivo. Strikingly, Wnt7a/Fzd7 activation of the PI3K/Akt/mTOR pathway was independent of IGF-receptor activation. Therefore, we have identified a so far undescribed non-canonical anabolic pathway for Wnt7a and its receptor Fzd7 in skeletal muscle.
Our results indicate that Wnt7a acts via distinct non-canonical pathways at two cellular levels in skeletal muscle. First, through PCP signalling in satellite cells to regulate the homeostatic levels of satellite cells4. Second, through the Akt/mTOR pathway in differentiated myofibres to regulate growth (summarized in Fig. S5). In this manner, Wnt7a functions to couple the expansion of the satellite cell pool to the mass of the muscle tissue. Loss of Wnt7a results in reduced numbers of satellite cells 3 weeks after injury4, thus demonstrating the physiological role of Wnt7a in regulating the homeostatic levels of satellite cells. Therefore, we predict that the dual function of Wnt7a in skeletal muscle has a role in the adaptation of muscle to different physiological stimuli.
We previously described a role for Wnt7a/Fzd7 signalling in satellite stem cells dependent on the PCP component Vangl24. By contrast, we found that Wnt7a/Fzd7 activation of the Akt/mTOR pathway in differentiated skeletal muscle was independent of Vangl2. Moreover, we observed that Gαs and PI3K were associated with the Fzd7 receptor complex specifically in differentiated myotubes, and not in proliferating myoblasts. These data therefore suggest that the Fzd7 receptor complex contains distinct components in satellite cells versus differentiated myofibres.
The presence of seven transmembrane domains in Fzd receptors is reminiscent of the structure of classical G protein receptors24. Frizzled proteins are considered a novel and separate family of G coupled receptors25. Therefore we speculated that the Fzd7 receptor complex in myotubes contains additional factors such as G proteins that are required to activate PI3K. Through mass spectrometry analyses we found the following G protein subunits associated with Fzd7: G protein alpha S (Gαs, encoded by the GNAS1 gene), G protein beta 1, G protein beta 2 and the G protein subunit O. We chose to investigate the function of Gαs since the GNAS1 gene is highly expressed during differentiation of C2C12 cells (see Fig. 4h,i).
Heteromeric G proteins consist of an alpha subunit which is regulated by guanine-nucleotide binding and the tightly associated beta-gamma subunit. Depending on the G protein family involved the alpha and beta-gamma subunits can activate diverse effectors. Our results suggest that Wnt7a activates the Gαs protein thereby leading to an activation of PI3K, presumably through a Gβγ complex involving G protein beta 1 or beta 2.
In differentiated myofibres, Wnt7a binds to Fzd7 resulting in the activation of PI3K, which in turn activates phosphatidyl inositol-dependent kinase1 (PDK1). PDK1 then activates Akt leading to the activation of its downstream targets S6kinase and S6 and the induction of hypertrophy26,27. Thus, Wnt7a binding to its receptor Fzd7 complex containing Gαs and PI3K activates the Akt/mTOR growth pathway in muscle entirely independently of IGF and IGFR.
Akt has the dual potential to drive the synthesis of protein while preventing proteolytic degradation. This makes Akt a key metabolic control point in diseases which are characterized by changes in muscle mass, for example cancer cachexia and sarcopenia28,29. Other catabolic states like malnutrition, critical illness and sepsis are associated with a profound reduction in the pool of IGF-1 resulting in muscle atrophy27, 30. Consequently IGF-1 treatment has been explored as a potential treatment to increase the lean body mass in patients suffering from these conditions31. However, limitation of this approach arises from the development of resistance to growth hormone, and due to the development of hypoglycemia32.
Muscle damage due to eccentric exercise as well as cachexia induces TNFα expression. This cytokine dramatically impairs insulin stimulation of IRS-133,34. It is believed that TNFα induces insulin resistance in skeletal muscle by activation of IKK, which then phosphorylates IRS-1, thereby precluding subsequent activation of PI3K and Akt35. Wnt7a activates the PI3K/Akt/mTOR signalling axis without an activation of IRS-1. Therefore, it is interesting to speculate that Wnt7a will be able to induce hypertrophy and/or prevent muscle wasting in the context of high TNFα levels without developing insulin resistance.
Our work has identified an additional non-canonical Wnt signalling pathway to the currently known variations of canonical and non-canonical pathways. The Wnt anabolic signalling pathway, where Wnt7a activates the Fzd7/PI3K/Akt/mTOR hypertrophy pathway, provides a promising new therapeutic focus for the amelioration of muscle wasting diseases like sarcopenia, cachexia or muscular dystrophies.
Supplementary Material
Acknowledgments
The authors thank Feodor Price and Vahab Soleimani for critical reading of the manuscript. M.A.R. holds the Canada Research Chair in Molecular Genetics and is an International Research Scholar of the Howard Hughes Medical Institute. C.F.B. is supported by the Swiss National Science Foundation. This work was funded by grants from the Muscular Dystrophy Association, Canadian Institutes of Health Research, National Institutes of Health, Howard Hughes Medical Institute, and the Canada Research Chair Program.
Footnotes
AUTHOR CONTRIBUTIONS
J.M. and M.A.R. planned the experimental design, analyzed data and wrote the paper. J.M. and C.F.B. conducted the experiments.
COMPETING FINANCIAL INTEREST
The authors declare a competing financial interest. M.A.R. is a founding scientist and consultant with Fate Therapeutics.
References
- 1.Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. The Journal of biological chemistry. 2006;281:22429–22433. doi: 10.1074/jbc.R600015200. [DOI] [PubMed] [Google Scholar]
- 2.Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annual review of cell and developmental biology. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 3.Montcouquiol M, Kelley MW. Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. J Neurosci. 2003;23:9469–9478. doi: 10.1523/JNEUROSCI.23-28-09469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Grand F, Jones AE, Seale V, Scime A, Rudnicki MA. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell stem cell. 2009;4:535–547. doi: 10.1016/j.stem.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tajbakhsh S, et al. Differential activation of Myf5 and MyoD by different Wnts in explants of mouse paraxial mesoderm and the later activation of myogenesis in the absence of Myf5. Development (Cambridge, England) 1998;125:4155–4162. doi: 10.1242/dev.125.21.4155. [DOI] [PubMed] [Google Scholar]
- 6.Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature. 2005;433:317–322. doi: 10.1038/nature03126. [DOI] [PubMed] [Google Scholar]
- 7.Borello U, et al. The Wnt/beta-catenin pathway regulates Gli-mediated Myf5 expression during somitogenesis. Development (Cambridge, England) 2006;133:3723–3732. doi: 10.1242/dev.02517. [DOI] [PubMed] [Google Scholar]
- 8.Anakwe K, et al. Wnt signalling regulates myogenic differentiation in the developing avian wing. Development (Cambridge, England) 2003;130:3503–3514. doi: 10.1242/dev.00538. [DOI] [PubMed] [Google Scholar]
- 9.Rochat A, et al. Insulin and wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Molecular biology of the cell. 2004;15:4544–4555. doi: 10.1091/mbc.E03-11-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Velden JL, et al. Inhibition of glycogen synthase kinase-3beta activity is sufficient to stimulate myogenic differentiation. American journal of physiology. 2006;290:C453–462. doi: 10.1152/ajpcell.00068.2005. [DOI] [PubMed] [Google Scholar]
- 11.Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell stem cell. 2008;2:50–59. doi: 10.1016/j.stem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. The international journal of biochemistry & cell biology. 2005;37:1974–1984. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Crescenzi M, Crouch DH, Tato F. Transformation by myc prevents fusion but not biochemical differentiation of C2C12 myoblasts: mechanisms of phenotypic correction in mixed culture with normal cells. The Journal of cell biology. 1994;125:1137–1145. doi: 10.1083/jcb.125.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins AR, Squires S, Johnson RT. Inhibitors of repair DNA synthesis. Nucleic acids research. 1982;10:1203–1213. doi: 10.1093/nar/10.4.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka S, Akiyoshi T, Mori M, Wands JR, Sugimachi K. A novel frizzled gene identified in human esophageal carcinoma mediates APC/beta-catenin signals. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10164–10169. doi: 10.1073/pnas.95.17.10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kengaku M, et al. Distinct WNT pathways regulating AER formation and dorsoventral polarity in the chick limb bud. Science (New York, N Y) 1998;280:1274–1277. doi: 10.1126/science.280.5367.1274. [DOI] [PubMed] [Google Scholar]
- 18.Harwood AJ. Regulation of GSK-3: a cellular multiprocessor. Cell. 2001;105:821–824. doi: 10.1016/s0092-8674(01)00412-3. [DOI] [PubMed] [Google Scholar]
- 19.Parrizas M, Gazit A, Levitzki A, Wertheimer E, LeRoith D. Specific inhibition of insulin-like growth factor-1 and insulin receptor tyrosine kinase activity and biological function by tyrphostins. Endocrinology. 1997;138:1427–1433. doi: 10.1210/endo.138.4.5092. [DOI] [PubMed] [Google Scholar]
- 20.Kandarian SC, Jackman RW. Intracellular signaling during skeletal muscle atrophy. Muscle & nerve. 2006;33:155–165. doi: 10.1002/mus.20442. [DOI] [PubMed] [Google Scholar]
- 21.Miyazaki M, Esser KA. Cellular mechanisms regulating protein synthesis and skeletal muscle hypertrophy in animals. J Appl Physiol. 2009;106:1367–1373. doi: 10.1152/japplphysiol.91355.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pietrangelo T, et al. Characterization of specific GTP binding sites in C2C12 mouse skeletal muscle cells. Journal of muscle research and cell motility. 2002;23:107–118. doi: 10.1023/a:1020288117082. [DOI] [PubMed] [Google Scholar]
- 23.Freissmuth M, et al. Suramin analogues as subtype-selective G protein inhibitors. Molecular pharmacology. 1996;49:602–611. [PubMed] [Google Scholar]
- 24.Schulte G, Bryja V. The Frizzled family of unconventional G-protein-coupled receptors. Trends in pharmacological sciences. 2007;28:518–525. doi: 10.1016/j.tips.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Foord SM, et al. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacological reviews. 2005;57:279–288. doi: 10.1124/pr.57.2.5. [DOI] [PubMed] [Google Scholar]
- 26.Timmers S, Schrauwen P, de Vogel J. Muscular diacylglycerol metabolism and insulin resistance. Physiology & behavior. 2008;94:242–251. doi: 10.1016/j.physbeh.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Frost RA, Lang CH. Protein kinase B/Akt: a nexus of growth factor and cytokine signaling in determining muscle mass. J Appl Physiol. 2007;103:378–387. doi: 10.1152/japplphysiol.00089.2007. [DOI] [PubMed] [Google Scholar]
- 28.Tisdale MJ. Cancer cachexia. Curr Opin Gastroenterol. 2010;26:146–151. doi: 10.1097/MOG.0b013e3283347e77. [DOI] [PubMed] [Google Scholar]
- 29.Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3:90–101. doi: 10.2174/1874609811003020090. [DOI] [PubMed] [Google Scholar]
- 30.Costelli P, et al. IGF-1 is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol. 2006;291:R674–683. doi: 10.1152/ajpregu.00104.2006. [DOI] [PubMed] [Google Scholar]
- 31.Botfield C, Ross RJ, Hinds CJ. The role of IGFs in catabolism. Bailliere’s clinical endocrinology and metabolism. 1997;11:679–697. doi: 10.1016/s0950-351x(97)80958-5. [DOI] [PubMed] [Google Scholar]
- 32.Lang CH, Hong-Brown L, Frost RA. Cytokine inhibition of JAK-STAT signaling: a new mechanism of growth hormone resistance. Pediatric nephrology (Berlin, Germany) 2005;20:306–312. doi: 10.1007/s00467-004-1607-9. [DOI] [PubMed] [Google Scholar]
- 33.Grounds MD, Radley HG, Gebski BL, Bogoyevitch MA, Shavlakadze T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clinical and experimental pharmacology & physiology. 2008;35:846–851. doi: 10.1111/j.1440-1681.2007.04868.x. [DOI] [PubMed] [Google Scholar]
- 34.Del Aguila LF, et al. Muscle damage impairs insulin stimulation of IRS-1, PI 3-kinase, and Akt-kinase in human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E206–212. doi: 10.1152/ajpendo.2000.279.1.E206. [DOI] [PubMed] [Google Scholar]
- 35.de Alvaro C, Teruel T, Hernandez R, Lorenzo M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. The Journal of biological chemistry. 2004;279:17070–17078. doi: 10.1074/jbc.M312021200. [DOI] [PubMed] [Google Scholar]
- 36.Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996;10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- 37.Parker MH, Perry RL, Fauteux MC, Berkes CA, Rudnicki MA. MyoD synergizes with the E-protein HEB beta to induce myogenic differentiation. Molecular and cellular biology. 2006;26:5771–5783. doi: 10.1128/MCB.02404-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.von Maltzahn J, Euwens C, Willecke K, Sohl G. The novel mouse connexin39 gene is expressed in developing striated muscle fibers. J Cell Sci. 2004;117:5381–5392. doi: 10.1242/jcs.01413. [DOI] [PubMed] [Google Scholar]
- 39.Gillespie MA, et al. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol. 2009;187:991–1005. doi: 10.1083/jcb.200907037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinnell IW, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nature cell biology. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.