

Abstract
There is considerable evidence to support the hypothesis that the blockade of nAChR is responsible for the antidepressant action of nicotinic ligands. The nicotinic acetylcholine receptor (nAChR) antagonist, mecamylamine, has been shown to be an effective add-on in patients that do not respond to selective serotonin reuptake inhibitors. This suggests that nAChR ligands may address an unmet clinical need by providing relief from depressive symptoms in refractory patients. In this study, a new series of nAChR ligands based on an isoxazole-ether scaffold have been designed and synthesized for binding and functional assays. Preliminary structure-activity relationship (SAR) efforts identified a lead compound 43, which possesses potent antidepressant-like activity (1 mg/kg, IP; 5 mg/kg, PO) in the classical mouse forced swim test. Early stage absorption, distribution, metabolism, excretion, and toxicity (ADME-Tox) studies also suggested favorable drug-like properties, and broad screening towards other common neurotransmitter receptors indicated that compound 43 is highly selective for nAChRs over the other 45 neurotransmitter receptors and transporters tested.
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) are pentamers assembled from varying combinations of subunits (α2–α10, β2–β4) and belong to the ligand-gated ion channel super-family of neurotransmitter receptors.1–3 These receptors are broadly distributed in the central and peripheral nervous systems, where they modulate many processes, such as ganglionic transmission regulated by α3β4*-nAChRs (the * indicates that subunits other than those specified are known or possible partners in the closed assembly), neuroprotection of dopaminergic pathways and nociception mediated by α4*-nAChRs, as well as learning, memory, and addiction by β2*-nAChR.3–6 Over the past two decades, many compounds targeting nAChRs have been tested in various stages of clinical trials.7 However, only one new chemical entity, varenicline (1), has been launched and marketed as a potent partial agonist at the α4β2-nAChR for smoking cessation (Figure 1).8, 9, 10
Figure 1.

Selected nicotinic acetylcholine receptor ligands.
Given nAChR subtype diversity and their involvement in the modulation of a host of neurotransmitter systems, nicotinic ligands have the potential to treat a multitude of central nervous system (CNS)-related dysfunctions, including chronic depression.8, 11 There is considerable evidence to support the hypothesis that the blockade(antagonism or receptor desensitization) of nAChR is responsible for the antidepressant action of nicotinic ligands.12–14 In particular, clinical studies have shown that the cholinesterase inhibitor, physostigmine, produces depressive symptoms in humans15 whereas mecamylamine16 and the muscarinic antagonist scopolamine17, 18 relieve depressive symptoms in humans. Additionally, preclinical studies provide support for the hypothesis that increased cholinergic activity leads to depressed mood states. Flinders sensitive rats, a line selectively bred for increased cholinergic sensitivity, exhibit several depressive-like behaviors19, 20 Moreover, administration of the nicotinic antagonist, mecamylamine elicits an antidepressant-like effect in the mouse forced swim test, and this effect is reduced when the β2 subunit gene is knocked out.11 The same effects were also observed in response to the tricyclic antidepressant amitriptyline, strongly suggesting that β2*-nAChRs are involved in the antidepressant efficacy of nicotinic ligands.21 The α4β2-nAChR is the predominant subtype in the vertebrate CNS, and the α4β2 nicotinic agonists cytisine (2)22, A-85380 (9)23 and compound 124 induce antidepressant-like effects in mice that are similar to the effects of the antagonist mecamylamine. The S-enantiomer of mecamylamine (TC-5214, 5) is an α4β2-nAChR modulator now in Phase II clinical trials for use in the treatment of depression.25 Therefore, the α4β2-nAChR is an attractive target for the development of novel antidepressants, although it is unclear whether nAChR activation, desensitization, or some combination of both is essential. It is also known from clinical studies that α3β4*-nAChRs contribute to adverse side effects in vivo, although roles in mood control also are possible, as exemplified by mecamylamine.26–28 Consequently, we chose to focus on developing potent agonists selective for the α4β2-nAChR, bearing in mind that activity at the α3β4*-nAChR subtype might be an attribute or a detriment.
Multiple modifications to the structure of natural nicotinic ligands, especially nicotine (3), epibatidine (4), and compound 2, have already been explored over the past twenty years. Most of the reported nicotinic ligands bear a substituted pyridine ring as the core scaffold. Some compound classes involving replacement of the pyridine ring by isosteres have been investigated, as exemplified by substituted phenyl derivatives29, quinolines30, furopyridines31, structurally-related chroman derivatives32, and most interestingly the five-membered heteroaromatic rings, isoxazole and isothiazole.33–37 The ability to replace the pyridine moiety of S-(−)-nicotine with an isoxazole ring was first investigated by Abbott Laboratories, leading to the clinical study of ABT-418 (6) for the treatment of both Alzheimer’s disease (AD) and attention deficit hyperactivity disorder (ADHD). Compound 6 is a selective, full agonist at the α4β2-nAChR with a Ki value of 7.4 nM.8, 38 The complete subtype selectivity profile was not determined. In preclinical studies, compound 6 demonstrated efficacy and potency similar to that of compound 3 in animal models of cognition while exhibiting reduced toxicity.38 Although compound 6 failed in clinical development due to the occurrence of nausea as a side effect, isoxazole-containing nicotinic ligands remain an exciting area of investigation. Substituted isoxazoles were also successfully applied in optimization studies of compound 4. Replacement of the chloropyridyl group in compound 4 with a methylisoxazolyl group led to epiboxidine (7),39, 40 which was approximately 10-fold less potent than compound 4 but 17-fold more potent than compound 6 in the displacement of [3H]nicotine at the α4β2-nAChR from rat cerebral cortical membranes. Epiboxidine retained analgesic activity in mice at 25 mg/kg compared to compound 4 at 10 mg/kg, with greatly reduced toxicity. Furthermore, a series of 3-(5-isoxazolyl)methylene-1-azabicyclic compounds (8) were synthesized as potent nicotinic ligands.41
Pyridyl ethers, in which a CH2O linker is inserted between compound 3’s pyrrolidine (or in analogues, azetidine) ring and its pyridine ring, have attracted considerable interest as α4β2-nAChR agonists because of their high potency.42, 43 For example, compound 9 possesses a Ki value of ca. 50 pM and a high efficacy of 163% compared to compound 3 at the human α4β2-nAChR44. ABT-594 (10), a nAChR agonist, was advanced to a Phase II clinical trial for the treatment of chronic and neuropathic pain with a potency 50 times that of morphine, but was discontinued due to an unacceptably narrow therapeutic index.45, 46 ABT-089 (11) is currently under investigation as a replacement for compound 6 that has an improved preclinical therapeutic index and a better pharmacokinetic profile. It has gone through Phase II clinical studies for cognitive dysfunction.47, 48 Based upon the precedent quoted above, we anticipated that an isoxazole moiety could be used as a readily accessible replacement for the pyridine core in the design of new nicotinic ligands. Herein, we report the synthesis and pharmacological evaluation of a novel series of nAChR ligands based on an isoxazole core in combination with the CH2O linker (“isoxazole ethers”; Figure 2). Selected compounds were further assessed in behavioral tests, in a broad screening panel of common CNS neurotransmitter receptors and transporters, as well as in preliminary in vitro ADME-Tox studies.
Figure 2.

General structure of the present series of isoxazole ether nAChR ligands.
Results and Discussion
Chemistry
First, we designed compounds that could be accessed from readily available starting materials to ascertain whether an isoxazole moiety could replace the pyridine core in the previously published pyridine ether nicotinics developed by Abbott. The 3-alkoxyisoxazoles 18–21 were synthesized in 3–6 steps utilizing the synthetic routes shown in Scheme 1. Intermediate 16 was formed via the Mitsunobu reaction of Boc-protected 2(S)-azetidinylmethanol (15) and 3-hydroxyisoxazole-5-carboxylic acid methyl ester (14), which was in turn prepared as described in the literature from dimethyl 2-butynedioate (13). The ester 16 was subsequently reduced with LiBH4 to furnish the primary alcohol 17, and this intermediate was carried on to the iodide. The phenyl ether 19 and the aliphatic ethers 20–21 were obtained through nucleophilic substitution following standard methods. After acidic deprotection and subsequent purification by HPLC, compounds 18–21 were obtained as trifluoro acetates. The number of equivalents of trifluoroacetic acid (TFA) in these non-stoichiometric compounds was determined by elemental analysis. The 5-methylated 3-alkoxyisoxazole 24 was synthesized in the same manner from commercially available 3-hydroxy-5-methylisoxazole 22 (Scheme 1).
Scheme 1a.

a Reagents and conditions: (a) N-hydroxyurea, 1,5-diazabicyclo[5.4.0]undec-5-ene, MeOH, 0 °C, then HCl; (b) 1-(tert-butoxycarbonyl)-2(S)-azetidinylmethanol (15), diisopropyl azodicarboxylate, PPh3, THF, 0 °C to rt; (c) LiBH4, THF, 0 °C to rt; (d) TFA, CH2Cl2; (e) I2, PPh3, imidazole, CH2Cl2, 0 °C to rt; (f) phenol, K2CO3, DMF, rt; (g) NaH, DMF, RBr, 0 °C to rt.
The preparation of 5-alkoxyisoxazole ligands proceeded through the common intermediates 29 and 30, which were in turn prepared from the commercially available ester 26 (Scheme 2). Compounds 31, 32, 33, and 43 were synthesized by employing the same strategy as described in Scheme 1. The primary alcohol 29 was transformed to an iodide, followed by nucleophilic substitution with aniline or 4-fluoroaniline to afford the precursors of amine derivatives 34 and 35. Carbamate analogues 34–38 were prepared by reaction of 29 with the corresponding isocyanates. The fluoromethyl derivative 39 was obtained by treatment of alcohol 29 with (diethylamino)sulfur trifluoride. Subsequent Boc deprotection of the precursors yielded the desired final compounds 34–39.
Scheme 2a.

a Reagents and conditions: (a) diisopropyl azodicarboxylate, PPh3, THF, 0 °C to rt; (b) LiBH4, THF, 0 °C to rt; (c) TFA, CH2Cl2; (d) I2, PPh3, imidazole, CH2Cl2, 0 °C to rt; (e) K2CO3, DMF, phenol, rt; (f) HNR1R2, CH3CN, rt; (g) RN=C=O, 4-(dimethylamino)pyridine, PhMe, 80 °C; (h) Et2NSF3, CH2Cl2, −78 °C.
In vitro Characterization—Radioligand Binding Studies
In vitro binding affinities of the five 3-alkoxyisoxazoles (18–21, 24) were determined by the standard [3H]epibatidine binding assay at seven rat nAChR subtypes (Table 1).49 While this initial set of compounds showed weak binding to all seven nAChR subtypes tested, compound 18 exhibited a moderate affinity for α4β2- and α4β2*-nAChRs.
Table 1.
Binding affinities of 3-alkoxyisoxazole ligands at seven rat nAChR subtypes
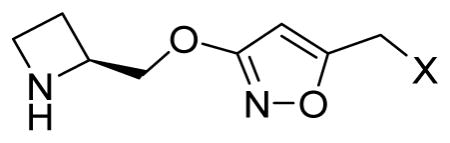 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | X |
Ki (nM)a |
||||||
| α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β2*b | α4β4 | ||
| 18 | OH | 337 | >104 | 990 | >104 | 176 | 405 | 7530 |
| 19 | OC6H5 | NAd | NA | NA | NA | NA | NA | NA |
| 20 | OCH3 | NA | NA | 4100 | NA | NA | NA | NA |
| 21 | OCH2CH(CH2)2 | NA | NA | NA | NA | NA | NA | NA |
| 24 | H | 3340 | >104 | 6670 | >104 | 1950 | 7380 | >104 |
| 3c | - | 5.5 | 70.0 | 29.0 | 260 | 4.9 | 9.8 | 23.0 |
See Experimental Section.
α4β2*, endogenous receptors prepared from rat forebrain. Besides α4 and β2, other unidentified subunits may also be present. Details are provided in the Experimental Section.
The Ki values for compound 3 are taken from the PDSP Assay Protocol Book.
not active, defined as < 50% binding in the primary assay at 10μM.
It is commonly accepted that the essential pharmacophore of nicotinic ligands consist of a cationic center (e.g., quaternized or protonated nitrogen) and a hydrogen-bond acceptor (e.g., a nitrogen atom in the case of pyridine-containing ligands).50 The inactivity of our first batch of isoxazole-ether compounds is possibly a result of misalignment of these two key elements. Therefore, to align these pharmacophoric elements differently, and hopefully more appropriately, isoxazoles with a reverse position of their N and O ring atoms were synthesized. As the alcohol 18 (Table 1) was the only congener that so far showed moderate affinity to the α4β2-nAChR, the reverse 5-hydroxyisoxazole 31 (Table 2) and its phenyl ether 32 were synthesized. Encouragingly, the binding affinities of 32 were dramatically improved, while compound 31 maintained its affinity in comparison to 18. Subsequently amines, carbamates, simple alkyl, and fluoromethyl derivatives were synthesized to further study the structure-activity relationship (SAR).
Table 2.
Binding affinities of 5-alkoxyisoxazole ligands at seven rat nAChR subtypes
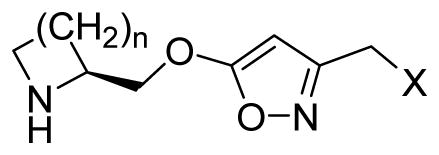 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | n | X |
Ki (nM)a |
||||||||
| α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β2*b | α4β4 | α7 | α7*b | |||
| 31 | 1 | OH | 197 | >104 | 521 | >104 | 137 | 637 | 4900 | NDd | ND |
| 32 | 1 | OC6H5 | 47.9 | 58.0 | 362 | 186 | 23.8 | 172 | 27.5 | ND | ND |
| 33 | 2 | OC6H5 | 176 | 320 | 2040 | 809 | 160 | 2120 | 55.1 | ND | ND |
| 34 | 1 | NHC6H5 | 150 | 144 | 462 | 771 | 75.9 | 386 | 33.2 | ND | ND |
| 35 | 1 | NHC6H4F-p | 201 | 30.3 | ND | 171 | 49.0 | 417 | 9.9 | ND | ND |
| 36 | 1 | OC(O)NHC6H5 | 42.3 | 162 | 123 | 1760 | 19.7 | 157 | 60.0 | ND | ND |
| 37 | 1 | OC(O)NHC2H5 | 157 | 6570 | 315 | NAe | 31.2 | 207 | 1240 | ND | ND |
| 38 | 1 | OC(O)NH-c-C5H11 | 126 | 9970 | 370 | NA | 13.1 | 149 | 3480 | ND | ND |
| 39 | 1 | F | 11.8 | 472 | 17.3 | 1270 | 7.3 | 11.9 | 163 | ND | ND |
| 43 | 1 | H | 4.3 | 311 | 8.7 | 692 | 4.6 | 12.0 | 86.0 | 2890 | 6790 |
| 44 | 2 | H | 616 | 5810 | 1030 | 8780 | 129 | 1100 | 4140 | ND | ND |
| 3c | - | - | 5.5 | 70.0 | 29.0 | 260 | 4.9 | 9.8 | 23.0 | ND | ND |
| 1f | - | - | - | - | - | 86 | 0.4 | - | 110 | 125 | - |
See Experimental Section.
α4β2* or α7*, endogenous receptors prepared from rat forebrain. Besides α4, β2 or α7, other unidentified subunits may also be present. Details are provided in the Experimental Section.
The Ki values for compound 3 are taken from the PDSP Assay Protocol Book.
ND: not detected.
not active, defined as < 50% binding in the primary assay at 10 μM.
The Ki values for compound 1 are from reference 51.
In general, most of the 5-alkoxyisoxazoles (Table 2) bind to α4β2-nAChRs with nanomolar affinity. The binding affinities of all amines (34, 35), carbamates (36, 37, and 38), the 3-methyl derivative 43, and the 3-fluoromethyl derivative 39 were increased 1.8- to 30-fold compared to the parent alcohol 31. Compound 43 carrying a 3-methyl substituent turned out to have the best affinity for α4β2- and α4β2*-nAChRs with Ki values of 4.6 nM and 12.0 nM, respectively. All of these compounds have a similar subtype selectivity profile as compound 3 and compound 151; they display selectivity for the α4β2- over the α3β4-nAChR in radioligand binding competition assays. The alcohol 31, methyl derivative 43, fluoromethyl derivative 39, and carbamates 36–38 actually exceed compound 3’s α4β2- over α3β4-selectivity. Among these ligands, the cyclopentylcarbamate 38 exhibits the best selectivity with a Ki value of 13.1 nM at the α4β2-nAChR and inactivity at the α3β4-nAChR. The aforementioned six compounds also exhibit selectivity for all the other β2- over β4-containing subtypes. Their affinities at the α4β2*-nAChR dropped 1.6- to 13.5- fold compared to those at the α4β2-nAChR, probably due to the existence of additional subunits in the former.
To determine the influence of different heteroalicyclic ring sizes, the pyrrolidine analogues 33 and 44 were also synthesized. Pyrrolidines 33 and 44 retained all of the selectivity exhibited by the corresponding azetidines (32 and 43), but their affinities were reduced at all seven rat nAChR subtypes, which is consistent with previous studies in the pyridine ether series.42
In vitro Functional Characterization
The most potent α4β2-nAChR ligands based on binding assays, 39 and 43, as well as pyrrolidine analogue 44 were selected for evaluation of functional activity using the 86Rb+ ion flux assay in SH-EP1-hα4β2, SH-SY5Y (α3β4*) and TE671/RD (α1β1γδ) cells (Figure 3; Tables 3 and 4). Consistent with the binding data, the azetidines 39 and 43 were found to be more potent than the pyrrolidine 44, both in agonism and functional inactivation at the α4β2-nAChR (Figure 3, Table 3). Compounds 39 and 43 had agonist efficacies at the α4β2-nAChR comparable to compound 3 and higher than that of compound 1. Compounds 39 and 43 have functional inactivation efficacies lower than those of compound 3 or compound 1. They were both full agonists at the α3β4*-nAChR, with similar potencies to those seen at the α4β2-nAChR, though they were less potent in the functional inactivation of the α3β4*-nAChR (Table 4, 39 and 43Figure 3). Whereas compounds have high selectivity for α4β2- over α3β4*-nAChRs (174- and 150-fold) in the binding affinity assays (Table 2), this selectivity does not translate to the functional assay (Tables 3 and 4, 31Figure 3). None of the compounds displayed significant activity at the α1β1γδ-nAChR. Compounds –38 were not potent in our preliminary functional screening.
Figure 3.

Functional activities of compounds 39, 43, and 44 at nAChR subtypes. Specific 86Rb+ efflux (ordinate; percentage of control ± SEM) was determined as described in the Experimental Section for intrinsic agonist activity over a 9.5 min period for compounds 39 (left), 43 (middle), or 44 (right) at the indicated concentrations (abscissa; log molar scale) on human α4β2-nAChR (●), α3β4*-nAChR (▲), or α1β1γδ-nAChR (◆) naturally or heterologously expressed by SH-EP1-hα4β2, SH-SY5Y, or TE671/RD cells, respectively. Also shown are functional inactivation effects of pre-treatment for 10 min with the same agents at the indicated concentrations on subsequent agonist action of an EC90 concentration of the full agonist, carbamylcholine (applied in the continuing presence of the indicated agents), acting at α4β2-nAChR (○), α3β4*-nAChR (△), or α1β1γδ-nAChR (◇). Results are normalized to responses to a fully efficacious concentration of carbamylcholine (see Experimental Section for details). Nanomolar agonist EC50 values and inactivation IC50 values are provided in Tables 3 and 4, as are agonism and inactivation efficacies (normalized to those for a full agonist or antagonist, respectively). SEM values were determined for each parameter and although not presented here, typically are less than 15% for efficacy measures and no more than a factor of 2 for molar EC50 or IC50 values. Compounds 39 and 43 have full agonist activity at α4β2-nAChRs and α3β4*-nAChRs (efficacies are similar to that of carbamylcholine) and little activity at α1*-nAChRs. Compound 44 was less potent than 39 and 43 by approximately one order of magnitude, so a full functional profile would have required 1 mM concentrations of the compound and was not pursued.
Table 3.
Potencies and efficacies of ligand agonism and inactivation of human α4β2-nAChRsa
| Compound | Agonism | Inactivation | Ki (nM) | ||
|---|---|---|---|---|---|
| EC50 (nM) | Efficacy (%) | IC50 (nM) | Efficacy (%) | α4β2 | |
| 39 | 1090 | 93 | 151 | 72 | 7.3 |
| 43 | 1180 | 108 | 169 | 78 | 4.6 |
| 44 | > 3000 | > 50 | 1100 | 69 | 129 |
| 3 | 290 | 88 | 430 | 92 | 4.9 |
| 1 | 1400 | 53 | ~ 110 | ~ 85 | 0.05 |
See Experimental Section for details. The term “inactivation” is used because compounds may be acting to desensitize receptors or as competitive or non-competitive antagonists, and further work is needed to make such a distinction. SEM values were determined for each parameter and, although not presented here, typically are less than 3% and frequently less than 1% of the maximal carbamylcholine response for efficacy measures for ligands potent enough to reach maximal efficacy at 10 μM. SEM values for EC50 and IC50 values were no more than a factor of 2. See Table 2 for structures.
For compounds that were not potent enough to cause maximal inhibition at the highest concentration tested, inactivation efficacy was fixed at 100% to allow IC50 values to be fit during graphical analysis.
Table 4.
Potencies and efficacies of ligand agonism and inactivation of α3β4*-nAChRsa
| Compound | Agonism | Inactivation | Ki (nM) | ||
|---|---|---|---|---|---|
| EC50 (nM) | Efficacy (%) | IC50 (nM) | Efficacy (%) | α3β4 | |
| 39 | 3400 | 120 | > 3000 | > 70 | 1270 |
| 43 | 1200 | 120 | > 3000 | > 70 | 692 |
| 44 | > 3000 | > 80 | > 3000 | > 30 | 8780 |
| 3 | 30000 | 90 | ND | ND | 260 |
| 1 | 2200 | 110 | ND | ND | ND |
See Experimental Section and the legend to Table 3 for details.
In addition, compounds 39 and 43 are similar in potency at the α4β2-nAChR (EC50 values of 1090 nM and 1180 nM, respectively) compared to compound 1 (EC50 value of 1400 nM). Both are inactive at the α1β1γδ-nAChR and have activity similar to compound 1 at the α3β4*-nAChR. Whereas in vitro binding and functional data are intriguing, a more proximal measure of therapeutic value is likely to be behavioral pharmacological activity in an animal model of the indication of interest. Therefore we decided to further test these compounds for their antidepressant profile in vivo.
In vivo Behavioral Studies—Mouse Forced Swim Test
Antidepressant efficacy was assessed with the mouse forced swim test, an assay in which a mouse is placed into a beaker of water, and the time it spends passively floating in the water (immobility) is recorded (Figure 4). Most traditional antidepressants decrease the amount of time the mouse spends immobile. Mice were administered compounds 43 (1 and 5 mg/kg ip) or the selective serotonin reuptake inhibitor (SSRI) antidepressant, sertraline, as a positive control (10 mg/kg). Drug administration produced a reduction in immobility. Fisher’s post hoc tests showed that compound 43 reduced immobility at both doses (1 and 5 mg/kg), suggestive of a potent antidepressant-like effect. Moreover, 43 was also active at an orally administered dose of 5 mg/kg.
Figure 4.

Compound 43 reduced immobility in the mouse forced swim test at low (1 and 5 mg/kg) doses. The SSRI, sertraline, produced the expected decrease in immobility. (ANOVAs: Fs > 6.845, ps < 0.0010. *Fisher’s PLSD post-hoc test: ps < 0.05 vs vehicle). All drugs were administered intraperitoneally (A) or orally (B); n = 9–10/group). Compound 43 reduced immobility in the forced swim test in mice at 5 mg/kg, but was inactive at 1 mg/kg when administered orally.
Broad Screening at Other Neurotransmitter Receptors and Transporters
A broad-range screening study was carried out for compound 43 to further determine its effects at 10 μM on about 45 other CNS neurotransmitter receptors and transporters, including serotonin receptors, dopamine receptors, GABA receptors, biogenic amine transporters, adrenergic receptors, muscarinic receptors, opioid receptors, sigma receptors, and histamine receptors (NIMH-PDSP, University of North Carolina, Chapel Hill). No inhibition caused by 10 μM of compound 43 was greater than 50% in the preliminary binding screen, indicating that 43 has no significant activity at these targets. (see Supporting Information)
Preliminary in vitro ADME-Tox Profile
Compound 43 showed good properties in assays for cytochrome P450 (CYP) inhibition, metabolic stability, and plasma protein binding (SRI, Stanford Research Institute), as well as hERG (human ether-ago-go-related gene) inhibition (NIMH-PDSP). (see the Supporting Infornation)
CYP Inhibition
The inhibitory effect of 43 on in vitro CYP activity in human liver microsomes was screened using a high-throughput multiple CYP assay for CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. In the presence of 1 μM and 10 μM of 43, none of these CYP isoforms’ activity was reduced to less than 70% of the control, suggesting that 43 will not alter the metabolism of other xenobiotics or endogenous compounds that are substrates for the CYP isoforms tested.
Metabolic Stability
The metabolic stability of 43 was studied using human and mouse liver microsomes. The test article was incubated at two concentrations (1 μM and 10 μM), and aliquots (100 μL) were removed at various time points (0, 15, 30, and 60 min) for analysis by LC-MS/MS. This study showed that liver microsomes from both species caused a time-dependent decrease in parent drug at both concentrations. Human liver microsomes metabolized 43 to a greater extent in 60 min than mouse liver microsomes; 53.4% and 67.2% of the parent drug remained unchanged at 1 μM and 10 μM, respectively. Incubation with mouse liver microsomes resulted in 67.5% and 73.7% of 43 remaining unchanged after 60 min incubation at 1μM and 10μM, respectively.
PPB (Plasma Protein Binding)
The binding of 43 to proteins in human and mouse plasma was determined using equilibrium dialysis. Binding of 43 was evaluated at concentrations of 0, 0.1, 1, and 10μM. The mean percentage of binding of this compound to human plasma ranged from 8.2% to 17.7%. Its mean percentage of binding to mouse plasma ranged from 12.9% to 19.3%.
hERG Inhibition
HEK293 cells stably expressing recombinant human hERG were used in a fluorescence-based membrane potential assay. The observed hERG inhibition of 43 at 10 μM was similar to that of the negative control, giving 0% hERG blockade.
Conclusions
In the present study, a new series of isoxazole ether nAChR ligands have been identified, and their preliminary SAR has been explored. In the PDSP binding study, most of the 5-alkoxyisoxazole ligands were found to bind to the rat α4β2-nAChR with a significantly higher affinity than to the α3β4*-nAChR. Compound 43 was identified as the lead compound from this series as it displayed favorable in vitro nAChR binding affinities. The functional potency of isoxazle 43 at the α4β2-nAChR is similar to that of compound 1, but it has higher efficacy, helping to distinguish it from compound 1. When tested in vivo, compound 43 demonstrated a potent antidepressant-like activity2 at both 5 mg/kg and 1 mg/kg ip, and was orally active at 5 mg/kg in the mouse forced swim test. Preliminary in vitro ADME-Tox characterization further suggested promising drug-like properties of 43. The PDSP broad range screening indicated that compound 43 was highly selective for nAChRs and did not have significant binding affinity to the other 45 neurotransmitter receptors and transporters tested.
It remains unclear whether activity at the α3β4*-nAChR subtype is an attribute or a detriment for treatment of depression. Further studies are required to fully understand the importance of α3β4*-nAChR involvement in depression. Our previously reported nicotinic ligands such as sazetidine-A and 3-alkoxy-5-aminopyridine derivatives, that contain appropriate substituents in the 5-position of their pyridine rings, exhibited improved selectivity for β2- over β4-containing nAChRs.6, 52–55 Considering that the present isoxazole ligands are easily accessible through substitution reactions performed upon a pre-assembled isoxazole building block, the 5-alkoxyisoxazole scaffold should be a useful starting point for a broader optimization campaign to discover more α4β2-selective nAChR ligands, perhaps importantly with higher efficacy than compound 1, that may in due course lead to a novel treatment option for depression.
Experimental Section
General Methods
Starting materials, reagents, and solvents were purchased from commercial suppliers and used without further purification, unless otherwise stated. Anhydrous THF and CH2Cl2 were obtained by distillation over sodium wire or CaH2, respectively. All non-aqueous reactions were run under an argon atmosphere with exclusion of moisture from reagents, and all reaction vessels were oven-dried. The progress of reactions was monitored by TLC on SiO2. Spots were visualized by their quenching of the fluorescence of an indicator admixed to the SiO2 layer, or by dipping into KMnO4 solution followed by heating. SiO2 for column chromatography (CC) was of 230–400 mesh particle size, and an EtOAc/hexane mixture or gradient was used unless stated otherwise. 1H NMR spectra were recorded at a spectrometer frequency of 300 or 400 MHz, 13C NMR spectra at 75 MHz or 100 MHz. 1H chemical shifts are reported in δ (ppm) using the δ 7.26 signal of CDCl3, the δ 4.80 signal of D2O, the δ 3.31 signal of CD3OD, or the δ 2.50 signal of DMSO-d6 as internal standards. 13C chemical shifts are reported in δ (ppm) using the δ 77.23 signal of CDCl3, the δ 49.15 signal of CD3OD, or the δ 39.51 signal of DMSO-d6 as internal standards. 13C NMR spectra in D2O were not adjusted. Purities of final compounds (>98%) were established by analytical HPLC, which was carried out on an Agilent 1100 HPLC system with a Synergi 4 μ Hydro-RP 80A column, with detection at 220 or 254 nm on a variable wavelength detector G1314A; flow rate = 1.4 mL/min; gradient of 0 to 100% methanol in water (both containing 0.05 vol% of TFA) in 18 min. Final products were purified by preparative HPLC under the following conditions: column, ACE AQ, 250 × 20 mm; flow, 17 mL/min; all solvents containing 0.05 vol% TFA; UV detection at 254 nm and 220 nm; Gradient I: 25–100% MeOH in water in 30 min, 100% for 5 min, return to 25% in 4 min, and equilibration at 25% for 1 min; Gradient II: 8–100% MeOH in water in 30 min, 100% for 5 min, return to 25% in 4 min, and equilibration at 8% for 1 min; Gradient III: 0–50% MeOH in water in 20 min, to 100% in 5 min, 100% for another 5 min, return to 0% in 5 min, and equilibration at 0% for 1 min.
General Procedure for the Deprotection of N-Boc-Amines to Afford TFA Salts (Method A)
To a solution of the N-Boc protected precursor (1 mmol) in CH2Cl2 (10 mL) was added TFA (1 mL) under argon with ice cooling. The mixture was stirred overnight at rt. After the solvent was evaporated, the residue was dissolved in distilled water (5 mL). The solution was filtered over a syringe filter (polytetrafluoroethylene, 17 mm diameter, 0.45 μm pore size), then concentrated to 2–3 mL under reduced pressure at 30 °C bath temperature. The crude product was purified by preparative HPLC. After the solvent was evaporated, the residue was dissolved in distilled water (about 2–3 mL). The solution was lyophilized to obtain the TFA salt.
General Procedure for the Mitsunobu Reaction of 1-(tert-Butoxycarbonyl)-2(S)-azetidinylmethanol (15) or 1-(tert-Butoxycarbonyl)-2(S)-pyrrolidinylmethanol (25) with Hydroxyisoxazoles to Afford Alkoxyisoxazoles (Method B)
To a stirred solution of a hydroxyisoxazole (1 mmol), alcohol 15 or 25 (1.2 mmol), and PPh3 (1.5 mmol) in anhydrous THF (20 mL) was added diethyl azodicarboxylate or diisopropyl azodicarboxylate (1.5 mmol) dropwise. After stirring overnight at rt, the solvent was evaporated, and the residue was dissolved in EtOAc. The solution was washed with water (20 mL) and brine (15 mL), dried over Na2SO4, filtered, and concentrated under vacuum. The residue was purified by CC on SiO2 to give the alkoxyisoxazole.
General Procedure for the Reduction of Isoxazolecarboxylic Acid Esters to Alcohols (Method C)
To a solution of an isoxazolecarboxylic acid ester (1 mmol) in anhydrous THF (20 mL) was added LiBH4 (4 mmol) with ice cooling under Ar. After stirring overnight at rt, saturated aqueous NH4Cl solution was added with ice cooling. Extraction with EtOAc, drying over Na2SO4, and CC on SiO2 gave the alcohol.
General Procedure for the Preparation of Iodides from Alcohols (Method D)
To a stirred solution of an isoxazolylmethanol (1 mmol), imidazole (1.5 mmol), and PPh3 (1.5 mmol) in anhydrous PhMe (8 mL) was added I2 (1.5 mmol) with ice cooling under Ar. After stirring overnight at rt, the solvent was evaporated. The residue was purified by CC on SiO2 to give the iodide.
General Procedure for the Preparation of Phenyl Ethers from Iodides (Method E)
To a stirred solution of an (iodomethyl)isoxazole (1 mmol) and phenol (2 mmol) in anhydrous DMF (4 mL) was added K2CO3 (6 mmol) under Ar. After stirring overnight at rt, saturated aqueous NH4Cl solution was added. The mixture was extracted with EtOAc (2 × 15 mL), and the combined organic phases were washed with water (3 × 10 mL), dried over Na2SO4, and evaporated. The residue was purified by CC on SiO2 to give the product.
General Procedure for the Preparation of Alkyl Ethers from Alcohols (Method F)
To a stirred solution of an isoxazolylmethanol (1 mmol) in anhydrous DMF (2 mL) was added NaH (60% dispersion in oil, 1.9 mmol) with ice cooling under Ar. After stirring for 30 min at rt, alkyl halide (1 mmol) was added. Stirring was continued for 2 h at rt, then the reaction was quenched with saturated aqueous NH4Cl solution with ice cooling. The mixture was extracted with EtOAc (2 × 15 mL), and the combined organic phases were washed with water (3 × 10 mL), dried over Na2SO4, and evaporated. The residue was purified by CC on SiO2 to give the product.
General Procedure for the Preparation of Amines from Iodides (Method G)
To a solution of an (iodomethyl)isoxazole (1 mmol) in anhydrous CH3CN (10 mL) were added at rt K2CO3 (6 mmol) and amine (4 mmol). After stirring overnight, the reaction mixture was concentrated. The residue was purified by CC on SiO2 (CH2Cl2/MeOH) to obtain the product.
General Procedure for the Preparation of Carbamates from Alcohol 29 (Method H)
A solution of 29 (1 mmol), isocyanate (2 mmol), and 4-(dimethylamino)pyridine (0.1 mmol) in anhydrous toluene (5 mL) was stirred at 80 °C under Ar for 5 h. The solvent was removed under reduced pressure, and the residue was purified by CC on SiO2 (acetone/hexane) to obtain the product.
3-[[1-(tert-Butoxycarbonyl)-2(S)-azetidinyl]methoxy]-5-isoxazolylmethanol (17)
This compound was obtained from 1-(tert-butoxycarbonyl)-2(S)-azetidinylmethanol and 3-hydroxyisoxazole-5-carboxylic acid methyl ester in two steps as a colorless oil in 84% yield employing Method B and Method C. 1H NMR (400 MHz, CDCl3) δ 6.57 (s, 1H), 4.58–4.54 (m, 1H), 4.48 (m, 1H), 4.35 (d, 2H, J = 10.8 Hz), 3.92 (s, 1H), 3.84 (t, 2H, J = 7.6 Hz), 2.33–2.28 (m, 1H), 2.22–2.18 (m, 1H), 1.39 (s, 9H).
3-[(2(S)-Azetidinyl)methoxy]-5-isoxazolylmethanol (18)
This compound was obtained from 17 employing Method A and Gradient III. Colorless oil; yield 51%; purity 99.8%. [α]D20 3.1 (c = 0.42, MeOH); 1H NMR (400 MHz, D2O) δ 6.20 (s, 1H), 4.95 (m, 1H), 4.66 (s, 2H), 4.60–4.52 (m, 2H), 4.15 (dd, 1H, J = 9.2, 18.8 Hz), 4.04 (dd, 1H, J = 9.2, 18.4 Hz), 2.67 (q, 2H, J = 8.8 Hz); 13C NMR (100 MHz, D2O) δ 172.8, 171.0, 162.3 (TFA), 115.5 (TFA), 93.0, 67.6, 58.5, 54.7, 43.1, 19.8. Anal. Calcd for C8H12N2O3•1.0TFA•0.25H2O (FW 305): C, 39.68; H, 4.49; N, 9.25; F, 18.98. Found: C, 39.94; H, 4.25; N, 8.92; F, 18.98.
3-[(2(S)-Azetidinyl)methoxy]-5-(phenoxymethyl)isoxazole (19)
This compound was obtained from 17 and phenol employing Methods D, E, and A and Gradient I. Colorless solid; yields of the individual steps 93%, 89%, and 81%; purity ~100%. [α]D20 1.2 (c = 0.68, MeOH); 1H NMR (400 MHz, D2O) δ 7.02–7.00 (m, 2H), 6.70 (m, 3H), 6.01 (s, 1H), 4.75–4.70 (m, 3H), 4.34 (s, 2H), 4.02 (m, 1H), 3.90 (m, 1H), 2.52-2.43 (m, 2H); 13C NMR (100 MHz, D2O) δ 170.7, 168.8, 161.9 (TFA), 156.8, 129.1, 121.3, 115.9 (TFA), 114.2, 94.7, 67.5, 60.3, 58.1, 43.0, 19.8. Anal. Calcd for C14H16N2O3•1.1TFA•0.55H2O (FW 398): C, 49.18; H, 4.64; N, 7.08; F, 15.85. Found: C, 48.89; H, 4.27; N, 7.06; F, 15.90.
3-[(2(S)-Azetidinyl)methoxy]-5-[(hexyloxy)methyl]isoxazole (20)
This compound was obtained from 17 and MeI employing Methods F and A and Gradient I. Colorless oil; yields of the individual steps 84% and 76%; purity ~100%. [α]D20 2.8 (c = 0.14, MeOH); 1H NMR (400 MHz, D2O) δ 6.10 (s, 1H), 4.90–4.86 (m, 1H), 4.47–4.43 (m, 2H), 4.41 (s, 2H), 4.09 (m, 1H), 4.02–3.95 (m, 1H), 3.45 (t, 2H, J = 6.8 Hz), 2.64–2.52 (m, 2H), 1.53–1.48 (m, 2H), 1.28–1.23 (m, 6H), 0.83–0.80 (m, 3H); 13C NMR (100 MHz, D2O) δ 170.8, 170.4, 161.8 (TFA), 120.2, 115.8 (TFA), 94.1, 70.7, 67.5, 62.8, 58.2, 43.1, 30.9, 28.6, 24.9, 21.8, 19.9, 13.0. Anal. Calcd for C14H24N2O3•1.0TFA•0.05H2O (FW 385): C, 50.14; H, 6.60; N, 7.31; F, 14.87. Found: C, 49.93; H, 6.26; N, 7.18; F, 14.56.
3-[(2(S)-Azetidinyl)methoxy]-5-[(cyclopropylmethoxy)methyl]isoxazole (21)
This compound was obtained from 17 and (bromomethyl)cyclopropane employing Methods F and A and Gradient II. Colorless oil; yields of the individual steps 77% and 59%; purity 99.8%. [α]D20 −0.1 (c = 2.8, MeOH); 1H NMR (400 MHz, D2O) δ 6.25 (s, 1H), 4.93 (m, 1H), 4.60 (s, 2H), 4.57–4.51 (m, 2H), 4.12 (m, 1H), 4.05 (m, 1H), 3.40 (d, 2H, J = 7.2 Hz), 2.69–2.62 (m, 2H), 1.04 (m, 1H), 0.56–0.52 (m, 2H), 0.22–0.18 (m, 2H); 13C NMR (100 MHz, D2O) δ 170.9, 170.2, 162.2 (TFA), 115.9 (TFA), 94.6, 75.7, 67.6, 62.2, 58.4, 43.2, 19.9, 9.2, 2.1. Anal. Calcd for C12H18N2O3•1.1TFA•0.65H2O (FW 378): C, 45.43; H, 5.48; N, 7.46; F, 16.70. Found: C, 45.20; H, 5.19; N, 7.31; F, 16.84.
3-[[1-(tert-Butoxycarbonyl)-2(S)-azetidinyl]methoxy]-5-methylisoxazole (23)
This compound was obtained from 1-(tert-butoxycarbonyl)-2(S)-azetidinylmethanol and 3-hydroxy-5-methylisoxazole as a pale-yellow solid in 67% yield employing Method B. 1H NMR (400 MHz, CDCl3) δ 5.63 (s, 1H), 4.48–4.42 (m, 2H), 4.29–4.26 (m, 2H), 3.83 (t, 2H, J = 9.4 Hz), 2.31–2.16 (m, 5H), 1.39 (s, 9H); 13C NMR (100 MHz, D2O) δ 172.3, 170.6, 156.3, 93.1, 79.8, 69.9, 60.2, 47.0, 28.5, 21.9, 19.0, 13.0.
3-[(2(S)-Azetidinyl)methoxy]-5-methylisoxazole (24)
This compound was obtained employing Method A and Gradient III. Colorless oil; yield 58%; purity 99.7%. [α]D20 −0.2 (c = 2.4, MeOH); 1H NMR (400 MHz, D2O) δ 5.90 (s, 1H), 4.98 (m, 1H), 4.46 (m, 1H), 4.12 (m, 1H), 4.00 (m, 1H), 2.66–2.58 (m, 2H), 2.30 (s, 3H); 13C NMR (100 MHz, D2O) δ 172.3, 171.2, 162.1 (TFA), 115.9 (TFA), 92.4, 67.3, 58.5, 43.2, 19.8, 11.6. Anal. Calcd for C8H12N2O2•1.15TFA•0.55H2O (FW 312): C, 40.01; H, 4.64; N, 9.06; F, 21.20. Found: C, 39.88; H, 4.43; N, 9.11; F, 21.41.
5-[(1-(tert-Butoxycarbonyl)-2(S)-azetidinyl)methoxy]isoxazole-3-carboxylic Acid Ethyl Ester (27)
This compound was obtained from 1-(tert-butoxycarbonyl)-2(S)-azetidinylmethanol and 5-hydroxyisoxazole-3-carboxylic acid ethyl ester (Princeton Bio) as a pale-yellow solid in 55% yield employing Method B. 1H NMR (400 MHz, CDCl3) δ 5.71 (s, 1H), 4.52 (m, 2H), 4.41 (q, 2H, J = 7.2 Hz), 4.31 (dd, 1H, J = 2.4, 10.0 Hz), 3.89-3.84 (m, 2H), 1.41 (s, 9H), 1.25 (t, 3H, J = 7.2 Hz).
5-[(1-(tert-Butoxycarbonyl)-2(S)-pyrrolidinyl)methoxy]isoxazole-3-carboxylic Acid Ethyl Ester (28)
This compound was obtained from 1-(tert-butoxycarbonyl)-2(S)-pyrrolidinylmethanol and 5-hydroxyisoxazole-3-carboxylic acid ethyl ester as a pale-yellow solid in 65% yield employing Method B. 1H NMR (400 MHz, CDCl3; two rotamers about the N-Boc bond in an approximate ratio of 1:1) δ 5.66 (br, 1H), 4.35 (q, 2H, J = 7.2 Hz), 4.28 (br, 1.5H), 4.07 (br, 1.5H), 3.29 (br, 2H), 2.01–1.83 (m, 4H), 1.41 (s, 9H), 1.34 (t, 3H, J = 7.2 Hz).
5-[(1-(tert-Butoxycarbonyl)-2(S)-pyrrolidinyl)methoxy]-3-isoxazolylmethanol (30)
This compound was obtained from 5-[[1-(tert-butoxycarbonyl)-2(S)-pyrrolidinyl]methoxy]isoxazole-3-carboxylic acid ethyl ester as a pale-yellow solid in 94% yield employing Method C. 1H NMR (400 MHz, CDCl3; two rotamers about the N-Boc bond in an approximate ratio of 1:1) δ 5.28 (br, 0.5H), 5.22 (br, 0.5H), 4.43 (d, 2H, J = 5.6 Hz), 4.25–3.97 (m, 4H), 3.22 (br, 2H), 1.90–1.75 (m, 4H), 1.32 (s, 9H).
5-[(2(S)-Azetidinyl)methoxy]-3-isoxazolylmethanol (31)
5-[[1-(tert-Butoxycarbonyl)-2(S)-azetidinyl]methoxy]-3-isoxazolylmethanol (29) was obtained from 27 as a pale-yellow solid in quantitative yield employing Method C. The title compound 31 was obtained from 29 employing Method A and Gradient III. Colorless oil; yield 67%; purity 100%. [α]D20 −3.0 (c = 1.2, MeOH); 1H NMR (400 MHz, D2O) δ 5.52 (s, 1H), 4.85 (m, 1H), 4.53–4.45 (m, 4H), 4.06 (m, 1H), 3.95 (m, 1H), 2.60–2.53 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.3, 166.1, 162.3 (TFA), 115.9 (TFA), 77.4, 69.8, 58.1, 55.5, 43.2, 19.8. Anal. Calcd for C8H12N2O3•1.4TFA (FW 344): C, 37.73; H, 3.93; N, 8.15; F, 23.21. Found: C, 37.82; H, 4.10; N, 8.47; F, 23.26.
5-[(2(S)-Azetidinyl)methoxy]-3-(phenoxymethyl)isoxazole (32)
The title compound was obtained from 29 employing Method A and Gradient II. Colorless solid; yield 38%; purity 98.9%. [α]D20 −1.0 (c = 1.1, MeOH); 1H NMR (400 MHz, CD3OD) δ 7.30–7.26 (m, 2H), 7.00–6.94 (m, 3H), 5.69 (s, 2H), 4.90 (m, 1H), 4.59 (m, 1H), 4.52 (m, 1H), 4.09 (m, 1H), 4.00 (m, 1H), 2.62 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 172.6, 163.0, 160.1 (TFA), 157.7, 128.8, 120.8, 115.9 (TFA), 114.1, 77.4, 69.9, 61.3, 58.0, 42.8, 19.9. Anal. Calcd for C14H16N2O3•1.55TFA•0.45H2O (FW 445): C, 46.14; H, 4.18; N, 6.29; F, 19.85. Found: C, 45.98; H, 4.04; N, 6.39; F, 20.03.
3-(Phenoxymethyl)-5-[(2(S)-pyrrolidinyl)methoxy]isoxazole (33)
This compound was obtained employing Method A and Gradient II. Colorless solid; yield 75%; purity 100%. [α]D20 −1.7 (c = 1.5, MeOH); 1H NMR (400 MHz, CD3OD) δ 7.30–7.26 (m, 2H), 7.00–6.94 (m, 3H), 5.69 (s, 2H), 5.04 (s, 2H), 4.62 (m, 1H), 4.40 (m, 1H), 4.06 (m, 1H), 3.36 (t, 2H, J = 7.2 Hz), 2.26 (m, 1H), 2.13–2.05 (m, 2H), 1.87 (m, 1H); 13C NMR (100 MHz, CD3OD) δ 172.3, 162.8, 160.0 (TFA), 157.5, 128.6, 120.6, 115.7 (TFA), 113.8, 77.1, 69.8, 61.1, 57.7, 45.0, 25.1, 22.7. Anal. Calcd for C15H18N2O3•1.2TFA•0.35H2O (FW 417): C, 50.06; H, 4.80; N, 6.71; F, 16.38. Found: C, 50.22; H, 4.73; N, 6.67; F, 16.14.
3-(Anilinomethyl)-5-[(2(S)-azetidinyl)methoxy]isoxazole (34)
The title compound was obtained from 29 employing Methods D, G, and A and Gradient III. Pale-yellow oil; yields of the individual steps 76%, quantitative and 35%; purity 99.9%. [α]D20 −2.7 (c = 0.79, MeOH); 1H NMR (400 MHz, D2O) δ 7.52 (t, 2H, J = 7.6 Hz), 7.44 (t, 1H, J = 7.4 Hz), 7.35 (d, 2H), 5.61 (s, 1H), 4.94–4.92 (m, 1H), 4.63 (s, 1H), 4.58–4.53 (m, 2H), 4.17–4.10 (m, 1H), 4.06–3.99 (m, 1H), 2.70–2.60 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.8, 162.5 (TFA), 159.0, 135.7, 129.8, 127.8, 120.7, 115.9 (TFA), 78.8, 69.9, 58.0, 44.6, 43.2, 19.7. Anal. Calcd for C14H17N3O2•1.8TFA•1.25 H2O (FW 487): C, 43.40; H, 4.41; N, 8.63; F, 21.06. Found: C, 43.34; H, 4.27; N, 8.76; F, 21.09.
5-[(2(S)-Azetidinyl)methoxy]-3-[(4-fluoroanilino)methyl]isoxazole (35)
This compound was obtained from 29 employing Methods D, G, and A and Gradient III. Pale-yellow oil; yields of the individual steps 76%, quantitative, and 74%; purity 99.5%. [α]D20 −2.0 (c = 1.4, MeOH); 1H NMR (400 MHz, D2O) δ 7.13–7.08 (m, 4H), 5.58 (s, 1H), 4.92 (m, 1H), 4.63 (s, 1H), 4.58–4.54 (m, 2H), 4.44 (s, 2H), 4.13 (m, 1H), 4.02 (m, 1H), 2.68–2.59 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.5, 162.2, 162.2 (TFA), 158.7 (d, JC-F = 20.3 Hz), 137.5, 119.1 (d, JC-F = 8.3 Hz), 115.9 (TFA), 115.9 (d, JC-F = 238.4 Hz), 78.3, 69.8, 58.0, 43.2, 42.5, 19.7. Anal. Calcd for C14H16FN3O2•1.4TFA•0.55H2O (FW 447): C, 45.16; H, 4.17; N, 9.40; F, 22.11. Found: C, 45.17; H, 4.13; N, 9.35; F, 22.09.
5-[(2(S)-Azetidinyl)methoxy]-3-[(N-phenylcarbamoyloxy)methyl]isoxazole (36)
This compound was obtained from 29 and phenyl isocyanate employing Methods H and A and Gradient II. Colorless oil; yields of the individual steps 93% and 40%; purity 99.8%. [α]D20 −3.7 (c = 3.4, MeOH); 1H NMR (400 MHz, D2O) δ 7.22 (d, 2H, J = 8.0 Hz), 7.09 (t, 2H, J = 8.0 Hz), 6.87 (t, 2H, J = 7.4 Hz), 5.39 (s, 1H), 4.93 (s, 2H), 4.74 (m, 1H), 4.31–4.25 (m, 2H), 4.01 (m, 1H), 3.90–3.85 (m, 1H), 2.55-2.36 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.2, 162.2, 162.0 (TFA), 153.8, 137.0, 128.5, 123.4, 118.8, 115.9 (TFA), 77.7, 69.7, 58.0, 57.8, 43.1, 20.0. Anal. Calcd for C15H17N3O4•1.55TFA•2.35 H2O (FW 552): C, 41.62; H, 4.49; N, 8.04; F, 16.91. Found: C, 41.51; H, 4.46; N, 8.19; F, 16.94.
5-[(2(S)-Azetidinyl)methoxy]-3-[(N-ethylcarbamoyloxy)methyl]isoxazole (37)
This compound was obtained from 29 and ethyl isocyanate employing Methods H and A and Gradient III. Colorless oil; yields of the individual steps 99% and 50%; purity 99.6%. [α]D20 −3.0 (c = 2.8, MeOH); 1H NMR (400 MHz, D2O) δ 5.62 (s, 1H), 5.04 (s, 2H), 4.96 (m, 1H), 4.63–4.55 (m, 2H), 4.15 (m, 1H), 4.04 (m, 1H), 3.13 (q, J = 7.2 Hz, 2H), 2.72–2.62 (m, 2H), 1.08 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, D2O) δ 172.4, 163.1, 162.2 (TFA), 157.0, 115.9 (TFA), 77.7, 69.9, 58.1, 58.0, 43.2, 35.2, 19.8, 13.6. Anal. Calcd for C11H17N3O4•1.4TFA•0.95H2O (FW 432): C, 38.37; H, 4.74; N, 9.73; F, 18.47. Found: C, 38.13; H, 4.86; N, 9.97; F, 18.63.
5-[(2(S)-Azetidinyl)methoxy]-3-[(N-cyclopentylcarbamoyloxy)methyl]isoxazole (38)
This compound was obtained from 29 and cyclopentyl isocyanate employing Methods H and A and Gradient III. Colorless oil; yields of the individual steps 94% and 40%; purity 99.6%. [α]D20 −2.2 (c = 1.7, MeOH); 1H NMR (400 MHz, D2O) δ 5.63 (s, 1H), 5.07 (s, 2H), 4.96 (m, 1H), 4.65–4.57 (m, 2H), 4.13 (m, 1H), 4.06 (m, 1H), 3.87 (m 1H), 2.70–2.64 (m, 2H), 1.91–1.85 (m, 2H), 1.66–1.57 (m, 4H), 1.49–1.43 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.4, 163.1, 162.4 (TFA), 156.7, 115.7 (TFA), 77.7, 69.8, 58.0, 57.9, 52.3, 43.2, 31.8, 22.7, 19.7. Anal. Calcd for C14H21N3O4•1.7TFA•1.65H2O (FW 522): C, 40.28; H, 5.05; N, 8.10; F, 18.67. Found: C, 40.09; H, 4.81; N, 8.28; F, 18.81.
5-[(2(S)-Azetidinyl)methoxy]-3-(fluoromethyl)isoxazole Trifluoroacetate (39)
To a solution of Et2NSF3 (0.22 mL, 1.7 mmol) in anhydrous CH2Cl2 (10 mL) was added at −78 °C 5-[[1-(tert-butoxycarbonyl)-2(S)-azetidinyl]methoxy]-3-isoxazolylmethanol (425 mg, 1.5 mmol) in CH2Cl2 (5 mL) under Ar. The solution was stirred for 2 h at −78 °C, then allowed to warm to rt. After quenching the reaction with saturated aqueous NaHCO3 solution, the phases were separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL), and the combined organic phases were washed with water (5 mL), dried over Na2SO4, and concentrated. The residue was subjected to CC on SiO2 to obtain 5-[(1-(tert-butoxycarbonyl)-2(S)-azetidinyl)methoxy]-3-(fluoromethyl)isoxazole (70 mg, 16%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.39 (s, 1H), 5.32 (s, 1H), 5.21 (s, 1H), 4.48–4.44 (m, 2H), 4.26 (m, 1H), 3.87–3.78 (m, 2H), 2.35–2.22 (m, 2H), 1.39 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 174.2, 161.9 (d, JC-F = 22.4 Hz), 156.1, 80.1, 77.2, 76.6 (d, JC-F = 165.9 Hz), 72.6, 59.1, 47.3, 28.4, 18.7.
This title compound was obtained employing Method A and Gradient III. Colorless oil; yield 46%; purity 99.4%. [α]D20 −2.8 (c = 1.9, MeOH); 1H NMR (400 MHz, D2O) δ 5.74 (s, 1H), 5.48 (s, 1H), 5.36 (s, 1H), 4.97 (m, 1H), 4.66–4.58 (m, 2H), 4.15 (m, 1H), 4.06 (m, 1H), 2.72–2.64 (m, 2H); 13C NMR (100 MHz, D2O) δ 172.6, 162.3 (TFA), 162.3 (d, JC-F = 20.3 Hz), 115.9 (TFA), 77.8, 76.2 (d, JC-F = 162.3 Hz), 70.0, 58.1, 43.2, 19.8; 19F NMR (376 MHz, D2O) δ −76, −222. Anal. Calcd for C8H11FN2O2•1.05TFA•0.85 H2O (FW 323): C, 37.77; H, 4.31; N, 8.72; F, 24.54. Found: C, 37.50; H, 4.03; N, 8.65; F, 24.32.
5-[[1-(tert-Butoxycarbonyl)-2(S)-azetidinyl]methoxy]-3-methylisoxazole (41)
This compound was obtained from 15 and 3-methylisoxazol-5(4H)-one morpholine salt (40; Fluka) as a pale-yellow solid in 67% crude yield employing Method B. 1H NMR (400 MHz, CDCl3) δ 5.07 (s, 1H), 4.45–4.39 (m, 2H), 4.18 (dd, 2H, J = 10.0, 1.5 Hz), 3.78 (t, 2H, J = 8.4 Hz), 2.30–2.21 (m, 2H), 2.13 (s, 3H), 1.36 (s, 9H).
5-[(2(S)-Azetidinyl)methoxy]-3-methylisoxazole (43)
This compound was obtained from 41 employing Method A and Gradient III. Colorless oil; yield 38%; purity 100%. [α]D20 −1.9 (c = 0.47, MeOH); 1H NMR (400 MHz, D2O) δ 5.46 (s, 1H), 4.56 (m, 1H), 4.39 (m, 1H), 4.10 (m, 1H), 3.40 (t, 2H, J = 7.2 Hz), 2.27 (m, 1H), 2.20 (s, 3H), 2.16–2.06 (m, 2H), 1.90 (m, 1H); 13C NMR (100 MHz, D2O) δ 171.9, 164.0, 162.3 (TFA), 115.9 (TFA), 79.2, 69.6, 58.1, 43.1, 19.8, 10.9. Anal. Calcd for C8H12N2O2•1.1TFA•0.25H2O (FW 300): C, 41.09; H, 4.60; N, 9.40; F, 21.03. Found: C, 40.94; H, 4.40; N, 9.23; F, 21.02.
3-Methyl-5-[(2(S)-pyrrolidinyl)methoxy]isoxazole (44)
The title compound was obtained from 25 and 40 employing Method B and A and Gradient III. Colorless oil; yields of the individual steps 58% and 38%; purity 99.5%. [α]D20 6.7 (c = 2.6, MeOH); 1H NMR (400 MHz, D2O) δ 5.46 (s, 1H), 4.56 (m, 1H), 4.39 (m, 1H), 4.10 (m, 1H), 3.40 (t, 2H, J = 7.2 Hz), 2.27 (m, 1H), 2.20 (s, 3H), 2.16–2.06 (m, 2H), 1.90 (m, 1H); 13C NMR (100 MHz, D2O) δ 171.9, 164.0, 162.3 (TFA), 115.9 (TFA), 79.2, 69.7, 57.9, 45.5, 25.2, 22.9, 10.9. Anal. Calcd for C9H14N2O2•1.1TFA•0.8H2O (FW 324): C, 41.77; H, 5.23; N, 8.70; F, 19.47. Found: C, 41.57, H, 4.85; N, 8.58; F, 19.13.
In vitro binding studies
[3H]Epibatidine competition studies and broad-range screening were carried out by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). For experimental details please refer to the PDSP web site http://pdsp.med.unc.edu/
Cell lines and culture
Cell lines naturally or heterologously expressing specific, functional, human nAChR subtypes were used. The human clonal cell line TE671/RD naturally expresses human muscle-type α1*-nAChRs, containing α1, β1, γ, and δ subunits, with function detectable using 86Rb+ efflux assays.56 The human neuroblastoma cell line SH-SY5Y naturally expresses autonomic α3β4*-nAChRs, containing α3, β4, probably α5, and sometimes β2 subunits, and also displays function detectable using 86Rb+ efflux assays.57 SH-SY5Y cells also express homopentameric α7-nAChRs, however, their function is not detected in the 86Rb+ efflux assay under the conditions used. SH-EP1 human epithelial cells stably transfected with human α4 and β2 subunits (SH-EP1-hα4β2 cells) have been established and characterized with both ion flux and radioligand binding assays.58
TE671/RD, SH-SY5Y, and transfected SH-EP1 cell lines were maintained as low passage number (1–26 from our frozen stocks) cultures to ensure stable expression of native or heterologously expressed nAChRs as previously described.56 Cells were passaged once a week by splitting just-confluent cultures 1/300 (TE671/RD), 1/10 (SH-SY5Y), or 1/40 (transfected SH-EP1) in serum-supplemented medium to maintain log-phase growth.
86Rb+ efflux assays
Function of nAChR subtypes was investigated using an established 86Rb+ efflux assay protocol.56 The assay is specific for nAChR function under the conditions used, for example, giving identical results in the presence of 100 nM atropine to exclude possible contributions of muscarinic acetylcholine receptors. Cells harvested at confluence from 100 mm plates under a stream of fresh medium only (SH-SY5Y cells) or after mild trypsinization (Irvine Scientific, USA; for TE671/RD or transfected SH-EP1 cells) were then suspended in complete medium and evenly seeded at a density of 1.25–2 confluent 100 mm plates per 24-well plate (Falcon; ~100–125 mg of total cell protein per well in a 500 μL volume; poly-l-lysine-coated for SH-SY5Y cells). After cells had adhered generally overnight, but no sooner than 4 h later, the medium was removed and replaced with 250 μL per well of complete medium supplemented with ~350000 cpm of 86Rb+ (NEN; counted at 40% efficiency using Cerenkov counting and the Packard TriCarb 1900 Liquid Scintillation Analyzer). After at least 4 h and typically overnight, 86Rb+ efflux was measured using the “flip-plate” technique. Briefly, after aspiration of the bulk of 86Rb+ loading medium from each well of the “cell plate,” each well containing cells was rinsed with 2 mL of fresh 86Rb+ efflux buffer (130 mM NaCl, 5.4 mM KCl, 2 mM CaCl2, 5 mM glucose, and 50 mM HEPES; pH 7.4) to remove extracellular 86Rb+. Following removal of residual rinse buffer by aspiration, the flip-plate technique was used again to simultaneously introduce 1.5 mL of fresh efflux buffer containing drugs of choice at indicated final concentrations from a 24-well “efflux/drug plate” into the wells of the cell plate. After a 9.5 min incubation, the solution was “flipped” back into the efflux/drug plate, and any remaining buffer in the cell plate was removed by aspiration. 10 min after the initiation of the first drug treatment, a second efflux/drug plate was used to reintroduce the same concentrations of drugs of choice with the addition of an ~EC90 concentration of the full agonist carbamylcholine for 5 min (~EC90 concentrations were 200 μM for SH-EP1-hα4β2 cells, 2 mM for SHSY5Y cells, and 464 mM for TE671/RD cells). The second drug treatment was then flipped back into its drug plate, and the remaining cells in the cell plate were lysed and suspended by addition of 1.5 mL of 0.1 M NaOH with 0.1% sodium dodecyl sulfate to each well. Suspensions in each well were then subjected to Cerenkov counting (Wallac Micobeta Trilux 1450; 25% efficiency) after placement of inserts (Wallac 1450–109) into each well to minimize cross-talk between wells.
For quality control and normalization purposes, the sum of 86Rb+ in cell plates and efflux/drug plates was defined to confirm material balance (i.e., that the sum of 86Rb+ released into the efflux/drug plates and 86Rb+ remaining in the cell plate were the same for each well). Similarly, the sum of 86Rb+ in cell plates and efflux/drug plates also determined the efficiency of 86Rb+ loading (the percentage of applied 86Rb+ actually loaded into cells). Furthermore, the sum of 86Rb+ in cell plates and the second efflux/drug plates defined the amount of intracellular 86Rb+ available at the start of the second, 5 min assay and were used to normalize nAChR function assessed.
For each experiment, in one set of control samples, total 86Rb+ efflux was assessed in the presence of a fully efficacious concentration of carbamylcholine alone (1mM for SH-EP1-hα4β2 and TE671/RD cells, or 3mM for SH-SY5Y cells). Nonspecific 86Rb+ efflux in another set of control samples was measured either in the presence of the fully efficacious concentration of carbamylcholine plus 100μM mecamylamine, which gave full block of agonist-induced and spontaneous nAChR-mediated ion flux, or in the presence of efflux buffer alone. Both determinations of nonspecific efflux were equivalent. Specific efflux was then taken as the difference in control samples between total and nonspecific 86Rb+-efflux. The same approaches were used to define total, nonspecific, and specific ion flux responses in samples subjected to the second, 5 min, exposure to test drug with or without carbamylcholine at its ~EC90 concentration.
Intrinsic agonist activity of test drugs was ascertained during the first 9.5 min of the initial 10 min exposure period using samples containing test drug only at different concentrations and was normalized, after subtraction of nonspecific efflux, to specific efflux in carbamylcholin control samples. Specific 86Rb+ efflux elicited by test drug as a percentage of specific efflux in carbamylcholine controls was the same in these samples whether measured in absolute terms or as a percentage of loaded 86Rb+. Even in samples previously giving an efflux response during the initial 10 min exposure to a partial or full agonist, residual intracellular 86Rb+ was adequate to allow assessment of nAChR function in the secondary, 5 min assay. However, care was needed to ensure that data were normalized to the amount of intracellular 86Rb+ available at the time of the assay, as absolute levels of total, nonspecific, or specific efflux varied in cells partially depleted of intracellular 86Rb+ due to action of any agonist present during the 10 min drug exposure period. That is, calculations of specific efflux as a percentage of loaded 86Rb+ typically were corrected for any variation in the electrochemical gradient of 86Rb+ created by intracellular ion depletion after the first (agonism/pretreatment) drug treatment.
Ion flux assays (n ≥ 3 separate studies for each drug and cell line combination) were fit to the Hill equation, F = Fmax/(1+(X/EC50)n), where F is the percentage of control, Fmax, for EC50 (n > 0 for agonists) or IC50 (n < 0 for antagonists) values using Prism 4 (GraphPad, San Diego, USA). Most ion flux data were fit allowing maximum and minimum ion flux values to be determined by curve fitting but in some cases, where antagonists or agonists had weak functional potency, minimum ion flux was set at 0% of control or maximum ion flux was set at 100% of control, respectively.
General Procedures for Behavioral Studies
Animals
BALB/cJ male mice (8–10 weeks old at testing) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed four to a cage in a colony room maintained at 22±2 °C on a 12 h light–dark cycle. All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the PsychoGenics Animal Care and Use Committee.
Drugs
Compounds 43 were synthesized as described above, and sertraline was purchased from Toronto Research Chemicals (Ontario, Canada). All compounds were dissolved in injectable water and administered by intraperitoneal (IP) injection or oral gavage (PO) in a volume of 10 mL/kg.
Mouse Forced Swim Test
Procedures were based on those previously described.25 Mice were individually placed into clear glass cylinders (15 cm tall × 10 cm wide, 1 L beakers) containing 23±1 °C water 12 cm deep (approximately 800 mL). Mice were administered vehicle, the SSRI sertraline (10 or 20 mg/kg) as a positive control, or compound 43 (1 or 5 mg/kg). Thirty minutes following IP or PO administration, mice were placed in the water, and the time the animal spent immobile was recorded over a 6 min trial. Immobility was defined as the postural position of floating in the water.
Statistical Analysis
Data were analyzed with Analysis of Variance (ANOVA) with treatment group (vehicle, sertraline, or compound 43 (1 and 5 mg/kg)) as the between group variable and total time immobile (seconds over the 6 min trial) as the dependent variable. Significant main effects were followed up with the post hoc Fisher’s test.
Supplementary Material
Acknowledgments
This research was supported by Award Number U19MH085193 from the National Institute of Mental Health. The Phoenix research component was also supported in part by the Barrow Neurological Foundation and was conducted in part in the Charlotte and Harold Simensky Neurochemistry of Alzheimer’s Disease Laboratory. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health. We thank the PDSP program for performing binding affinity assays, and SRI for performing ADME studies.
Abbreviations
- CNS
central nervous system
- AD
Alzheimer’s disease
- ADHD
attention deficit hyperactivity disorder
- NIMH-PDSP
National Institute of Mental Health Psychoactive Drug Screening Program
- nAChR(s)
nicotinic acetylcholine receptor(s)
- SAR
structure-activity relationship
- ADME-Tox
absorption, distribution, metabolism, excretion, and toxicity
- SSRI
selective serotonin reuptake inhibitor
- CYP
cytochrome P450
- PPB
plasma protein binding
- hERG
human ether-a-go-go-related gene
- CC
column chromatography
- rt
room temperature
- TFA
trifluoroacetic acid
Footnotes
Supporting Information. Broad screening data and detailed preliminary in vitro ADME-Tox profile. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 2.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 3.Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 4.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–4745. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 5.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Romanelli MN, Gratteri P, Guandalini L, Martini E, Bonaccini C, Gualtieri F. Central nicotinic receptors: structure, function, ligands, and therapeutic potential. ChemMedChem. 2007;2:746–767. doi: 10.1002/cmdc.200600207. [DOI] [PubMed] [Google Scholar]
- 7.Taly A, Corringer PJ, Guedin D, Lestage P, Changeux JP. Nicotinic receptors: allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov. 2009;8:733–750. doi: 10.1038/nrd2927. [DOI] [PubMed] [Google Scholar]
- 8.Arneric SP, Holladay M, Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem Pharmacol. 2007;74:1092–1101. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 9.Mihalak KB, Carroll FI, Luetje CW. Varenicline is a partial agonist at alpha4beta2 and a full agonist at alpha7 neuronal nicotinic receptors. Mol Pharmacol. 2006;70:801–805. doi: 10.1124/mol.106.025130. [DOI] [PubMed] [Google Scholar]
- 10.Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD, 3rd, O’Neill BT. Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- 11.Philip NS, Carpenter LL, Tyrka AR, Price LH. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl) 2010;212:1–12. doi: 10.1007/s00213-010-1932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mineur YS, Picciotto MR. Nicotine receptors and depression: revisiting and revising the cholinergic hypothesis. Trends Pharmacol Sci. 2010;31:580–586. doi: 10.1016/j.tips.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR. Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry. 2002;7:525–535. doi: 10.1038/sj.mp.4001035. [DOI] [PubMed] [Google Scholar]
- 14.Caldarone BJ, Wang D, Paterson NE, Manzano M, Fedolak A, Cavino K, Kwan M, Hanania T, Chellappan SK, Kozikowski AP, Olivier B, Picciotto MR, Ghavami A. Dissociation between duration of action in the forced swim test in mice and nicotinic acetylcholine receptor occupancy with sazetidine, varenicline, and 5-I-A85380. Psychopharmacology (Berl) 2011;217:199–210. doi: 10.1007/s00213-011-2271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janowsky DS, el-Yousef MK, Davis JM, Sekerke HJ. A cholinergic-adrenergic hypothesis of mania and depression. Lancet. 1972;2:632–635. doi: 10.1016/s0140-6736(72)93021-8. [DOI] [PubMed] [Google Scholar]
- 16.George TP, Sacco KA, Vessicchio JC, Weinberger AH, Shytle RD. Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J Clin Psychopharmacol. 2008;28:340–344. doi: 10.1097/JCP.0b013e318172b49e. [DOI] [PubMed] [Google Scholar]
- 17.Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67:432–438. doi: 10.1016/j.biopsych.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overstreet DH. Selective breeding for increased cholinergic function: development of a new animal model of depression. Biol Psychiatry. 1986;21:49–58. doi: 10.1016/0006-3223(86)90007-7. [DOI] [PubMed] [Google Scholar]
- 20.Pucilowski O, Overstreet DH, Rezvani AH, Janowsky DS. Chronic mild stress-induced anhedonia: greater effect in a genetic rat model of depression. Physiol Behav. 1993;54:1215–1220. doi: 10.1016/0031-9384(93)90351-f. [DOI] [PubMed] [Google Scholar]
- 21.Caldarone BJ, Harrist A, Cleary MA, Beech RD, King SL, Picciotto MR. High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol Psychiatry. 2004;56:657–664. doi: 10.1016/j.biopsych.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Mineur YS, Somenzi O, Picciotto MR. Cytisine, a partial agonist of high-affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice. Neuropharmacology. 2007;52:1256–1262. doi: 10.1016/j.neuropharm.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buckley MJ, Surowy C, Meyer M, Curzon P. Mechanism of action of A-85380 in an animal model of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:723–730. doi: 10.1016/j.pnpbp.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 24.Rollema H, Guanowsky V, Mineur YS, Shrikhande A, Coe JW, Seymour PA, Picciotto MR. Varenicline has antidepressant-like activity in the forced swim test and augments sertraline’s effect. Eur J Pharmacol. 2009;605:114–116. doi: 10.1016/j.ejphar.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lippiello PM, Beaver JS, Gatto GJ, James JW, Jordan KG, Traina VM, Xie J, Bencherif M. TC-5214 (S-(+)-mecamylamine): a neuronal nicotinic receptor modulator with antidepressant activity. CNS Neurosci Ther. 2008;14:266–277. doi: 10.1111/j.1755-5949.2008.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong CL, Chiu YT, Lin NN, Cheng CC, Lin SZ, Lee TJ, Kuo JS. Regulation of the common carotid arterial blood flow by nicotinic receptors in the medulla of cats. Br J Pharmacol. 2006;149:206–214. doi: 10.1038/sj.bjp.0706844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shytle RD, Silver AA, Sheehan KH, Sheehan DV, Sanberg PR. Neuronal nicotinic receptor inhibition for treating mood disorders: preliminary controlled evidence with mecamylamine. Depress Anxiety. 2002;16:89–92. doi: 10.1002/da.10035. [DOI] [PubMed] [Google Scholar]
- 28.Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. J Pharmacol Exp Ther. 2001;297:646–656. [PubMed] [Google Scholar]
- 29.Elliott RL, Ryther KB, Anderson DJ, Raszkiewicz JL, Campbell JE, Sullivan JP, Garvey DS. Phenyl pyrrolidine analogues as potent nicotinic acetylcholine receptor (nAChR) ligands. Bioorganic & Medicinal Chemistry Letters. 1995;5:991–996. [Google Scholar]
- 30.Guandalini L, Martini E, Dei S, Manetti D, Scapecchi S, Teodori E, Romanelli MN, Varani K, Greco G, Spadola L, Novellino E. Design of novel nicotinic ligands through 3D database searching. Bioorganic & Medicinal Chemistry. 2005;13:799–807. doi: 10.1016/j.bmc.2004.10.039. [DOI] [PubMed] [Google Scholar]
- 31.Elliott RL, Ryther KB, Anderson DJ, Piattoni-Kaplan M, Kuntzweiler TA, Donnelly-Roberts D, Arneric SP, Holladay MW. Novel 2-(2′-furo[3,2-b]pyridinyl) pyrrolidines: potent neuronal nicotinic acetylcholine receptor ligands. Bioorganic & Medicinal Chemistry Letters. 1997;7:2703–2708. [Google Scholar]
- 32.Efange SM, Tu Z, von Hohenberg K, Francesconi L, Howell RC, Rampersad MV, Todaro LJ, Papke RL, Kung MP. 2-(2-Piperidyl)- and 2-(2-pyrrolidyl)chromans as nicotine agonists: synthesis and preliminary pharmacological characterization. J Med Chem. 2001;44:4704–4715. doi: 10.1021/jm010129z. [DOI] [PubMed] [Google Scholar]
- 33.Garvey DS, Wasicak JT, Decker MW, Brioni JD, Buckley MJ, Sullivan JP, Carrera GM, Holladay MW, Arneric SP, Williams M. Novel isoxazoles which interact with brain cholinergic channel receptors have intrinsic cognitive enhancing and anxiolytic activities. J Med Chem. 1994;37:1055–1059. doi: 10.1021/jm00034a002. [DOI] [PubMed] [Google Scholar]
- 34.Garvey DS, Wasicak JT, Elliott RL, Lebold SA, Hettinger AM, Carrera GM, Lin NH, He Y, Holladay MW, Anderson DJ, et al. Ligands for brain cholinergic channel receptors: synthesis and in vitro characterization of novel isoxazoles and isothiazoles as bioisosteric replacements for the pyridine ring in nicotine. J Med Chem. 1994;37:4455–4463. doi: 10.1021/jm00052a005. [DOI] [PubMed] [Google Scholar]
- 35.Lin NH, He Y, David JA, Wasicak JT, Kasson R, Sweeny D, James PS. Synthesis and structure-activity relationships of pyrrolidine-modified analogs of the potent cholinergic channel activator, ABT 418. Bioorganic & Medicinal Chemistry Letters. 1994;4:2389–2394. [Google Scholar]
- 36.Lin NH, He Y, Arneric SP, Sullivan JP. Synthesis and structure-activity relationships of 2′-(R) and (S) pyrrolidine-modified analogs of the cholinergic channel activator, ABT-418. Bioorganic & Medicinal Chemistry Letters. 1995;5:1141–1146. [Google Scholar]
- 37.Avenoza A, Busto JH, Cativiela C, Dordal A, Frigola J, Peregrina JM. Synthesis, activity and theoretical study of ABT-418 analogues. Tetrahedron. 2002;58:4505–4511. [Google Scholar]
- 38.Potter A, Corwin J, Lang J, Piasecki M, Lenox R, Newhouse PA. Acute effects of the selective cholinergic channel activator (nicotinic agonist) ABT-418 in Alzheimer’s disease. Psychopharmacology (Berl) 1999;142:334–342. doi: 10.1007/s002130050897. [DOI] [PubMed] [Google Scholar]
- 39.Badio B, Garraffo HM, Plummer CV, Padgett WL, Daly JW. Synthesis and nicotinic activity of epiboxidine: an isoxazole analogue of epibatidine. Eur J Pharmacol. 1997;321:189–194. doi: 10.1016/s0014-2999(96)00939-9. [DOI] [PubMed] [Google Scholar]
- 40.Fitch RW, Pei XF, Kaneko Y, Gupta T, Shi D, Federova I, Daly JW. Homoepiboxidines: further potent agonists for nicotinic receptors. Bioorg Med Chem. 2004;12:179–190. doi: 10.1016/j.bmc.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 41.Tonder JE, Hansen JB, Begtrup M, Pettersson I, Rimvall K, Christensen B, Ehrbar U, Olesen PH. Improving the nicotinic pharmacophore with a series of (Isoxazole)methylene-1-azacyclic compounds: synthesis, structure-activity relationship, and molecular modeling. J Med Chem. 1999;42:4970–4980. doi: 10.1021/jm9910627. [DOI] [PubMed] [Google Scholar]
- 42.Abreo MA, Lin NH, Garvey DS, Gunn DE, Hettinger AM, Wasicak JT, Pavlik PA, Martin YC, Donnelly-roberts DL, Anderson DJ, Sullivan JP, Williams M, Arneric SP, Holladay MW. Novel 3-Pyridyl ethers with subnanomolar affinity for central neuronal nicotinic acetylcholine receptors. J Med Chem. 1996;39:817–825. doi: 10.1021/jm9506884. [DOI] [PubMed] [Google Scholar]
- 43.Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Piattoni-Kaplan M, Campbell JE, McKenna DG, Molinari E, Hettinger AM, Garvey DS, Wasicak JT, Holladay MW, Williams M, Arneric SP. A-85380 [3-(2(S)-azetidinylmethoxy) pyridine]: in vitro pharmacological properties of a novel, high affinity alpha 4 beta 2 nicotinic acetylcholine receptor ligand. Neuropharmacology. 1996;35:725–734. doi: 10.1016/0028-3908(96)84644-2. [DOI] [PubMed] [Google Scholar]
- 44.Koren AO, Horti AG, Mukhin AG, Gundisch D, Kimes AS, Dannals RF, London ED. 2-, 5-, and 6-Halo-3-(2(S)-azetidinylmethoxy)pyridines: synthesis, affinity for nicotinic acetylcholine receptors, and molecular modeling. J Med Chem. 1998;41:3690–3698. doi: 10.1021/jm980170a. [DOI] [PubMed] [Google Scholar]
- 45.Decker MW, Meyer MD, Sullivan JP. The therapeutic potential of nicotinic acetylcholine receptor agonists for pain control. Expert Opin Investig Drugs. 2001;10:1819–1830. doi: 10.1517/13543784.10.10.1819. [DOI] [PubMed] [Google Scholar]
- 46.Decker MW, Curzon P, Holladay MW, Nikkel AL, Bitner RS, Bannon AW, Donnelly-Roberts DL, Puttfarcken PS, Kuntzweiler TA, Briggs CA, Williams M, Arneric SP. The role of neuronal nicotinic acetylcholine receptors in antinociception: effects of ABT-594. J Physiol Paris. 1998;92:221–224. doi: 10.1016/s0928-4257(98)80014-4. [DOI] [PubMed] [Google Scholar]
- 47.Wilens TE, Decker MW. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: focus on cognition. Biochem Pharmacol. 2007;74:1212–1223. doi: 10.1016/j.bcp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marks MJ, Wageman CR, Grady SR, Gopalakrishnan M, Briggs CA. Selectivity of ABT-089 for alpha4beta2* and alpha6beta2* nicotinic acetylcholine receptors in brain. Biochem Pharmacol. 2009;78:795–802. doi: 10.1016/j.bcp.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ki determinations were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA.
- 50.Bunnelle WH, Dart MJ, Schrimpf MR. Design of ligands for the nicotinic acetylcholine receptors: the quest for selectivity. Curr Top Med Chem. 2004;4:299–334. doi: 10.2174/1568026043451438. [DOI] [PubMed] [Google Scholar]
- 51.Rollema H, Shrikhande A, Ward KM, Tingley FD, 3rd, Coe JW, O’Neill BT, Tseng E, Wang EQ, Mather RJ, Hurst RS, Williams KE, de Vries M, Cremers T, Bertrand S, Bertrand D. Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol. 2010;160:334–345. doi: 10.1111/j.1476-5381.2010.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Eaton JB, Caldarone B, Lukas RJ, Kozikowski AP. Chemistry and pharmacological characterization of novel nitrogen analogues of AMOP-H-OH (Sazetidine-A, 6-[5-(azetidin-2-ylmethoxy)pyridin-3-yl]hex-5-yn-1-ol) as alpha4beta2-nicotinic acetylcholine receptor-selective partial agonists. J Med Chem. 2010;53:6973–6985. doi: 10.1021/jm100765u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei ZL, Xiao Y, Yuan H, Baydyuk M, Petukhov PA, Musachio JL, Kellar KJ, Kozikowski AP. Novel pyridyl ring C5 substituted analogues of epibatidine and 3-(1-methyl-2(S)-pyrrolidinylmethoxy)pyridine (A-84543) as highly selective agents for neuronal nicotinic acetylcholine receptors containing beta2 subunits. J Med Chem. 2005;48:1721–1724. doi: 10.1021/jm0492406. [DOI] [PubMed] [Google Scholar]
- 54.Bunnelle WH, Tietje KR, Frost JM, Peters D, Ji J, Li T, Scanio MJ, Shi L, Anderson DJ, Dyhring T, Gronlien JH, Ween H, Thorin-Hagene K, Meyer MD. Octahydropyrrolo[3,4-c]pyrrole: a diamine scaffold for construction of either alpha4beta2 or alpha7-selective nicotinic acetylcholine receptor (nAChR) ligands. Substitutions that switch subtype selectivity. J Med Chem. 2009;52:4126–4141. doi: 10.1021/jm900249k. [DOI] [PubMed] [Google Scholar]
- 55.Gatto GJ, Bohme GA, Caldwell WS, Letchworth SR, Traina VM, Obinu MC, Laville M, Reibaud M, Pradier L, Dunbar G, Bencherif M. TC-1734: an orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long-lasting cognitive effects. CNS Drug Rev. 2004;10:147–166. doi: 10.1111/j.1527-3458.2004.tb00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukas RJ, Fryer JD, Eaton JB, Gentry CL. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levin ED, editor. Nicotinic receptors and the nervous system. CRC Press; Boca Raton, FL: 2002. pp. 3–27. [Google Scholar]
- 57.Lukas RJ, Norman SA, Lucero L. Characterization of Nicotinic Acetylcholine Receptors Expressed by Cells of the SH-SY5Y Human Neuroblastoma Clonal Line. Mol Cell Neurosci. 1993;4:1–12. doi: 10.1006/mcne.1993.1001. [DOI] [PubMed] [Google Scholar]
- 58.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.