

Abstract
To identify the disease-causing gene responsible for an autosomal dominantly inherited Charcot–Marie–Tooth neuropathy subtype in a family excluded for mutations in the common Charcot–Marie–Tooth genes, we used array-based sequence capture to simultaneously analyse the disease-linked protein coding exome at chromosome 14q32. A missense mutation in fibulin-5, encoding a widely expressed constituent of the extracellular matrix that has an essential role in elastic fibre assembly and has been shown to cause cutis laxa, was detected as the only novel non-synonymous sequence variant within the disease interval. Screening of 112 index probands with unclassified Charcot–Marie–Tooth neuropathies detected two further fibulin-5 missense mutations in two families with Charcot–Marie–Tooth disease and hyperextensible skin. Since fibulin-5 mutations have been described in patients with age-related macular degeneration, an additional 300 probands with exudative age-related macular degeneration were included in this study. Two further fibulin-5 missense mutations were identified in six patients. A mild to severe peripheral neuropathy was detected in the majority of patients with age-related macular degeneration carrying mutations in fibulin-5. This study identifies fibulin-5 as a gene involved in Charcot–Marie–Tooth neuropathies and reveals heterozygous fibulin-5 mutations in 2% of our patients with age-related macular degeneration. Furthermore, it adumbrates a new syndrome by linking concurrent pathologic alterations affecting peripheral nerves, eyes and skin to mutations in the fibulin-5 gene.
Keywords: age-related macular degeneration, CMT, cutis laxa, fibulin-5, neuropathy
Introduction
Fibulin-5 (FBLN5) is an extracellular matrix calcium-binding glyco-protein expressed in elastic fibre-rich tissues (Yanagisawa et al., 2009). It is critical for correct deposition of elastin, mediates cell-matrix communication and regulates organogenesis, fibrogenesis, vascular remodelling and tumour metastasis (Zheng et al., 2007; Yanagisawa et al., 2009). Three homozygous missense mutations (p.C217R, p.S227P and p.R284X) in FBLN5 have been reported in patients with an autosomal recessive, generalized form of the connective tissue disorder cutis laxa (Loeys et al., 2002; Elahi et al., 2006; Hu et al., 2006; Claus et al., 2008; Nascimento et al., 2010), and a heterozygous in-frame tandem duplication of exons 5–8 in FBLN5 was seen in a sporadic patient with cutis laxa suggesting that the large protein acts in a dominant negative way (Markova et al., 2003). Ten distinct, heterozygous FBLN5 missense mutations have been associated with age-related macular degeneration, the leading cause of severe visual loss among patients older than 50 years in the western world (Stone et al., 2004; Lotery et al., 2006; Jones et al., 2009, 2010; Schneider et al., 2010). The p.S227P, the p.C217R FBLN5 mutants causing cutis laxa, and the age-related macular degeneration-associated p.G412E, p.G267S, p.I169T and p.Q124P FBLN5 mutants showed decreased secretion suggesting compromised elastic fibre formation as a likely mechanism of the disease (Lotery, 2006; Jones, 2010). In eyes with age-related macular degeneration FBLN5 has been localized to pathologic basal deposits beneath the retinal pigment epithelium as well as in some small drusen (Mullins et al., 2007). The molecular interactions of FBLN5 and the biochemical basis underlying pathogenic effects of FBLN5 alterations still remain incompletely understood (Zheng et al., 2007; Jones et al., 2009, 2010; Schneider et al., 2010).
The inherited peripheral neuropathies, also known as Charcot–Marie–Tooth disease, comprise a clinically and genetically heterogeneous group of neuromuscular disorders characterized by lifelong disability due to distal muscle weakness and sensory disturbances in upper and lower limbs. Diverse molecular pathways involved in the pathogenesis of Charcot–Marie–Tooth disease have been elucidated in the past and mutations in >30 genes have been identified (Reilly and Shy, 2009). It has also been speculated that extracellular matrix genes might be good candidates for Charcot–Marie–Tooth neuropathies as interactions between Schwann cells and extracellular matrix are required for myelination in the developing peripheral nervous system (Berti et al., 2006).
To identify the causative gene responsible for an autosomal dominant Charcot–Marie–Tooth disease subtype 1, we mapped the disease locus to chromosome 14q32 by single nucleotide polymorphism array-based linkage analysis, and applied sequence capture array-based resequencing to simultaneously analyse all protein encoding genes within the disease interval. A novel missense mutation in FBLN5 was detected and subsequently two further FBLN5 mutations were identified in families with Charcot–Marie–Tooth disease. In each Charcot–Marie–Tooth disease family one patient was also diagnosed with age-related macular degeneration, and four patients had hyperelastic skin. Based on these findings we expanded our study to examine a further 300 patients with exudative age-related macular degeneration to delineate the characteristics of a novel oculo-neuro-cutaneous syndrome caused by mutations in FBLN5.
Materials and methods
Study participants
A total of 424 patients were enrolled in this study. One hundred and twenty-four probands (including all index patients and additional affected family members) had genetically unclassified Charcot–Marie–Tooth disease and 300 patients were diagnosed with exudative age-related macular degeneration. In addition, direct sequencing was performed in 317 healthy control subjects, 85 of which—all being >50 years—were excluded for age-related macular degeneration by ophthalmological examination. The particular c.268 G>A FBLN5 variant (encoding p.G90S FBLN5) was tested in 200 additional healthy control individuals.
Study participants were ascertained at the Medical University of Graz, Austria and gave written informed consent. The study was approved by the local Ethical Committee of the Medical University of Graz.
Clinical, electrophysiological and ophthalmological evaluation
After obtaining medical and family history, a full neurological examination was carried out by an experienced neurologist (M.A.-G.) in all patients with Charcot–Marie–Tooth disease, healthy relatives and in age-related macular degeneration probands carrying FBLN5 mutations.
Electrophysiological studies followed standard methods. Motor nerve conduction velocities were used for further subclassification of the patients with Charcot–Marie–Tooth disease: a cut-off of 38 m/s for the median motor nerve conduction velocity established a diagnosis of Charcot–Marie–Tooth disease type 1 (<38 m/s, demyelinating Charcot–Marie–Tooth disease subtype), or Charcot–Marie–Tooth disease type 2 (>38 m/s, axonal Charcot–Marie–Tooth disease subtype). Patients lacking sensory disturbances both clinically and electrophysiologically were classified as spinal Charcot–Marie–Tooth disease (also known as distal hereditary motor neuropathy) (Reilly and Shy, 2009). Based on these criteria, 63 patients with Charcot–Marie–Tooth disease were diagnosed as Charcot–Marie–Tooth disease type 1, 38 as Charcot–Marie–Tooth disease type 2 and 23 as spinal Charcot-Marie-Tooth disease. Electromyography was performed in selected patients only.
Patients with age-related macular degeneration and Charcot–Marie–Tooth disease probands carrying FBLN5 mutations received an opthalmological examination by experienced opthalmologists (M.W., A.H.) including visual acuity testing, slit lamp and fundus examination. Also, optical coherence tomography and fluorescein angiography were performed in most patients.
Molecular genetic studies
Linkage studies
DNA was isolated from peripheral blood samples according to standard methods. Initially, genetic linkage studies were performed in two families (Families A and B, Fig. 1) whose ancestors originated from the same small village but in whom blood relationship could not be established by genealogical studies. Index patients of both families were excluded for mutations in PMP22, MPZ, GJB1, LITAF, EGR2 and NEFL. In Family A GDAP1 also tested negative. Samples from eight affected, three unaffected individuals and two spouses from Families A and B were hybridized to Affymetrix GeneChip® Human Mapping 10 K arrays XbaI 142 2.0 (Affymetrix). For fine mapping of the locus, five of these affected individuals were subsequently genotyped using the Affymetrix Human SNP Array 6.0. Parametric multipoint log of odds score calculations and haplotypes were obtained with the ALLEGRO program (Gudbjartsson et al., 2005) and an autosomal-dominant, fully penetrant model.
Figure 1.
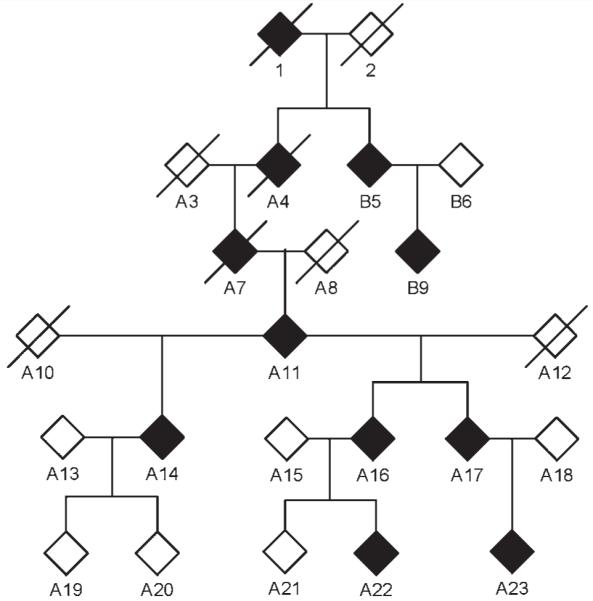
Simplified and virtual pedigree, as used for the linkage study, including Families A and B. The gender of the family members is not shown for privacy. Filled and crossed out symbols indicate affected and deceased persons, respectively. Individuals denominated with a ‘B’ have been added to the pedigree for linkage studies, because they exhibit the same phenotype as individuals of Family A, are excluded for mutations in the known Charcot-Marie-Tooth disease type 1 genes, and originate from the same small village. For linkage analyses, samples from Patients B5, B9, A11, A14, A15, A16, A17, A18, A19, A20, A21, A22 and A23 were genotyped using the Affymetrix GeneChip® Human Mapping 10K XbaI 142 2.0 array. Individuals B5, B9, A11, A17 and A23 were later genotyped using the high-density Affymetrix GeneChip® Human Mapping 6.0 array for high-resolution confirmation of a common haplotype in Families A and B and fine mapping of the candidate region.
Sequence capture array design
A custom tiling 385 K sequence capture array targeting the exonic sequences of all protein-encoding genes (a total of 120.4 kb) within chromosome 14: bp 87 435 655–91 913 433 was designed using BioMart EnsEMBL release 54—May 2009 based on the NCBI 36 assembly of the human genome (November 2005) and manufactured by Roche NimbleGen. Sequence Capture followed the manufacturer’s instructions (Roche NimbleGen). A GFF- or BED-formatted file allowing visualization of the tiled intervals by the Genome Browser (http://genome.ucsc.edu/) is available on request.
Sequence capture library construction
Genomic DNA (20 μg) from Patient A16 was processed into a GS FLX Titanium chemistry compatible capture library according to the NimbleGen Arrays User’s Guide v.3.0, December 2008 (Roche NimbleGen, Inc.).
Capture array handling
Hybridization was performed using microarrays merged with X1 mixer on the NimbleGen Hybridization System for 3 days at 42°C following the manufacturer’s recommended conditions (NimbleGen Arrays User’s Guide vs.3.0, Dec. 2008; Roche NimbleGen, Inc.). Quantitative polymerase chain reaction (SYBR-Green based; LC480 instrument) using four internal NimbleGen control loci (NSC-0237, NSC-0247, NSC-0268 and NSC-0272) was performed to estimate relative fold-enrichment (data not shown).
GS FLX sequencing
The amplified capture libraries (4 μg per sample) were processed into sequencing libraries for the 454 GS-FLX using the GS FLX Titanium General Library Preparation protocol (454 Life Sciences) according to the manufacturer’s recommended conditions (without the nebulization step). The captured sample library was sequenced using a quarter of a Titanium PicoTiterplate (70 × 75) run on the GS-FLX platform using Titanium chemistry and standard settings.
Data analysis: variant detection and annotation
High confidence differences were calculated using the GS Reference Mapper software (version 2.3; Roche Diagnostics, 454 Life Sciences) and USCS genome annotation database for the March 2006 Genbank freeze assembled by NCBI (hg18, build 36.1) as reference sequence.
The GS Reference Mapper application uses a combination of flow signal information, quality score information and difference type information to determine if a difference is high confidence (GS Reference Mapper Manual version 2.3; Roche Diagnostics, 454 Life Sciences).
Mutation confirmation
The FBLN5 coding region and splice sites were polymerase chain reaction-amplified using 11 primer pairs that were designed based on National Centre for Biotechnology Information annotations for FBLN5 genomic reference (NC_000014.8) and messenger RNA (NM_006329.3) sequences (www.ncbi.nlm.nih.gov, Supplementary Table 1) and sequenced on an ABI 3730 DNA sequencer.
Skin, muscle and nerve biopsies
Biopsies of sun-protected skin were taken from Patients C1, C2, C3 and B9 and from a healthy control. Tissues from muscle and nerve biopsies were available from routine diagnostic studies in Patients C1, C2 and B5. Immunohistochemical detection of FBLN5 and elastin followed standard protocols (Kadoya et al., 2005).
Sections from paraffin embedded material were cut, de-paraffinized and rehydrated. Four antigen retrieval sections were incubated with proteinase K (Dako REAL™ diluted 1:40 with Dako REAL™ proteinase K diluent) for 30 min at room temperature. Subsequently, endogenous peroxidase activity was blocked by immersion of the sections in 1% H2O2 in methanol. Staining for elastin was performed according to the protocol of the UltraVison LP Large Volume Detection System (Thermo Scientific) starting with Ultra V Block for 5 min at room temperature, incubation with anti-elastin (rabbit, 1:3000, Sigma-Aldrich), antibody for 30 min at room temperature FBLN5 was detected with mouse monoclonal (1G6A4) antibody (1:500, Abcam), followed by incubation with primary antibody enhancer for 10 min at room temperature, horseradish peroxidase polymer for 30 min at room temperature and detection with liquid DAB + Substrate Chromogen System for Autostainer (Dako Diagnostica) for 5 min at room temperature. Sections were counterstained with hemalaun. Between all incubation steps sections were rinsed three times in phosphate-buffered saline. Sections were dehydrated and mounted in Tissue Tek® Mounting Media 4494 Coverslipping Resin (Sanova). For negative controls rabbit IgG (1:3000, Linaris) was used instead of the primary antibody.
Ultrathin sections were counterstained with lead citrate and uranyl acetate and examined in a Philips EM 10 electron microscope.
Results
Clinical, electrophysiological and molecular genetic studies
Clinical and electrophysiological findings in eight patients of Families A and B were consistent with late-onset Charcot–Marie–Tooth disease type 1 (Table 1, Supplementary Table 2). The oldest Charcot–Marie–Tooth disease patient in Family A (Patient A11) had received a diagnosis of age-related macular degeneration at 80 years of age.
Table 1.
Clinical and electrophysiological characteristics of probands carrying FBLN5 mutations
| Family/ Individual number |
Age at disease onset/ Clinical presentation (years) |
FBLN5 sequence variation |
Initial diagnosis |
Distal muscle involvement LL/UL |
Patellar tendon reflexes |
Peripheral neuropathy LL/ULa |
Hyper- elastic skin |
Distinct additional features/ diseases |
Age- related macular degeneration |
|---|---|---|---|---|---|---|---|---|---|
| Family A | |||||||||
| A11 | 50/81 | p.R373C | CMT1 | + + + / + + + | 1 | Yes/Yes | No | - | Yesb |
| A14 | 50/64 | p.R373C | CMT1 | + /− | 2 | NE/Yes | No | CTS-like symptoms |
No |
| A16 | 35/47 | p.R373C | CMT1 | + / + + | 0 | Yes/NE | No | CTS-like symptoms |
NE |
| A17 | 45/55 | p.R373C | CMT1 | + / + | 1 | Yes/Yes | No | CTS-like symptoms |
No |
| A22 | AS/30 | p.R373C | CMT1 | −/− | 1 | NE/Yes | No | - | No |
| A23 | 25/31 | p.R373C | CMT1 | −/+ | 1 | Yes/Yes | No | CTS-like symptoms |
NE |
| Family B | |||||||||
| B5 | 50/74 | p.R373C | CMT1 | + + + / + + | 0 | NE/NE | No | - | No |
| B9 | 37/43 | p.R373C | CMT1 | + + + / + + + | 0 | Yes/Yes | NE | Distal sensory loss in LL |
No |
| Family C | |||||||||
| C1 | 4/21 | p.G90S | Spinal CMT | + + + / + + | 2 | Yes/Yes | Yes | Joint hypermobilityc |
No |
| C2 | 4/19 | p.G90S | Spinal CMT | + + + / + + | 3 | Yes/Yes | Yes | Joint hypermobilityc |
No |
| C3 | AS/47 | p.G90S | AS | −/− | 1 | No/Yes | Yes | - | No |
| C4 | 71/73 | p.G90S | Age-related macular degeneration |
+ / + + | 0 | Yes/No | No | Scoliosis, lordosis Yesd | |
| C5 | AS/70 | p.G90S | AS | + /− | 0 | Yes/No | No | - | |
| Family D | No | ||||||||
| D1 | 20/46 | p.V126M | Spinal CMT | + + / + | 1 | No/Yes | Yes | Mild scapula alata |
No |
| D2 | 82/83 | p.V126M | AS | −/− | 0 | Yes/No | No | Alcohol abuse | Yesb |
| Age-related macular degeneration-Patients | |||||||||
| E | NE | p.V126M | Age-related macular degeneration |
NE | NE | NE/NE | NE | NE | Yese |
| F | 79/87 | p.V126M | Age-related macular degeneration |
−/− | 2 | Yes/NE | No | Asthma bronchiale |
Yesf |
| G | 79/88 | p.G90S | Age-related macular degeneration |
+ +/+ | 2 | Yes/Yes | No | - | Yesf |
| H | 80/85 | p.T48I | Age-related macular degeneration |
+ +/+ + | 0 | No/No | No | - | Yesf |
| K | 82/83 | p.G267S | Age-related macular degeneration |
NE | NE | NE/NE | NE | NE | Yesf |
| L | 82/89 | p.G267S | Age-related macular degeneration |
−/ + | 0 | Yes/No | No | - | Yesf |
| Controls | |||||||||
| I | AS/82 | p.V126M | Control | + + / + | 0 | Yes/Yes | No | Distal sensory loss in LL |
No |
| J | AS/84 | p.V126M | Control | −/ + | 0 | Yes/No | No | - | No |
AS = asymptomatic; CMT1 = Charcot–Marie–Tooth disease type 1; CTS = carpal tunnel syndrome; LL = lower limbs; NE = not examined; UL = upper limbs; − = not present; + = mild; + + = moderate; + + + = severe; 0 = absent; 1 = diminished; 2 = normal; 3 = brisk.
For detailed results and normal values of NCS see Supplementary Table 3
Bilateral, late, dry.
For further features refer to ‘Results’ section
Bilateral, early, drusen
Bilateral, late, dry and exudative in right/left eye
Bilateral late exudative
Whole genome, multipoint linkage analysis including six affected and three unaffected family members as well as two spouses of Family A revealed two chromosomal intervals on chromosomes 3 p and 14q tentatively linked to the disease, with maximal log of odds scores of 2.1032 and 2.1071, respectively (data not shown). However, calculation of the enlarged virtual pedigree including Families A and B with probable relationship (Fig. 1) revealed a significant maximum log of odds score of 3.31 on chromosome 14q32 only (Fig. 2) with all affected individuals of Families A and B sharing a haplotype consisting of 17 single-nucleotide polymorphism alleles over a region of 5.9 Mb (bp 87 435 655–93 339 833). Subsequent genotyping of five patients (Patients A11, A17, A23, B5, B9) with high density 6.0 Affymetrix arrays confirmed a common haplotype between Families A and B containing 1534 single nucleotide polymorphism markers and covering a refined region of 4.5 Mb (bp 87 435 655–91 913 433). After exclusion of pathogenic mutations in four candidate genes selected on putative function and expression profile (CALM1, GALC, PTPN21, KCNK13) by direct sequencing we simultaneously analysed all 35 protein coding genes within the disease region using array-based sequence capture followed by parallel resequencing (DNA obtained from Patient A16). We generated ~72.2 Mb of sequence information with a >10-fold sequence coverage for >91% of the coding nucleotides and exon–intron boundaries within the disease-linked target interval (Supplementary Table 3, Supplementary Fig. 1). Applying high-confidence sequence variant filtering using the standard GS Reference Mapper tool (reference: UCSC genome annotation database for the March 2006 GenBank freeze assembled by NCBI hg18 build 36.1, dbSNP build130) identified 12 non-synonymous sequence variants, 11 of which were known single-nucleotide polymorphisms. A single novel missense variant was located in exon 10 of FBLN5, segregated with the disease and causes an exchange of arginine at position 373 to cysteine (c.1117 C>T, p.R373C; Fig. 3 and Supplementary Fig. 2). The R373 residue is highly conserved throughout evolution (Supplementary Fig. 3) and was absent in 317 control individuals making causality of the disease likely.
Figure 2.

Mapping a Charcot–Marie–Tooth disease type 1 locus in Families A and B. The graph represents a parametric log of odds score (LOD) on the y-axis in relation to genetic position on the x-axis. Human chromosomes are concatenated from p-ter (left) to q-ter (right) on the x-axis, and the genetic distance is given in centimorgans (cM). This scan identified a single region on chromosome 14q32 segregating with the disease based on the hypothesis of autosomal dominant inheritance and 100% penetrance of the mutation.
Figure 3.

Mutation detection by direct sequencing. Electropherograms derived from FBLN5 sequencing of exons 4, 8 and 10 from patients compared to unaffected individuals (wild-type). The positions of the heterozygous mutations are indicated with an arrow above the sequence. The effect of the mutation is shown to the right of each sequence.
To investigate the prevalence of FBLN5 mutations as a potential cause of Charcot–Marie–Tooth disease neuropathies, we screened 112 further index probands with genetically unclassified Charcot–Marie–Tooth disease and identified two further sequence variations. Patient C1 carried a c.268 G>A change (p.G90S) in exon 4 (Fig. 3), affecting a well, although not fully conserved residue (Supplementary Fig. 3). This sequence change was not detected in our 517 healthy control individuals, and was not reported in previous studies including 1204 patients with age-related macular degeneration and 766 control individuals (Stone et al., 2004; Lotery et al., 2006) or in the 1000 genome project (www.1000genomes.org/). Patient C1 and his brother who also carried the same c.268 G>A FBLN5 mutation (Patient C2) exhibited a spinal Charcot–Marie–Tooth disease phenotype with slowly progressive distal muscle weakness and wasting pre-dominating in the lower limbs from early childhood (Fig. 4A). Nerve conduction velocities revealed a pure axonal motor neuropathy (Supplementary Table 2). EMG and muscle biopsy were consistent with chronic neurogenic disturbances. Sural nerve biopsy was normal. Known pathogenic mutations in several Charcot–Marie–Tooth disease genes (PMP22, MPZ, GJB1, NEFL, LITAF, GDAP1, YARS, HSPB1, HSPB8, TRPV4, KIAA1985, exons 13–16 of DNM2, BSCL2, GARS, MFN2) and also in ATP7A, which is known to cause both X-linked cutis laxa and spinal Charcot–Marie–Tooth disease (Kennerson et al., 2010), had been excluded previously in either Patient C1 or C2 by direct sequencing. A novel sequence variation was identified in YARS (c.1015 G>A; p.A339T) in Patients C2 and C3 but not in the severely affected Patient C1. Reported polymorphisms leading to amino acid changes were detected in LITAF (rs4280262), GARS (rs1049402), MFN2 (rs41278630; Lawson et al., 2005) and in ATP7A (rs4826245). Both Patients C1 and C2 demonstrated hyperstretchable skin with normal elastic recoil (Fig. 4B). Skin was not fragile and wound healing was inconspicuous. Additional features included hypermobility of distal joints, a high palatum, and mild skeletal abnormalities consisting of pectus excavatus, prominent chin and dolichocephaly (Fig. 4A, 4B). Growth retardation had been diagnosed in childhood but the patients reached a height of 178 and 180 cm as adults. Patient C1 complained of chronic diarrhoea of unknown aetiology and Patient C2 was reported to have limited lung function. Three further family members (parent C3, grand-aunt C4 and grandparent C5), found to carry the heterozygous c.268 G>A FBLN5 mutation, were mildly affected with respect to neurological and/or connective tissue involvement (Fig. 4C), however Patient C4 was also diagnosed with early age-related macular degeneration at age 71 (Table 1, Supplementary Table 2).
Figure 4.

Clinical findings. (A) Distal muscle atrophy in the lower limbs and pes cavus foot deformity in Patient C1, and pectus excavatus in Patient C2, both carrying the p.G90S FBLN5 variant. (B) Hypermobility of joints and hyperextensibility of the skin in Patients C1 and C2 carrying the p.G90S FBLN5 variant. Note also muscle wasting in the small hand muscles. (C) Hyperextensibility of the skin in Proband D1 carrying the p.V126M and in Patient C3 carrying the p.G90S FBLN5 sequence variant.
A third FBLN5 variation (c.376 G>A; p.V126M; Fig. 3) was detected in a 46-year-old patient (D1) diagnosed with sporadic spinal Charcot–Marie–Tooth disease, who had initially been excluded for mutations in GARS, exon 3 of BSCL2, HSPB1 and HSPB8. Examination revealed atrophy of the small hand and foot muscles and hyperstretchable skin (Fig. 4C) but no macular degeneration. The mutation was inherited from the 83-year-old father (Patient D2) who had progressive visual loss due to dry age-related macular degeneration and bilateral cataracts (Fig. 5), and an axonal neuropathy. The p.V126M FBLN5 variant is highly conserved (Supplementary Fig. 3) but was also found in three of our 317 control individuals, in one patient with Charcot–Marie–Tooth disease type 1 where segregation with the disease in the family was excluded, and in 2 of 300 patients with age-related macular degeneration. Re-examination of three of these individuals revealed normal clinical and mildly abnormal electrophysiological results in Patient F with age-related macular degeneration, whereas Controls I and J presented with marked axonal neuropathy without age-related macular degeneration at age 82 and 84 years, respectively (Table 1, Supplementary Table 2).
Figure 5.
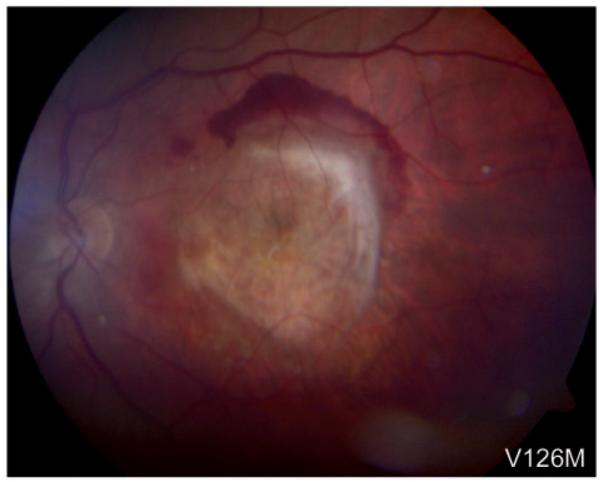
Age-related macular degeneration. The left eye of Patient F carrying the p.V126M FBLN5 sequence variant shows subretinal fibrosis caused by a choroidal neovascularization due to age-related macular degeneration (exudative).
Four further FBLN5 missense variants were detected in the 300 patients with age-related macular degeneration. Proband G with exudative age-related macular degeneration from age 79 carried the c.268 G>A (p.G90S) FBLN5 mutation and had mild to moderate weakness and wasting in the small hand and foot muscles and an axonal neuropathy in upper and lower limbs (Table 1, Supplementary Table 2). Other causes of peripheral neuropathies were not known. There was no family history of age-related macular degeneration or Charcot-Marie-Tooth disease but the patient had neither sibs nor children. At the FBLN5 locus Patient G shares 71 single nucleotide polymorphism markers with Patients C1 and C2 along 1.23 Mb making a common founder of this mutation conceivable.
Proband H with exudative age-related macular degeneration carried the novel highly-conserved c.143 C>T (p.T48I) FBLN5 variant (Fig. 3 and Supplementary Fig. 3) that was also absent in controls. She presented with moderate atrophy in the small hand muscles and severe gait disturbance that was attributed to joint degeneration. Nerve conduction velocities were within the normal range (Table 1, Supplementary Table 2). The deceased father had had visual loss since age 60 and severe deformity of the hands by history. The patient had no children.
Further physical examination of Patient K with exudative age-related macular degeneration carrying the known, highly conserved p.G267S FBLN5 variant (c.799 G>A) was not possible because the patient was already deceased at the time of genetic diagnosis, whereas neurological examination in Patient L carrying the same variant revealed a pronounced axonal neuropathy in the lower limbs (Table 1, Supplementary Table 2).
All known FBLN5 mutations, their frequencies and associated phenotypes are compiled in Table 2. All FBLN5 sequence variants identified in this study were additionally reviewed for their existence in the full project single nucleotide polymorphism call release data from the 1000 Genomes project (www.1000genomes.org/) November 2010 Data Release. These single nucleotide polymorphism calls are based on 628 individuals from the 20100804 sequence and alignment release of the project. The c.376 G>A; FBLN5 variant (rs61734479 dbSNP; p.V126M) was the only known variant cited in this data set.
Table 2.
Frequency of known and novel sequence variations in FBLN5 identified in this study compared to the literature
| Number of individuals screened for mutations in FBLN5 | p.T48I | p.G90S | p.V126M | p.R373C | p.G267S | ||
|---|---|---|---|---|---|---|---|
| Controls | This study | 317 | 0 | 0a | 3 | 0 | 0b |
| Stone et al., 2004 | 429 | 0 | 0 | 0 | 0 | 0 | |
| Lotery et al., 2006 | 337 | 0 | 0 | 5 | 0 | 0 | |
| Patients with initial diagnosis of age-related macular degeneration | This study | 300 | 1 | 2 | 3 | 0 | 2 |
| Stone et al., 2004 | 402 | 0 | 0 | 0 | 0 | 0 | |
| Lotery et al., 2006 | 802 | 0 | 0 | 1 | 0 | 1 | |
| Patients with initial diagnosis of neuropathyc | This study | 124 | 0 | 4 | 2 | 8 | 0 |
| Stone et al., 2004 | NE | NE | NE | NE | NE | NE | |
| Lotery et al., 2006 | NE | NE | NE | NE | NE | NE | |
| Total | 2711 | 1 | 6 | 14 | 8 | 3 |
At this position 200 additional control individuals were tested.
During this study no control samples were tested at this position.
Three patients carrying the p.G90S FBLN5 variant and one patient carrying the p.V126M FBLN5 variant had hyperextensible skin.
NE = not examined.
Skin, muscle and nerve biopsies
In the healthy skin, FBLN5 staining showed fibres of small calibre in the upper dermis (Fig. 6Aa). In the dermis of Patients C1 and C2 (data not shown) carrying the p.G90S FBLN5 variant a marked increase in FBLN5 immunoreactivity was apparent, particularly recognized as short plump fibres. Immunoreactive fibres were more pronounced in the lower dermis (Fig. 6Ae). In Patient B9 carrying the p.R373C and in Proband C3 with the p.G90S FBLN5 variant, no prominent changes in the structure of the fibres were evident (Fig. 6Ac, g).
Figure 6.


Skin biopsies. (A) Immunhistochemical detection of FBLN5 (red) and elastin (brown) in skin samples from a healthy individual (a, b), Probands C3 (c, d) and C1 (e, f) carrying the p.G90S FBLN5 mutation and Patient B9 carrying the p.R373C FBLN5 mutation (g, h). Fibulin-immunoreactive fibres in the upper dermis of the healthy control are thin (a, arrowhead). In the patient carrying the p.G90S FBLN5 mutation these fibres are plump (e, arrowhead). Elastin-immunoreactive fibres in the dermis of the healthy control are long (b, arrowhead) in contrast to the short and fragmented fibres of the patient carrying the p.G90S FBLN5 mutation (f, arrowhead). Elastin-immunoreactivity in the dermis of the patients, in general, is much lower than that in healthy dermis and virtually absent in the papillary dermis. Scale bar = 150 μm (FBLN5), 75 μm (elastin). (B) Electron microscopy of skin samples from the Probands C1, C2 and C3 carrying the p.G90S FBLN5 mutation and Patient B9 carrying the p.R373C FBLN5 mutation (see text). The arrows shown in C1a and C2a indicate prominent elastotubules detached from the elastin.
In the healthy skin, elastic fibres stained by anti-elastin antibody formed long predominantly horizontal fibres. Below the epidermis a reticular pattern with candelabra-like structures in the papillae was observed (Fig. 6Ab). In Patients C1 and C2 (data not shown) staining was generally absent in the papillary dermis and reduced in the lower dermis (Fig. 6Af). In all patients, especially in Patients C1 and C2, elastin-immunoreactive fibres were fragmented and short. In Probands B9 and C3 only mild changes in structure and arrangement of these fibres were observed (Fig. 6Ad, h).
Ultrathin skin biopsy sections of Patients C1 and C2 showed that elastic fibres were reduced in number, small in size and disorderly shaped. Elastotubules were prominent and appeared to be detached from the elastin. (Fig. 6B, C1a and C2a). Collagen fibres contained small fibrils (Fig. 6B and C1a, b) and showed some twisted, rope like arrangement (Fig. 6B and C1b). Isolated collagen fibrils revealed bending (Fig. 6B and C2b), and cross-banding was preserved. In Patient C3 number and size of elastic fibres seemed to be within normal range, but still they were moderately disorderly shaped and elastotubules were prominent (Fig. 6B and C3). In Patient B9 number and size of elastic fibres seemed to be within the normal range. They were moderately disorderly shaped but the structure seemed preserved (Fig. 6B and B9). Dermal-epidermal junction, papillary dermis and ground structure appeared normal in all patients (data not shown).
In nerve and muscle biopsies from Patients B5, C1 and C2 no obvious changes in the immunohistochemical staining patterns for FBLN5 and elastin were noted (data not shown).
Discussion
We ascertained a novel disease-linked region for a Charcot–Marie–Tooth disease type 1 neuropathy and identified mutations in FBLN5 as a likely cause of Charcot–Marie–Tooth disease. This study is the first demonstrating the concomitant occurrence of peripheral neuropathy, age-related macular degeneration and hyperelasticity of the skin based on mutations in FBLN5 and delineates the fundamental clinical characteristics of an independent hitherto unknown syndrome displaying wide interindividual variability, even within families.
The power of next-generation sequencing to achieve an accurate genetic diagnosis has been demonstrated (Hodges et al., 2007; Ng et al., 2010; Nikopoulos et al., 2010). In a patient with Charcot-Marie-Tooth disease, whole-genome sequencing enabled the identification of the genetic subtype, but the same study pointed out considerable limitations addressing the interpretation of the numerous variants detected in the genome (Lupski et al., 2010). Furthermore, diagnostic implications of such comprehensive, complete or exome-wide genetic analyses for genetic counselling must be carefully appraised. Our study emphasizes disease-linked protein coding exome sequencing in families for which ‘restricted’ loci can be defined as an attractive, rapid and economic alternative.
During this screen, we gained at least 5-fold high quality coverage of >95% of the disease-linked targeted sequence making it less likely that another disease-causing variant located within the residual 5% of the target region was missed. As for the majority of previous linkage studies, functional variants located within the non-exonic sequences within the disease-linked interval would not have been detected by using this targeted exon sequencing approach.
We identified five different heterozygous missense mutations in FBLN5, three of which are novel. Each mutation was associated with age-related macular degeneration in at least one proband, each being >70 years. In a cohort of 300 patients with age-related macular degeneration, 2% (6/300) carried FBLN5 mutations. Opthalmoscopic changes consisted of both early and late (exudative or dry) age-related macular degeneration. FBLN5 is a secreted extracellular matrix protein and consists of six calcium-binding epidermal growth factor-like modules and a FBLN-type C-terminus (Fig. 7). Mutations located in the first epidermal growth factor domain appeared to be associated with a spinal Charcot–Marie–Tooth disease phenotype whereas patients carrying the p.R373C FBLN5 variant located in the FBLN-type C-terminus domain presented with a demyelinating neuropathy in upper and lower limbs. Presently, we cannot explain how distinct FBLN5 mutations may cause different neuropathic phenotypes. However, this situation is reminiscent of other Charcot–Marie–Tooth disease genes. Mutations in the MPZ gene may result in both severe early onset Charcot–Marie–Tooth disease type 1 and very late onset axonal Charcot–Marie–Tooth disease. Moreover, associated features such as pupillary abnormalities or deafness have been described for particular MPZ mutations (De Jonghe et al., 1999; Shy et al., 2004; Kabzinska et al., 2007). Similarly, allelic mutations in HSPB1, HSPB8 or BSCL2 have been associated with Charcot–Marie–Tooth disease type 2, distal hereditary motor neuropathy or hereditary spastic paraplegia (Evgrafov et al., 2004; Irobi et al., 2004, Auer-Grumbach et al., 2005; Tang et al., 2005), and allelic ATL1 mutations cause hereditary sensory neuropathy type I and hereditary spastic paraplegia (Guelly et al., 2011). An even more complex spectrum of phenotypes affecting diverse tissues has been reported for allelic mutations in TRPV4, LMNA, ATP7A or DMN2 (Rankin and Ellard, 2006; Auer-Grumbach et al., 2010; Durieux et al., 2010; Kennerson et al., 2010). Finally, mutations in the GDAP1, HSPB1 and MFN2 genes have been shown to cause both autosomal dominant and autosomal recessive Charcot–Marie–Tooth disease (Houlden et al., 2008; Nicholson et al., 2008; Crimella et al., 2010). It currently remains unclear whether the carrier parents of children with cutis laxa due to homozygous FBLN5 mutations are prone to develop any of the features of our patients described here. Alternatively, the cutis laxa-related FBLN5 mutations might either produce more functional protein than Charcot–Marie–Tooth disease-associated FBLN5 mutations or do not interfere with normal elastic fibre assembly from the wild-type allele.
Figure 7.

Schematic model of FBLN5 showing epidermal growth factor calcium-binding domains (EGF_Ca) in blue, the low complexity domain in green (domain boundaries are indicated with amino acid positions above the exons), and the FBLN-type carboxy-terminus (FC) in yellow; exons 1–11 and disease-associated mutations. Mutations found in patients with cutis laxa or hyperelastic skin are underlined. Variants found in our probands are indicated in red colour. *Putative polymorphisms.
The most complex although variable phenotype was seen in patients carrying the p.G90S FBLN5 variant. Predominant distal motor neuropathy, mild skeletal abnormalities, hyperstretchable skin and hypermobility of joints indicating some clinical overlap with Ehlers-Danlos syndrome were striking features in two sibs with the p.G90S FBLN5 variant. (Savasta et al., 2010). Interestingly, neuromuscular involvement including axonal neuropathies has also been reported in patients with various types of Ehlers–Danlos syndrome (Voermans et al., 2009). Though we cannot completely rule out the possibility that the severe phenotype observed in the two youngest patients carrying the p.G90S FBLN5 variant underlies further disease modifying genes or is aggravated by an effect of one of the polymorphisms identified in the Charcot–Marie–Tooth disease genes, it appears unlikely that this variant is a rare polymorphism considering the fact that it was not detected in a large number of control subjects. Also, FBLN5 immunoreactivity, indicating short plump elastic fibres in these patients, is reminiscent of the recessive form of FBLN5-associated cutis laxa (Van Maldergem et al., 2009). However, in contrast to the loss of anti-elastin and reduced FBLN5 immunoreactivity as observed for patients with cutis laxa with a homozygous FBLN5 mutation (Hu et al., 2006), we found an increase of FBLN5 protein staining in our probands carrying heterozygous FBLN5 mutations (Fig. 6A) with an accumulation of abnormal elastin in the dermis and a reduction of FBLN5 in the papillary dermis, but increased FBLN5 deposition in the deep dermis. This pattern resembles age-related changes in patients with solar elastosis that occur after repeated, prolonged and excessive sun exposure (Kadoya et al., 2005).
Of special interest is the p.V126M FBLN5 variant that was initially reported as a rare polymorphism (Lotery et al., 2006). Two later studies however presented controversial data designating this variant as pathogenic and absent in control individuals (Jones et al., 2009, 2010). In our study the p.V126M FBLN5 variant was detected in a proband with distal hereditary motor neuropathy and in three patients with age-related macular degeneration, but also in three control individuals, and in a patient with Charcot–Marie–Tooth disease type 1, in whom segregation with the disease was excluded, preliminarily suggesting that this variant might be a rare polymorphism. This is also supported by the data provided through the 1000 Genomes project (www.1000genomes.org/). However, five individuals in this study carrying the p.V126M FBLN5 variant exhibited a disease phenotype of either age-related macular degeneration, peripheral neuropathy and/or hyperelasticity of the skin (Table 1, Fig. 4C) pointing to at least increased susceptibility of carriers for any of these diseases. We therefore conclude that further clinical and genetic studies are needed to make any firm conclusions concerning the status of this FBLN5 variant.
The important role of FBLN5 in formation and repair of elastic fibres has been studied extensively and its interactions with fibrillin, tropoelastin and lysil oxidase-like proteins have been demonstrated (Choudhury et al., 2009) (Supplementary Fig. 4). Lysil oxidase is an extracellular copper enzyme that catalyzes the covalent cross-link of collagens and elastin, thus being responsible for stabilization of collagen fibrils and for the integrity and elasticity of mature elastin (Rodríguez et al., 2008). Altered lysil oxidase plays a crucial role in X-linked forms of cutis laxa due to ATP7A mutations and in types of Ehlers-Danlos syndrome (Tümer and Møller, 2010). Our study identifies novel FBLN5 mutants that lead to ultrastructural features as in cutis laxa, probably by disturbed interaction with lysil oxidase-like proteins, and also suggests a role of FBLN5 and the extracellular matrix in the maintenance of the peripheral nervous system.
It has been postulated that variants of FBLN5 identified in patients with age-related macular degeneration change its biophysical and biochemical properties and consequently cause weakening of Bruch’s membrane, a thin elastic fibre-enriched membrane that limits the retinal pigmented epithelium. FBLN5 has also been supposed to act as an endogenous angiogenesis inhibitor, suggesting that mutations causing a loss of FBLN5 function contribute to the choroidal neovascularization seen in age-related macular degeneration (Sullivan et al., 2007).
It is beyond the scope of this study to elucidate the pathogenic molecular mechanisms producing the diverse peripheral neuropathy phenotypes seen in our patients. Uncovering further patients with Charcot–Marie–Tooth disease and families carrying mutations in FBLN5 is needed to confirm its pathogenic role in the development of peripheral neuropathies. It is conceivable that the alterations identified in our patients cause misfolding of FBLN5 resulting in decreased secretion and/or impairment of extracellular matrix protein interactions (Schneider et al., 2010). Failure of FBLN5 to activate β1 integrin receptors led to cell spreading migration and proliferation of primary smooth muscle cells (Lomas et al., 2007). Critical neuronal cell-cell interactions might be directly or indirectly damaged by the abnormal matrix composition thereby leading to typical peripheral neuropathy phenotypes. According to the reported predominant expression of FBLN5 in multiple organs like the heart, colon, kidney, lung, pancreas and placenta, one could expect an even more complex phenotype. However, overall penetrance with multi-organ involvement might be modulated by tissue-specific interaction partners and mechanisms. The detection of such mechanisms and molecular pathways will be a challenge of future studies and will once help to develop reasonable therapies for affected patients.
Web resources and accession numbers
GenBank: human fibulin-5 cDNA, NM_006329.3, human fibulin-5, NP_006320.2.
The Human Gene Mutation database: www.hgmd.cf.ac.uk/ac/index.php.
1000 Genomes database: www.1000genomes.org (Data Release November 2010).
Supplementary Material
Supplementary material: Supplemental material is available at Brain online.
Acknowledgements
We are grateful for the participation of the patients and families in this study. We thank Claudia Meindl, Ulrike Schmidbauer, Carina Fischer, Martina Hatz, Theresa Maierhofer and Gabriele Michelitsch for expert technical assistance.
Funding
This work was supported by the Austrian Science Fund (FWF, P19455-B05), the Land Steiermark (A3-16.Z-24/2009-1, 2010-2) and the Oesterreichische Nationalbank (ÖNB, project 13010).
Abbreviation
- FBLN5
fibulin-5
Footnotes
For Permissions, please journals.permissions@oup.com
References
- Auer-Grumbach M, Schlotter-Weigel B, Lochmüller H, Strobl-Wildemann G, Auer-Grumbach P, Fischer R, et al. Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol. 2005;57:415–24. doi: 10.1002/ana.20410. [DOI] [PubMed] [Google Scholar]
- Auer-Grumbach M, Olschewski A, Papic L, Kremer H, McEntagart ME, Uhrig S, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42:160–4. doi: 10.1038/ng.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti C, Nodari A, Wrabetz L, Feltri ML. Role of intergrins in peripheral nerves and hereditary neuropathies. Neuromol Med. 2006;8:191–204. doi: 10.1385/nmm:8:1-2:191. [DOI] [PubMed] [Google Scholar]
- Choudhury R, McGovern A, Ridley C, Cain SA, Baldwin A, Wang MC, et al. Differential regulation of elastic fiber formation by fibulin-4 and -5. J Biolog Chem. 2009;284:24553–67. doi: 10.1074/jbc.M109.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus S, Fischer J, Mégarbané H, Mégarbané A, Jobard F, Debret R, et al. A p.C217R mutation in fibulin-5 from cutis laxa patients is associated with incomplete extracellular matrix formation in a skin equivalent model. J Invest Dermatol. 2008;128:1442–50. doi: 10.1038/sj.jid.5701211. [DOI] [PubMed] [Google Scholar]
- Crimella C, Tonelli A, Airoldi G, Baschirotto C, D’Angelo MG, Bonato S, et al. The GST domain of GDAP1 is a frequent target of mutations in the dominant form of axonal Charcot Marie Tooth type 2K. J. Med Genet. 2010;47:712–6. doi: 10.1136/jmg.2010.077909. [DOI] [PubMed] [Google Scholar]
- De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Lofgren A, et al. The thr124-to-met mutation in peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122:281–90. doi: 10.1093/brain/122.2.281. [DOI] [PubMed] [Google Scholar]
- Durieux AC, Prudhon B, Guicheney P, Bitoun M. Dynamin 2 and human diseases. J Mol Med. 2010;88:339–50. doi: 10.1007/s00109-009-0587-4. [DOI] [PubMed] [Google Scholar]
- Elahi E, Kalhor R, Banihosseini SS, Torabi N, Pour-Jafar H, Houshmand M, et al. Homozygous missense mutation in fibulin-5 in an Iranian autosomal recessive cutis laxa pedigree and associated haplotype. J Invest Dermatol. 2006;126:1506–9. doi: 10.1038/sj.jid.5700247. [DOI] [PubMed] [Google Scholar]
- Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602–6. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A. Allegro version 2. Nat Genet. 2005;37:1015–6. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- Guelly C, Zhu PP, Leonardis L, Papić L, Zidar J, Schabhüttl M, et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet. 2011;88:99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges E, Xuan Z, Balija V, Kramer M, Molla MN, Smith SW, et al. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–7. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- Houlden H, Laura M, Wavrant-De Vrièze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71:1660–8. doi: 10.1212/01.wnl.0000319696.14225.67. [DOI] [PubMed] [Google Scholar]
- Hu Q, Loeys BL, Coucke PJ, De Paepe A, Mecham RP, Choi J, et al. Fibulin-5 mutations: mechanisms of impaired elastic fiber formation in recessive cutis laxa. Hum Mol Genet. 2006;15:3379–86. doi: 10.1093/hmg/ddl414. [DOI] [PubMed] [Google Scholar]
- Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36:597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- Jones RP, Ridley C, Jowitt TA, Wang MC, Howard M, Bobola N, et al. Structural effects of fibulin 5 missense mutations associated with age-related macular degeneration and cutis laxa. Invest Ophthalmol Vis Sci. 2010;51:2356–62. doi: 10.1167/iovs.09-4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RP, Wang MC, Jowitt TA, Ridley C, Mellody KT, Howard M, et al. Fibulin 5 forms a compact dimer in physiological solutions. J Biol Chem. 2009;284:25938–43. doi: 10.1074/jbc.M109.011627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabzinska D, Korwin-Piotrowska T, Dreschler H, Drac H, Hausmanowa-Petrusewicz I, Kochanski A. Late-onset Charcot-Marie-Tooth type 2 disease with hearing impairment associated with a novel Pro105Thr mutation in the MPZ gene. Am. J. Med. Genet. 2007;143A:2196–9. doi: 10.1002/ajmg.a.31908. [DOI] [PubMed] [Google Scholar]
- Kadoya K, Sasaki T, Kostka G, Timpl R, Matsuzaki K, Kumagai N, et al. Fibulin-5 deposition in human skin: decrease with ageing and ultraviolet B exposure and increase in solar elastosis. Br J Dermatol. 2005;153:607–12. doi: 10.1111/j.1365-2133.2005.06716.x. [DOI] [PubMed] [Google Scholar]
- Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am J Hum Genet. 2010;86:343–52. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005;65:197–204. doi: 10.1212/01.wnl.0000168898.76071.70. [DOI] [PubMed] [Google Scholar]
- Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, et al. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet. 2002;11:2113–8. doi: 10.1093/hmg/11.18.2113. [DOI] [PubMed] [Google Scholar]
- Lomas AC, Mellody KT, Freeman LJ, Bax DV, Shuttleworth CA, Kielty CM. Fibulin-5 binds human smooth-muscle cells through alpha5beta1 and alpha4beta1 integrins, but does not support receptor activation. Biochem J. 2007;405:417–28. doi: 10.1042/BJ20070400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotery AJ, Baas D, Ridley C, Jones RP, Klaver CC, Stone E, et al. Reduced secretion of fibulin 5 in age-related macular degeneration and cutis laxa. Hum Mutat. 2006;27:568–74. doi: 10.1002/humu.20344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–91. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova D, Zou Y, Ringpfeil F, Sasaki T, Kostka G, Timpl R, et al. Genetic heterogeneity of cutis laxa: a heterozygous tandem duplication within the fibulin-5 (FBLN5) gene. Am J. Hum Genet. 2003;72:998–1004. doi: 10.1086/373940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Olvera MA, Clark AF, Stone EM. Fibulin-5 distribution in human eyes: relevance to age-related macular degeneration. Exp Eye Res. 2007;84:378–80. doi: 10.1016/j.exer.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento GM, Nunes CS, Menegotto PF, Raskin S, Almeida N. Cutis laxa: case report. An Bras Dermatol. 2010;85:684–6. doi: 10.1590/s0365-05962010000500013. [DOI] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson GA, Magdelaine C, Zhu D, Grew S, Ryan MM, Sturtz F, et al. Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology. 2008;70:1678–81. doi: 10.1212/01.wnl.0000311275.89032.22. [DOI] [PubMed] [Google Scholar]
- Nikopoulos K, Gilissen C, Hoischen A, van Nouhuys CE, Boonstra FN, Blokland EA, et al. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:240–7. doi: 10.1016/j.ajhg.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin J, Ellard S. The laminopathies: a clinical review. Clin Genet. 2006;70:261–74. doi: 10.1111/j.1399-0004.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80:1304–14. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- Rodríguez C, Rodríguez-Sinovas A. Martínez-González J Lysil oxidase as a potential therapeutic target. Drug News Perspect. 2008;21:218–24. doi: 10.1358/dnp.2008.21.4.1213351. [DOI] [PubMed] [Google Scholar]
- Savasta S, Merli P, Ruggieri M, Bianchi L, Spartà MV. Ehlers-Danlos syndrome and neurological features: a review. Childs Nerv Syst. 2010;27:365–371. doi: 10.1007/s00381-010-1256-1. [DOI] [PubMed] [Google Scholar]
- Schneider R, Jensen SA, Whiteman P, McCullagh JS, Redfield C, Handford PA, et al. Biophysical characterisation of fibulin-5 proteins associated with disease. J Mol Biol. 2010;401:605–17. doi: 10.1016/j.jmb.2010.06.039. [DOI] [PubMed] [Google Scholar]
- Shy ME, Jáni A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain. 2004;127:371–84. doi: 10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- Stone EM, Braun TA, Russell SR, Kuehn MH, Lotery AJ, Moore PA, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. N Engl J Med. 2004;351:346–53. doi: 10.1056/NEJMoa040833. [DOI] [PubMed] [Google Scholar]
- Sullivan KM, Bissonnette R, Yanagisawa H, Hussain SN, Davis EC. Fibulin-5 functions as an endogenous angiogenesis inhibitor. Lab Invest. 2007;87:818–27. doi: 10.1038/labinvest.3700594. [DOI] [PubMed] [Google Scholar]
- Tang B, Zhao G, Luo W, Xia K, Cai F, Pan Q, et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet. 2005;116:222–4. doi: 10.1007/s00439-004-1218-3. [DOI] [PubMed] [Google Scholar]
- Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet. 2010;18:511–8. doi: 10.1038/ejhg.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maldergem L, Loeys B. FBLN5-related cutis laxa. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet] University of Washington, Seattle; Seattle (WA): 1993–2009. [Google Scholar]
- Voermans NC, van Alfen N, Pillen S, Lammens M, Schalkwijk J, Zwarts MJ, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687–97. doi: 10.1002/ana.21643. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Schluterman MK, Brekken RA. Fibulin-5, an integrin-binding matricellular protein: its function in development and disease. J Cell Commun Signal. 2009;3:337–47. doi: 10.1007/s12079-009-0065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Davis EC, Richardson JA, Starcher BC, Li T, Gerard RD, et al. Molecular analysis of fibulin-5 function during de novo synthesis of elastic fibres. Mol Cell Biol. 2007;27:1083–95. doi: 10.1128/MCB.01330-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.