

Abstract
Mannose 6-phosphate (Man6P) residues represent a recognition signal required for efficient receptor-dependent transport of soluble lysosomal proteins to lysosomes. Upon arrival, the proteins are rapidly dephosphorylated. We used mice deficient for the lysosomal acid phosphatase Acp2 or Acp5 or lacking both phosphatases (Acp2/Acp5−/−) to examine their role in dephosphorylation of Man6P-containing proteins. Two-dimensional (2D) Man6P immunoblot analyses of tyloxapol-purified lysosomal fractions revealed an important role of Acp5 acting in concert with Acp2 for complete dephosphorylation of lysosomal proteins. The most abundant lysosomal substrates of Acp2 and Acp5 were identified by Man6P affinity chromatography and mass spectrometry. Depending on the presence of Acp2 or Acp5, the isoelectric point of the lysosomal cholesterol-binding protein Npc2 ranged between 7.0 and 5.4 and may thus regulate its interaction with negatively charged lysosomal membranes at acidic pH. Correspondingly, unesterified cholesterol was found to accumulate in lysosomes of cultured hepatocytes of Acp2/Acp5−/− mice. The data demonstrate that dephosphorylation of Man6P-containing lysosomal proteins requires the concerted action of Acp2 and Acp5 and is needed for hydrolysis and removal of degradation products.
INTRODUCTION
Lysosomes are acidic organelles (pH ≤ 5) capable of degrading macromolecules such as proteins, glycosaminoglycans, glycogen, nucleic acids, and lipids as well as extracellular material and pathogenic organisms delivered to lysosomes by autophagocytosis (41). These catabolic functions of lysosomes are catalyzed by more than 50 different soluble acid hydrolases and accessory activator proteins (29). The hydrolases are separated from the cytoplasm by a lysosomal membrane composed of about 140 highly glycosylated membrane proteins (2, 42, 43). In addition, the lipid composition of lysosomal membranes differs from that of plasma membranes or other limiting membranes of subcellular compartments and is characterized by a low cholesterol and an enriched anionic lipid content (44). Deficiencies and alterations in lysosomal proteins are associated with numerous human diseases (3, 39).
Many soluble lysosomal proteins are synthesized as inactive precursor proteins that are glycosylated in the endoplasmic reticulum (ER). Upon arrival in the Golgi apparatus, the lysosomal proteins can be phosphorylated at the C-6 position of selected mannoses of high-mannose-type oligosaccharides, a process catalyzed by two enzymes. First, the GlcNAc-1-phosphotranferase transfers GlcNAc-1-phosphate from UDP-GlcNAc to mannoses in the α-1,6 and/or α-1,3 branch of the oligosaccharide chains (17), resulting in a phosphodiester intermediate (25). In a second step, the covering GlcNAc is removed by the GlcNAc-1-phosphodiester-α-N-acetylglucosaminidase, resulting in mono- and/or bisphosphorylated oligosaccharides (26, 51). Depending on the lysosomal protein and the cell type studied, 2 to 7 mannose 6-phosphate (Man6P) residues per polypeptide have been described (37, 46). The Man6P residues serve as recognition markers for two types of Man6P-specific receptors mediating the vesicular transport of lysosomal proteins from the trans Golgi network to endosomes. After pH-induced dissociation of the receptor-ligand complexes, lysosomal proteins are delivered to lysosomes (7). In addition, extracellular lysosomal proteins can be internalized and transported along the endocytic pathway in a Man6P-dependent manner, which represents the therapeutic principle of enzyme replacement therapy of selected lysosomal storage disorders (38). Upon arrival in lysosomes, many lysosomal proteins undergo further modifications such as proteolytic activation and oligosaccharide processing (13, 54). In addition to the Man6P-dependent transport, cell type-specific and distinct Man6P-independent pathways for the transport of lysosomal enzymes have been reported (7).
Limited dephosphorylation of the Man6P recognition marker on lysosomal proteins has been observed in the prelysosomal/endosomal compartment, converting bisphosphorylated oligosaccharides to monophosphorylated forms (15, 16) followed by final dephosphorylation in dense lysosomes (9). There are two known acid lysosomal phosphatases, Acp2 and Acp5 (also called tartrate-resistant acid phosphatase or uteroferrin). Acp2, the enzyme which allowed Christian de Duve to discover the lysosomal compartment (14), is synthesized as a membrane-bound precursor protein that is C-terminally cleaved in two steps upon arrival in lysosomes, generating the mature and soluble phosphatase (19). Acp5 is a soluble protein that is transported in a Man6P-dependent manner to lysosomes and can be actively secreted (5). The role of Acp2 and Acp5 in the dephosphorylation of Man6P-containing lysosomal proteins has been a matter of debate. Neither overexpression nor deficiency of Acp2 affected the dephosphorylation of the Man6P-containing arylsulfatase A (ASA), suggesting that Acp5 may be responsible for the removal of Man6P residues on lysosomal enzymes (8, 9). This is in agreement with recent findings showing the accumulation of Man6P-containing proteins in several organs of Acp5-deficient (Acp5−/−) mice (47). However, the Man6P recognition marker is removed from the endocytosed arylsulfatase A in mouse fibroblasts doubly deficient for Acp2 and Acp5 (Acp2/Acp5−/−), as in control cells (48), indicating that neither Acp2 nor Acp5 is crucial for dephosphorylation of arylsulfatase A in these cells and implying the existence of further enzymes involved in this process.
In this study, we used a single-chain antibody fragment against Man6P residues (33) to examine the content of phosphorylated lysosomal proteins in lysosome-enriched fractions (tritosomes) from livers of mice singly and doubly deficient for acid phosphatase (Acp2−/−, Acp5−/−, and Acp2/Acp5−/−). The data demonstrate a role of both acid phosphatases in the removal of the Man6P recognition marker from lysosomal proteins. Additionally, we show that the different protein species of the Npc2 cholesterol binding protein are dephosphorylated in a concerted manner by both Acp2 and Acp5, which appears to be important for the cholesterol export out of lysosomes.
MATERIALS AND METHODS
Animals.
Homozygous Acp2-deficient (Acp2−/−), Acp5-deficient (Acp5−/−), and Acp2/Acp5 doubly deficient (Acp2/Acp5−/−) mice in a mixed genetic background were described previously (21, 40, 48) and were maintained under standard housing conditions in a homozygous breeding colony. Acp2−/−, Acp5−/−, and Acp2/Acp5−/− animals (2 to 3 months of age) and appropriate age-matched wild-type animals were used throughout the studies. Animals were maintained in accordance with institutional guidelines as approved by local authorities.
Subcellular fractionation.
Fractions highly enriched in lysosomal marker enzymes were obtained by differential centrifugation and subsequent isopycnic density gradient centrifugation (14, 53) as described previously (13). In brief, mice were injected 4 days prior to sacrifice with 17% (wt/vol) tyloxapol (Triton WR1339; Sigma-Aldrich) (5 μl per gram body weight) in saline solution. Livers were removed, homogenized in isotonic sucrose solution (250 mM sucrose), and used for differential centrifugation (14). The mitochondrion/lysosome (ML) pellet resuspended in 45% sucrose was layered beneath a discontinuous gradient of 14.3% and 34.5% sucrose, and lysosome-enriched fractions were collected after isopycnic centrifugation at the 14.3%/34.5% interface (53).
2D electrophoresis.
Prior to the one-dimensional (1D) isoelectric focusing (IEF), 60 μg (for Western blotting) or 200 μg (for Coomassie-stained gels) of lysosome-enriched fractions was subsequently precipitated by addition of 2% sodium deoxycholate and a 1/100 vol of 100% trichloroacetic acid. Protein pellets were resuspended in 100 μl of lysis buffer {7 M urea, 2 M thiourea, 2% 3-[(3-cholamidylpropyl)-dimethylammonio]-1-propanesulfonate (CHAPS)}, and 1 vol of rehydration buffer (lysis buffer supplemented with 2% [wt/vol] dithiothreitol [DTT] and 2% [vol/vol] immobilized pH gradient [IPG] buffer [GE Healthcare] [pH 4 to 7]) was added. Samples at the final volume of 200 μl were applied to 11-cm-long precast IPG strips (GE Healthcare) (pH 4 to 7) by passive reswelling over night. 1D IEF was carried out at 20°C with a 5-step program (15,000 V-h total) on IPGPhor II (GE Healthcare). After IEF, IPG strips were reduced and alkylated by incubation in equilibration buffer (6 M urea, 75 mM Tris-HCl [pH 8.8], 30% glycerol, 2% [wt/vol] sodium dodecyl sulfate [SDS]) containing DTT (10 mg/ml) or iodacetamide (25 mg/ml) for 15 min at room temperature. 2D polyacrylamide gel electrophoresis (PAGE) was carried out with 5 mA overnight on self-cast 15% SDS-polyacrylamide gels. 2D gels were stained with colloidal Coomassie dye (34) or electroblotted on nitrocellulose membrane.
Mass spectrometry.
For identification of proteins in Coomassie-stained gels by peptide mass fingerprinting (PMF), protein spots were excised (2-mm-diameter punches), washed, reduced, and subsequently carbamidomethylated (45). Finally, proteins were subjected to in-gel digestion with trypsin overnight at 37°C. After gel extraction with 1% trifluoroacetic acid (TFA), peptides were desalted on C18 ZipTips (Millipore). Desalted samples were applied to a dihydroxybenzoic acid matrix for subsequent matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS). Mass spectra were obtained with a Reflex III time-of-flight mass spectrometer (Bruker Daltonik); after assignment of monoisotopic peptide masses, proteins were identified by the Mascot search algorithm (Matrix Science Inc.) with the NCBI nonredundant protein database.
For identification of proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis using gels stained with a FireSilver staining kit (Proteome Factory), the protein bands were excised, washed twice in swelling buffer (100 mM NH4HCO3 in high-performance LC [HPLC] H2O) and shrinking buffer (50 mM NH4HCO3 and 60% acetonitrile [ACN] in HPLC H2O) for 30 min each time. After gel spots were dried, trypsin was added at a final concentration of 10 ng/μl in digestion buffer (50 mM NH4HCO3 and 10% ACN in HPLC H2O) for overnight incubation at 37°C. Peptides were extracted twice by 30 min of incubation with 65% ACN and 5% formic acid (FA) in HPLC H2O and a subsequent ultrasonic bath for 5 min. The samples were dried in a SpeedVac (Thermo Fisher). Before the analysis, tryptic peptides were dissolved in 50% ACN–0.1% FA–HPLC H2O and diluted with 14 μl of 0.2% FA–HPLC H2O.
LC-MS/MS was performed on an Agilent HPLC-Chip-Cube MS interface equipped with a 1100 LC/MSD trap XCT Ultra ESI-ion trap mass spectrometer (Agilent Technologies). The large-capacity HPLC chip (Agilent Technologies) integrates two on-chip columns (an enrichment column [internal volume, 160 nl] and a separation column [150 mm], both with 5 μm of Zorbax 300 SB-C18 material) and a nanospray emitter. A capillary pump attached to a microwell plate autosampler was used for sample injection. Gradient elution was performed with a nanoflow LC pump (1100 series Nanoflow LC system for MS; Agilent Technologies). Agilent ChemStation and MSD Trap Control software was used for system control and data acquisition. Mobile-phase gradients consisted of 0.2% FA (solvent A) and ACN (solvent B). For analysis of tryptic bovine serum albumin (BSA) peptides, 1 μl of a sample (100 fmol/μl) in solvent A was injected. A 10-μl volume of each spot sample was injected. Samples were loaded from the autosampler onto the enrichment column of the HPLC-chips with a mobile phase of 2% solvent B at a flow rate of 3 μl/min. The separation was performed with a gradient of 2% to 40% solvent B for 40 min at a flow rate of 400 nl/min. Data were acquired in the positive-ion mode, applying a voltage of −1.8 kV at the electrospray inlet capillary, a nitrogen drying gas flow of 4 liters/min, and a temperature of 325°C at the transfer capillary for desolvation. The mass spectrometer was operated in a data-dependent mode in which the three most intense ions in the precursor ion scan were subjected to subsequent automated MS/MS. Doubly charged ions were preferentially selected for fragmentation. The isolation width was set to 4 m/z and the MS/MS fragmentation amplitude to 1.25 V. Active exclusion was enabled after three cycles of MS/MS; the precursor ion was released from the exclusion after 1 min. The generic files for database searching were generated by data analysis software for 6300 Series Ion Trap LC/MS version 3.4; for precursor ion selection, a threshold of 100,000 and a retention time window of 0.5 min were applied and the absolute number of compounds was restricted to 1,000 per MS/MS experiment.
Protein identification was performed with a Mascot online search (version 2.3.01.241) (35). MS/MS data sets were used to search the spectra against the subset “Mus musculus” of the Swiss-Prot database (SwissProt_2011_08.fasta; [6]). Parameters were used as given in Table SA1 in the supplemental material.
Purification of Man6P-containing proteins from enriched lysosomal fractions.
Extracts from lysosome-enriched fractions (150 μg in 300 μl) supplemented with inhibitor cocktail (Sigma-Aldrich) and 0.2% (vol/vol) Triton X-100 were incubated with scFv M6P-1 antibody (1 mg/ml) immobilized to AminoLink Plus Gel beads (Pierce) for 30 min at 4°C in a column. Unbound material was collected (flowthrough). The column was washed with 10 vol each of 10 mM phosphate-buffered saline (PBS) (pH 7.4)–0.2% Triton X-100, 10 mM PBS containing 10 mM mannose (Sigma-Aldrich), and 10 mM glucose 6-phosphate (G6P; Sigma-Aldrich). Man6P-containing proteins were eluted with 3 vol of 10 mM PBS containing 10 mM Man6P (Sigma-Aldrich) and inhibitor cocktail. Fifty percent of each fraction was separated by SDS-PAGE (10% acrylamide) and transferred to a nitrocellulose membrane and analyzed by Western blotting.
Antibodies.
Man6P-containing proteins were detected using the scFv M6P-1 single-chain antibody fragment described previously (33). Murine cathepsin Z (Ctsz), cathepsin B (Ctsb), and cathepsin D (Ctsd) were detected by a polyclonal goat antibody against Ctsz (R&D Systems), polyclonal goat IgG against Ctsb (Neuromics/Acris), and polyclonal rabbit serum against Ctsd (10), respectively. Polyclonal rabbit anti-Npc2 serum was a kind gift from Shutish C. Patel, Neurology Service, Newington, CT. The polyclonal rabbit anti-Npc1 IgG was obtained from Abcam. The monoclonal mouse anti-myc and polyclonal rabbit anti-myc IgG were purchased from Cell Signaling and Sigma-Aldrich, respectively. Monoclonal antibody 1D4B against mouse Lamp1 was obtained from the NICHD Developmental Studies Hybridoma Bank (University of Iowa). Horseradish peroxidase-conjugated goat anti-rabbit IgG, rabbit anti-goat IgG, goat anti-mouse IgG, and goat anti-rat IgG were used as secondary antibodies (Jackson ImmunoResearch Laboratories Inc.). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL) (Pierce). Alexa Fluor 488–goat anti-rabbit and Alexa Fluor 594–goat anti-mouse antibodies, used for immunofluorescence microscopy, were from Invitrogen.
Cultivation of hepatocytes, immunofluorescence microscopy, cholesterol measurement, and expression analysis.
Primary cultured hepatocytes were isolated from Acp2/Acp5 doubly deficient and wild-type mice 2 months of age as described previously (31). Cells were cultured in 5% lipoprotein-deficient medium plated on glass coverslips, fixed with 4% paraformaldehyde, and incubated with filipin (Sigma-Aldrich) (500 μg/ml)–PBS for 1 h at room temperature. After several washes, cells were incubated for 1 h with specific primary antibodies and with secondary antibodies conjugated to Alexa Fluor 546 and Alexa Fluor 488 for 1 h at room temperature, respectively. After five washes, the cells were embedded in Mowiol. In the absence of filipin treatment, fixed cells were permeabilized with 0.1% Triton X-100 for 5 min and incubated with specific antibodies according the procedure described above.
Images were acquired with a Leica TCS SP2 or Perkin Elmer Ultra-View VoX spinning-disc confocal microscope (Leica Microscope and Scientific Instruments Group) and processed using a Leica TCS NT, Velocity (PerkinElmer), and Adobe Photoshop software. For cholesterol measurement and mRNA expression level determinations, primary hepatocytes were cultured for 24 h in lipoprotein-deficient media. Cells were washed with PBS and lysed in lysis buffer (50 mM Tris/HCl [pH 8.0], 2 mM CaCl2, 80 mM NaCl, 1% Triton X-100). Cellular cholesterol levels were determined using commercial kits (Invitrogen). Protein concentrations were measured by a Lowry method, which was modified for lipid-containing samples by the addition of 0.1% SDS (28).
Real-time PCR.
Real-time RT-PCR was performed as described previously (4, 36). For all genes, Assay-on-Demand primer/probe sets supplied by Applied Biosystems were used (assay catalog numbers are available upon request). Relative expression levels were calculated by normalization to selected housekeeper mRNAs (tbp or actB) by the ΔΔCt method. Data are reported as copy numbers relative to housekeeper numbers.
Acid phosphatase activity assays.
Acp2 and Acp5 activity in aliquots (1 μg of protein) of lysosome-enriched fractions was determined using p-nitrophenylphosphate as the substrate in the presence or absence of tartrate as described previously (48).
RESULTS
Acp2 and Acp5 are involved in the dephosphorylation of Man6P-containing proteins.
To evaluate the role of Acp2 and Acp5 in dephosphorylation of Man6P-containing proteins, liver extracts of wild-type, Acp5-deficient (Acp5−/−), and Acp2/Acp5 doubly deficient (Acp2/Acp5−/−) mice were analyzed by anti-Man6P Western blotting (33). In total extracts of wild-type liver, the level of Man6P-containing polypeptides is low (Fig. 1) and comparable to the level determined with extracts from Acp2−/− mice (Fig. 2). The intensity of Man6P-containing polypeptides increases significantly in liver extracts of Acp5−/− mice (Fig. 1) as shown previously (47). In Acp2/Acp5−/− mice, the amount of Man6P-containing proteins is further increased. The most predominant proteins exhibit molecular masses of 100, 78, 51, 46, 40 and 28 kDa. The total intensities of all Man6P-positive polypeptides as determined by densitometry were 45- and 54-fold higher in liver tissue of Acp5−/− and Acp2/Acp5−/− mice compared to wild-type mice, respectively. Equal loading of the gel was demonstrated by Western blotting of the lysosomal protease cathepsin D. Of note, the proteolytic processing of cathepsin D precursor to the mature 31-kDa form is impaired in Acp2/Acp5−/− lysosomes and resembles the proteolytic pattern previously observed in GlcNAc-1-phosphotransferase-deficient cells that fail to form Man6P residues on lysosomal enzymes (20, 50).
Fig 1.
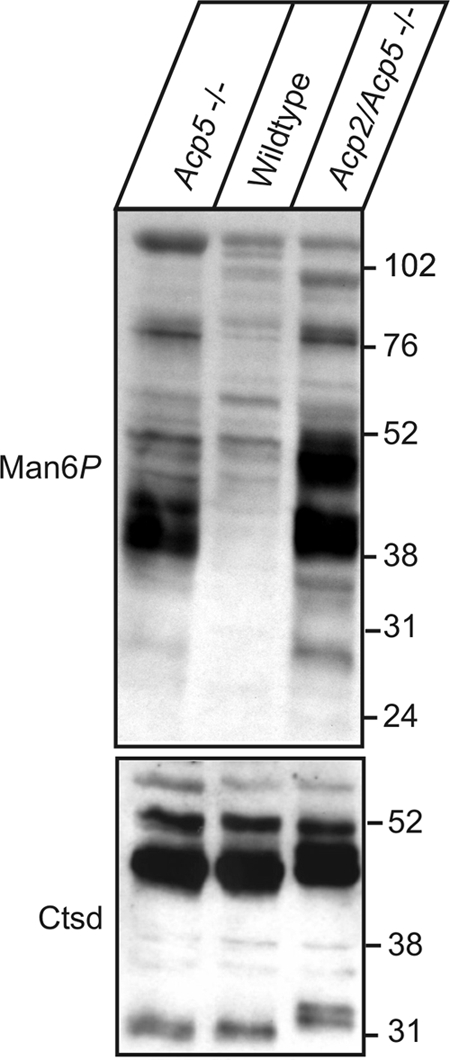
Man6P-containing proteins in liver of Acp5−/− and Acp2/Acp5−/− mice. Eighty micrograms of protein extracts prepared from liver tissue of 8-week-old wild-type, Acp5−/−, and Acp2/Acp5−/− mice was separated by SDS-PAGE and analyzed by anti-Man6P Western blotting (1 μg of scFv M6P-1/ml). The positions of the molecular mass marker proteins (in kilodaltons) are indicated. As a control for equal loading, Western blotting of the lysosomal protein cathepsin D is shown.
Fig 2.
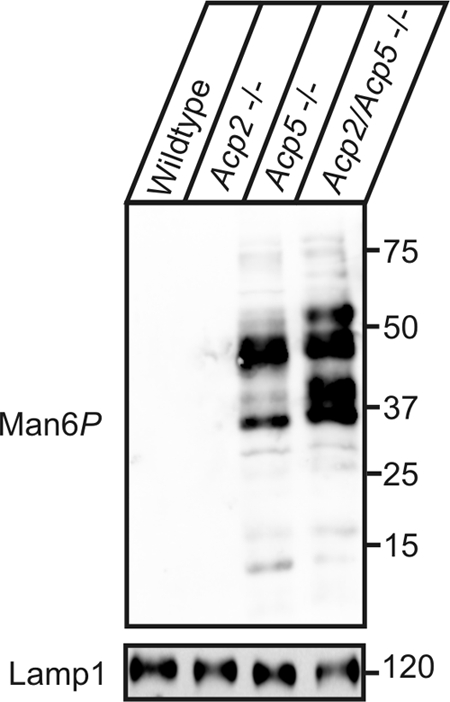
Man6P-containing proteins in lysosome-enriched fractions of Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mice. Extracts (5 μg) of lysosome-enriched fractions from wild-type, Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mice were separated by SDS-PAGE and analyzed by anti-Man6P Western blotting. The lysosome-associated Lamp1 membrane protein was used as a loading control. The positions of the molecular mass marker proteins (in kilodaltons) are indicated.
To investigate the role of Acp2 and Acp5 in dephosphorylation of lysosomal proteins in more detail, lysosome-enriched fractions were isolated from wild-type, Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mouse liver tissue after injection of Triton WR-1339 (tyloxapol), resulting in a selective density shift of lysosomes (13, 53). Aliquots of the lysosome-enriched fractions were separated by two-dimensional electrophoresis (2DE) and either stained with Coomassie blue or processed by anti-Man6P Western blotting. The patterns of the most abundant lysosomal polypeptides in wild-type, Acp2−/−, and Acp5−/− mice were similar, whereas the intensities of both the groups of protein species and the single proteins were altered in Acp2/Acp5−/− lysosome-enriched fractions (Fig. 3, right panels). Almost all lysosomal proteins of wild-type and Acp2−/− mice lacked Man6P residues as shown by anti-Man6P Western blotting (Fig. 3, left panels). In Acp5−/− mouse lysosomes, the amount of Man6P-containing proteins was greatly increased. These polypeptides mainly had isoelectric points in the pH range of 5.8 to 6.6. In lysosome-enriched fractions of Acp2/Acp5−/− mice, however, the proportion of Man6P-containing proteins was further elevated across the IEF gradient and an accumulation of Man6P-containing polypeptides of different molecular masses was observed at the most acidic pH of 4.0. The data clearly show that, in addition to acid phosphatase Acp5, Acp2 is also involved in the dephosphorylation of lysosomal proteins.
Fig 3.

2DE and Man6P Western blot analysis of lysosome-enriched fractions from Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mice. Lysosome-enriched fractions (200 μg) prepared from wild-type, Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mouse liver were separated by isoelectric focusing (IEF; pH gradient, 4 to 7) in the first dimension followed by SDS-PAGE with a 15% gel in the second dimension and Coomassie blue staining (right panels). In parallel, lysosome-enriched fractions (60 μg) of wild-type, Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mice were separated by 2DE and analyzed by anti-Man6P Western blotting (left panels). The positions of the molecular mass marker proteins (in kilodaltons) are indicated.
Man6P affinity chromatography of lysosome-enriched fractions.
To verify the presence of phosphorylated lysosomal enzymes in lysosome-enriched fractions of Acp2/Acp5−/− mice, extracts of those fractions were analyzed by Man6P affinity chromatography. Material specifically bound through Man6P residues to the column was eluted by Man6P and examined by cathepsin Z and B Western blot analysis. In lysosome-enriched fractions of wild-type mice, none of the lysosomal proteases contained Man6P; therefore, none bound to the affinity matrix (Fig. 4). In contrast, about 72% of the total cathepsin Z (Ctsz) applied in lysosome-enriched fractions of Acp2/Acp5−/− mice contained Man6P residues and bound to the matrix. A distinct proportion of cathepsin Z could be eluted only with 0.1 M glycine (pH 2.4) (Fig. 4). In this approach, cathepsin B (Ctsb) was used as a negative control that did not bind to the affinity matrix due to the proteolytic removal of the Man6P-containing propeptide in endosomes (49).
Fig 4.
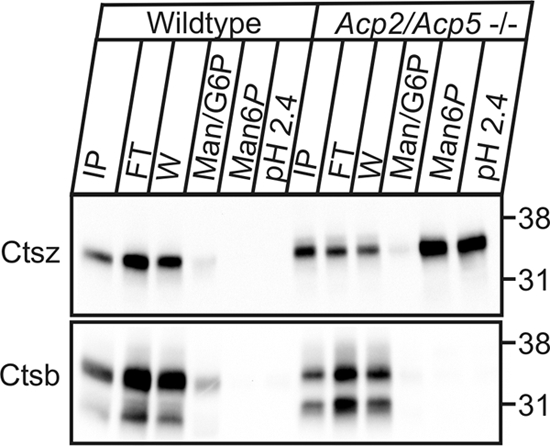
Man6P-containing cathepsin Z in Acp2/Acp5−/− lysosomes. Extracts (150 μg) of lysosome-enriched fractions of wild-type and Acp2/Acp5−/− mice were applied to scFv M6P-1 antibody covalently immobilized on beads. After loading for 30 min at 4°C, the beads were washed sequentially with 10 vol of PBS–Triton X-100, 10 vol of PBS containing 10 mM mannose (Man) and 10 mM glucose 6-phosphate (G6P), and 3 vol of 10 mM Man6P, followed by 3 vol of acidic wash buffer (pH 2.4). Aliquots of the input (IP; 7%), 50% of the flowthrough fraction (FT) and wash fraction 1 (W), and the Man/Glc6P, Man6P, and pH 2.4 fractions were separated by SDS-PAGE and analyzed by cathepsin Z (Ctsz) and cathepsin B (Ctsb) Western blotting (0.1 μg/ml each). The positions of the molecular mass marker proteins (in kilodaltons) are given.
Mass spectrometry analyses of lysosome-enriched fractions.
The tryptic digest of Man6P eluate after Man6P affinity chromatography purification of 150 μg of protein of lysosome-enriched fractions of Acp2/Acp5−/− mice was analyzed by LC-MS/MS. A total of 34 proteins were identified, of which 26 were known soluble lysosomal proteins (see Table SA1 in the supplemental material). Only a few of these known lysosomal proteins have been detected in the nonbound flowthrough fraction of the affinity chromatography, representing most likely nonphosphorylated polypeptides or lysosomal proteins with low affinity for the immobilized anti-Man6P antibody (see Table SA1 in the supplemental material). After the 2DE separation of the lysosome-enriched fraction from liver of Acp2/Acp5−/− mice, MALDI-TOF MS analyses of excised Coomassie-stained spots and comparison with blots of the Man6P proteome (Fig. 3) resulted in the identification of 10 known soluble lysosomal proteins in multiple spots (Fig. 5; see also Table SA1 in the supplemental material). Among these proteins, cathepsin D, Npc2, cathepsin Z, and legumain represent the most abundant substrates of Acp2 and Acp5.
Fig 5.

2D map of Man6P-containing proteins in Acp2/Acp5−/− lysosome-enriched fraction. Lysosome-enriched fractions from Acp2/Acp5−/− mice (60 μg of proteins) were separated by IEF (pH gradient, 4 to 7) in the first dimension followed by SDS-PAGE on a 15% gel in the second dimension, blotted onto a polyvinylidene difluoride (PVDF) membrane, and probed with an scFv M6P-1 antibody fragment followed by ECL. The positions of the molecular mass marker proteins (in kilodaltons) are given. Arbitrary annotated protein spots correspond to excised Coomassie-stained spots used for MALDI-TOF MS analysis; numbered spots are given in Table SA1 in the supplemental material.
Man6P residues differentially affect the isoelectric point of lysosomal proteins.
To examine the effect of Man6P residues on individual lysosomal proteins in more detail, lysosome-enriched fractions prepared from wild-type, Acp5−/−, and Acp2/Acp5−/− mice and containing lysosomal proteins without Man6P modification, with low-level Man6P modification, and with increasing amounts of Man6P residues, respectively, were analyzed (Fig. 3). After separation by 2DE and Western blotting, Npc2 and Ctsz were specifically detected. In wild-type lysosomes, the majority of the Npc2 protein exhibited an isoelectric point (pI) of 7.0 (Fig. 6, left upper panel). In the lysosome-enriched fraction of Acp5−/− mice, three major immunoreactive spots with pI 6.6, 6.2, and 6.1 and three minor spots with pI 7.0, 6.3, and 5.7 were observed (Fig. 6, left middle panel). The Npc2 protein pattern was further shifted to acidic pI of 6.2, 5.7, and 5.4 in fractions of Acp2/Acp5−/− mice (Fig. 6, left lower panel). For comparison, the protein pattern of Ctsz was examined, showing more-acidic pI values of 5.3, 5.1, and 4.9 in fractions of wild-type mice (Fig. 6, right upper panel). This pattern was changed only marginally in fractions of Acp5−/− mice. In lysosome-enriched fractions of Acp2/Acp5−/− mice, at least seven Ctsz species were detected, with pI ranging from 5.3 to 4.0 (Fig. 6, right lower panel). These data unequivocally show that both acid phosphatases are involved in the dephosphorylation of lysosomal proteins. In the absence of Acp2 alone, lysosomal proteins appear to be dephosphorylated by another phosphatase, most likely Acp5. The specificity of Acp2, however, seems to be insufficient to hydrolyze all Man6P residues from lysosomal proteins in the absence of Acp5. However, the further increases in the numbers of Man6P-containing proteins and their pI in lysosomal fractions of Acp2/Acp5−/− mouse liver tissue strongly suggest that the two lysosomal phosphatases act in concert. The most acidic species of Man6P-containing proteins appear to be substrates of Acp2, and the intermediate pI protein species are dephosphorylated by Acp5.
Fig 6.

2D Western blotting of Npc2 and cathepsin Z in lysosome-enriched fractions prepared from Acp5−/− and Acp2/Acp5−/− mice. Sixty micrograms of wild-type, Acp5−/−, and Acp2/Acp5−/− F2 fractions were separated by IEF (pH gradient, 4 to 7) in the first dimension followed by SDS-PAGE on a 15% gel in the second dimension, blotted onto a PVDF membrane, and probed with anti-Npc2 antibodies (1:500; left panel) and anti-cathepsin Z antibodies (0.1 μg/ml; right panel) followed by ECL. The approximate isoelectric points (pI) are given.
Accumulation of cholesterol in Acp2/Acp5−/− hepatocytes.
Intralysosomal membranes are enriched in bis(monoacylglycerol)phosphate (BMP) and other anionic lipids such as phosphatidylinositols and dolichol phosphate, whereas cholesterol is almost absent (44). Cholesterol liberated from endocytosed lipoproteins in lysosomes binds to Npc2 for export out of lysosomes (23). The neutral pI of Npc2 in lysosome-enriched fractions of wild-type mouse liver (Fig. 6) suggested that, at acidic lysosomal pH, the interaction of Npc2 with negatively charged lysosomal membranes is facilitated (55). In lysosomes of Acp2/Acp5−/− mice, the Man6P residues result in an acidic pI of Npc2 and should interfere with an electrostatic interaction with intralysosomal membranes and the Npc2-mediated egress of cholesterol. When primary cultured hepatocytes of Acp2/Acp5−/− mice were costained with filipin for unesterified cholesterol and the lysosomal Niemann-Pick type C1 membrane protein (Npc1), substantial lysosomal accumulation of unesterified cholesterol was observed (Fig. 7A). In wild-type hepatocytes, the filipin staining was faint and diffusely distributed through the cells. Concomitant visualization of Man6P-containing proteins with the single-chain antibody fragment scFv M6P-1 demonstrated a high level of Man6P-containing proteins only in Acp2/Acp5−/− hepatocytes (Fig. 7B). Biochemical quantification showed that the total cellular cholesterol was increased by ∼30% in Acp2/Acp5−/− hepatocytes compared to those of wild-type cells, underlining the importance of dephosphorylation for optimal Npc2 function (Table 1). Impaired cholesterol transport from lysosomes to ER membranes should cause cholesterol depletion in ER membranes, thereby activating sterol-responsive transcriptions factors which stimulate the expression of key regulatory proteins for de novo cholesterol synthesis and cholesterol uptake. In comparison to wild-type cells, mRNA expression of hydroxymethyl-glutaryl coenzyme A (CoA) reductase (Hmgr) and low-density lipoprotein (LDL) receptor (Ldlr) was increased in Acp2/Acp5−/− hepatocytes, demonstrating that impaired removal of Man6P residues from lysosomal proteins can affect cholesterol metabolism.
Fig 7.

Colocalization of accumulating free cholesterol with Niemann-Pick type C1 (Npc1) protein in Acp2/Acp5−/− hepatocytes. Primary cultured hepatocytes isolated from wild-type and Acp2/Acp5−/− mice were fixed and costained with filipin (blue) (500 μg/ml) and anti-Npc1 antibody (green) (1:100) (A) or stained for Man6P (red, 1 μg/ml) and the lysosomal protease cathepsin D (Ctsd; green) (1:2,000) (B) and analyzed by confocal microscopy. In the merged pictures, yellow indicates the colocalization. Bars, 10 μm.
Table 1.
Cholesterol homeostasis in cultured Acp2/Acp5−/− hepatocytesa
| Mouse strain | Cholesterol level (μg/mg protein) ± SD | Total copy no. (± SD) ofb: |
|
|---|---|---|---|
| Ldlr | Hmgr | ||
| Wild type | 14.7 ± 1.2 | 38,433 ± 3,175 | 20,583 ± 771 |
| Acp2/Acp5−/− | 22.4 ± 1.8 | 43,005 ± 4,976 | 33,571 ± 7,562 |
Hepatocytes were prepared and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 5% fetal calf serum (FCS). Four hours after seeding, cells were incubated in DMEM supplemented with 5% lipoprotein-deficient serum (LPDS) for 24 h (n = 4).
Total copy numbers of Lldr and Hmgr genes in relation to the housekeeping gene Tbp as determined by quantitative TaqMan analysis.
DISCUSSION
Using lysosome-enriched fractions from liver tissue after tyloxapol treatment of mice single or doubly deficient for acid phosphatases, we have shown that both the classical lysosomal Acp2 acid phosphatase and the tartrate-resistant Acp5 acid phosphatase are involved in the mannose 6 dephosphorylation of lysosomal proteins. In particular, comparative Man6P immunoblotting of lysosome-enriched fractions of Acp2−/−, Acp5−/−, and Acp2/Acp5−/− mice separated by 2DE demonstrated that only the concerted action of both lysosomal phosphatases resulted in a complete removal of the Man6P recognition marker on lysosomal proteins as observed in lysosomes of wild-type mice (Fig. 2 and 3). Mass spectrometric analysis of the most abundant Man6P-containing proteins accumulating in lysosomes of Acp2/Acp5−/− mice identified the first in vivo Man6P substrates of both phosphatases (see Table SA1 in the supplemental material). In previous studies, in vitro dephosphorylation of lysosomal arylsulfatase A (ASA) with purified human ACP2 failed and 100-fold overexpression of ACP2 did not affect the removal of Man6P residues from internalized ASA (8). Additionally, in Acp2−/− fibroblasts, Man6P residues of endocytosed ASA can still be dephosphorylated (9), suggesting that ASA is not a specific substrate of Acp2 but can be dephosphorylated by Acp5 exhibiting low substrate specificity. This conclusion was supported by recent observations demonstrating increased levels of Man6P-containing proteins in various tissues of Acp5−/− mice (47). On the other hand, β-glucuronidase internalized by fibroblasts from an inclusion cell (I-cell) disease patient was completely dephosphorylated (16). I-cell disease is caused by the loss of GlcNAc-1-phosphotransferase activity and results in missorting of lysosomal hydrolases lacking Man6P residues (50). In these fibroblasts, no soluble Acp5 activity is detectable whereas the membrane-bound Acp2 is present (27), suggesting that Acp2 is sufficient for dephosphorylation of β-glucuronidase. The data suggest that Acp2 and Acp5 differ in their substrate specificities, which might be determined by the lysosomal protein or by the presence and number of mono- and/or bisphorylated oligosaccharides and may explain how the Man6P residues are removed in liver lysosomes of Acp2−/− mice. Quantitative real-time PCR revealed no or marginal changes in the relative mRNA expression levels of Acp5 and Acp2 in the liver of Acp2−/− and Acp5−/− mice, respectively (Table 2). Additionally, no compensatory alterations in the activities of acid phosphatases were found in lysosome-enriched fractions of Acp2−/− and Acp5−/− mice in comparison with fractions from wild-type mice (Table 3), which supports the idea of differences in the substrate specificities rather than the transcriptional regulation of Acp2 and Acp5 expression. Although not in the focus of the present study, the various ratios of expression of Acp2 and Acp5 may also affect the steady-state concentrations of Man6P-containing proteins in lysosomes of different cells and tissues. Thus, loss of Acp5 has little or no significant effect on the pattern of Man6P-containing proteins in mouse brain and heart tissue whereas increased and tissue-dependent accumulation of Man6P-positive proteins in spleen, kidney, and lung of Acp5−/− mice has been previously reported (47). Another explanation might be the existence of a third acid phosphatase that can compensate for Acp2 or for both phosphatase activities. Because of the high number of Man6P-containing proteins in lysosome-enriched fractions of Acp2/Acp5−/− mice, this speculative phosphatase, however, does not appear to play a role in lysosomes of the liver.
Table 2.
Relative mRNA expression levels in wild-type and Acp-deficient mouse livera
| Gene | Mean fold change in Acp expression ± SD in indicated mouse strain |
|||
|---|---|---|---|---|
| Wild type | Acp2−/− | Acp5−/− | Acp2/Acp5−/− | |
| Acp2 | 1.0 ± 0.4 | 0.1 ± 0.0 | 1.3 ± 0.0 | 0.0 ± 0.0 |
| Acp5 | 1.0 ± 0.0 | 1.0 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 |
The mRNA levels were normalized to β-actin expression. The values represent the means of the results of triplicate PCRs performed with four independent RNA preparations and are expressed as the mean fold changes in expression of the indicated Acp genes (± standard deviations) compared to wild-type mice.
Table 3.
Specific phosphatase activities in lysosomal-enriched fractions of Acp-deficient mice
| Enzyme | Phosphatase activity (U/mg protein ± SD) in indicated mouse straina |
|||
|---|---|---|---|---|
| Wild type | Acp2−/− | Acp5−/− | Acp2/Acp5−/− | |
| Acp2 | 22.3 ± 5.4 | 2.0 ± 0.3 | 18.4 ± 2.0 | 0.0 ± 1.1 |
| Acp5 | 14.5 ± 2.0 | 15.0 ± 0.1 | 2.3 ± 1.0 | 1.5 ± 0.2 |
Data represent enzyme activities measured in triplicate experiments performed with two independent preparations.
Analysis of the different phenotypes of Acp2−/− and Acp5−/− mice indicated that the two lysosomal phosphatases use distinct substrates (21, 40). The severe phenotype of Acp2/Acp5−/− doubly deficient mice, which consists of more than a mere addition of the clinical signs observed in singly deficient Acp2−/− and Acp5−/− mice, suggested that the two acid phosphatases can substitute for each other for a distinct number of substrates (48). The accumulation of these common substrates, such as osteopontin, however, can be observed in the absence of both acid phosphatases. 2DE separation of purified lysosome-enriched fractions and Western blot analysis using a single-chain anti-Man6P antibody fragment provided evidence that more than 70 lysosomal polypeptides containing Man6P residues can be dephosphorylated by Acp2 and Acp5. About 40 abundant substrates of Acp2 and Acp5 have been isolated by Man6P affinity chromatography and identified by MALDI-TOF MS (Fig. 4; see also Table SA1 in the supplemental material). More than half of these proteins are known as typical lysosomal acid hydrolases (29). Included among these are dipeptidyl peptidase 2, which was found in Man6P receptor-purified secretions of cultured osteoclasts (12), and cathepsin Z, which has recently been shown to be transported in a Man6P-dependent manner to lysosomes (30).
One important observation made in this study was the occurrence of various forms of Npc2 that depend on the presence or absence of acid phosphatases. Npc2 lacking Man6P residues as found in lysosomes from wild-type mice exhibits a pI of ≥7.0, which corresponds to the calculated pI of 7.5. In the absence of Acp5, the pI values of the four major Npc2 species shifted between 6.6 and 6.1, which changed further to pIs between 6.2 and 5.4 upon the concomitant absence of Acp2 and Acp5 (Fig. 6). This provides the most striking evidence that Acp2 is involved in highly specific steps during the dephosphorylation of lysosomal proteins. It is likely that the more acidic Acp2-sensitive Npc2 forms contain a higher percentage of bisphosphorylated oligosaccharide chains. Newly synthesized lysosomal proteins contain a heterogenous population of phosphorylated oligosaccharides containing a single phosphomonoester or two phosphomonoesters in the α-1,6 and/or α-1,3 branch of the oligosaccharide chains (52). Since oligosaccharides that contain phosphomonoester units bind more strongly to Man6P receptors than phosphodiester-containing oligosaccharides (11, 22), they may also differ in their preferences with respect to substrates of Acp2 and Acp5. As a functional consequence, the most acidic and anionic Man6P-containing Ncp2 forms may be unable to interact with negatively charged intralysosomal membranes at a lysosomal pH of <5.0. These membranes are highly enriched in bis(monoacylglycero)phosphate (BMP; also called lysobisphosphatidic acid) (32), preventing the extraction of cholesterol from lysosomal membranes by Man6P-containing Npc2 and the subsequent cholesterol transfer to the Npc1 membrane protein (24). Furthermore, Npc2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes (23). This is in agreement with our findings demonstrating the highly elevated amount of filipin-positive unesterified cholesterol in lysosomes of Acp2/Acp5−/− hepatocytes (Fig. 7A). This redistribution of unesterified cholesterol to lysosomes was accompanied by an approximately 30% increase in total cholesterol levels. The idea of a disturbance of cholesterol homeostasis and intracellular distribution in Acp2/Acp5−/− hepatocytes is also supported by increased low-density lipoprotein (LDL) receptor and hydroxymethyl-glutaryl coenzyme A (CoA) reductase mRNA levels (Table 1). Both genes are regulated by the cholesterol content of ER membranes via SREBP cleavage-activating protein (SCAP). This cholesterol sensor protein controls the transport of sterol regulatory element-binding proteins (SREBPs) to the Golgi apparatus, where the active SREBP transcription factor is liberated to activate genes such as Ldlr or Hmgr for cholesterol synthesis (18). Thus, higher Ldlr and Hmgr mRNA levels indicate lower cholesterol concentrations in ER membranes, which can be explained by decreased cholesterol delivery from lysosomes in Acp2/Acp5−/− hepatocytes.
The importance of Man6P residues appears not to be restricted to Npc2, because several other soluble lysosomal hydrolases have neutral or even basic calculated pI values, especially those enzymes involved in the degradation of lipids such as lysosomal acid lipase (pI 7.7), acid ceramidase (pI 8.7), and palmitoyl protein thioesterase 1 (pI 8.3), and the removal of Man6P residues from these enzymes may directly affect the efficiency and degradation rates of cholesterol esters and triglycerides, ceramide, and fatty acid-modified proteins, respectively, or, secondarily, the cholesterol transfer to Npc2 (1). The majority of lysosomal proteases and glycosidases exhibit acidic pI values and may be marginally affected by the presence of Man6P residues, as demonstrated by the isoform pattern of cathepsin Z (Fig. 5). However, the pI and therefore the activity of distinct lysosomal enzymes such as cathepsin H (pI 8.6), lysosomal α-mannosidase (pI 8.3), and β-hexosaminidase subunit β (pI 8.3) might depend on the removal of Man6P residues in lysosomes by Acp2 and Acp5, which can be demonstrated, e.g., by the occurrence of unusual proteolytically processed or glycosylated forms of cathepsin D in lysosomal fractions of Acp2/Acp5−/− mice (Fig. 1).
In conclusion, the presented data provide evidence that, in addition to the signal structure for efficient lysosomal transport, Man6P residues are important for the electrochemical properties of soluble lysosomal proteins prerequisite for at least intralysosomal protein-lipid interactions at acidic pH. Furthermore, this study clearly demonstrated the importance of the concerted action of the lysosomal phosphatases Acp2 and Acp5 in the removal of Man6P residues for the function of lysosomes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (SFB877/B3 and Research Training Group 1459).
We thank Maike Langer and Sandra Ehret for help in supplying mice and expert technical assistance on isolating hepatocytes, respectively. We also thank Timothy M. Cox (University of Cambridge) and Alison Hayman (University of Bristol) for providing the Acp5−/− mice.
Footnotes
Published ahead of print 12 December 2011
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1. Abdul-Hammed M, et al. 2010. Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J. Lipid Res. 51:1747–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagshaw RD, Mahuran DJ, Callahan JW. 2005. Lysosomal membrane proteomics and biogenesis of lysosomes. Mol. Neurobiol. 32:27–41 [DOI] [PubMed] [Google Scholar]
- 3. Ballabio A, Gieselmann V. 2009. Lysosomal disorders: from storage to cellular damage. Biochim. Biophys. Acta 1793:684–696 [DOI] [PubMed] [Google Scholar]
- 4. Bartelt A, et al. 2011. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17:200–205 [DOI] [PubMed] [Google Scholar]
- 5. Baumbach GA, Saunders PT, Bazer FW, Roberts RM. 1984. Uteroferrin has N-asparagine-linked high-mannose-type oligosaccharides that contain mannose 6-phosphate. Proc. Natl. Acad. Sci. U. S. A. 81:2985–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boeckmann B, et al. 2003. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31:365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Braulke T, Bonifacino JS. 2009. Sorting of lysosomal proteins. Biochim. Biophys. Acta 1793:605–614 [DOI] [PubMed] [Google Scholar]
- 8. Bresciani R, Peters C, von Figura K. 1992. Lysosomal acid phosphatase is not involved in the dephosphorylation of mannose 6-phosphate containing lysosomal proteins. Eur. J. Cell Biol. 58:57–61 [PubMed] [Google Scholar]
- 9. Bresciani R, von Figura K. 1996. Dephosphorylation of the mannose-6-phosphate recognition marker is localized in later compartments of the endocytic route. Identification of purple acid phosphatase (uteroferrin) as the candidate phosphatase. Eur. J. Biochem. 238:669–674 [DOI] [PubMed] [Google Scholar]
- 10. Claussen M, et al. 1997. Proteolysis of insulin-like growth factors (IGF) and IGF binding proteins by cathepsin D. Endocrinology 138:3797–3803 [DOI] [PubMed] [Google Scholar]
- 11. Creek KE, Sly WS. 1983. Biosynthesis and turnover of the phosphomannosyl receptor in human fibroblasts. Biochem. J. 214:353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Czupalla C, et al. 2006. Proteomic analysis of lysosomal acid hydrolases secreted by osteoclasts. Mol. Cell. Proteomics 5:134–143 [DOI] [PubMed] [Google Scholar]
- 13. Damme M, et al. 2010. Impaired lysosomal trimming of N-linked oligosaccharides leads to hyperglycosylation of native lysosomal proteins in mice with alpha-mannosidosis. Mol. Cell. Biol. 30:273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. 1955. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60:604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gabel CA, Foster SA. 1986. Mannose 6-phosphate receptor-mediated endocytosis of acid hydrolases: internalization of beta-glucuronidase is accompanied by a limited dephosphorylation. J. Cell Biol. 103:1817–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gabel CA, Foster SA. 1987. Postendocytic maturation of acid hydrolases: evidence of prelysosomal processing. J. Cell Biol. 105:1561–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldberg DE, Kornfeld S. 1981. The phosphorylation of beta-glucuronidase oligosaccharides in mouse P388D1 cells. J. Biol. Chem. 256:13060–13067 [PubMed] [Google Scholar]
- 18. Goldstein JL, DeBose-Boyd RA, Brown MS. 2006. Protein sensors for membrane sterols. Cell 124:35–46 [DOI] [PubMed] [Google Scholar]
- 19. Gottschalk S, Waheed A, Schmidt B, Laidler P, von Figura K. 1989. Sequential processing of lysosomal acid phosphatase by cytoplasmic thiol proteinase and lysosomal aspartyl proteinase. EMBO J. 8:3215–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasilik A, Neufeld EF. 1980. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J. Biol. Chem. 255:4946–4950 [PubMed] [Google Scholar]
- 21. Hayman AR, et al. 1996. Mice lacking tartrate-resistant acid phosphatase (Acp5) have disrupted endochondral ossification and mild osteopetrosis. Development 122:3151–3162 [DOI] [PubMed] [Google Scholar]
- 22. Hoflack B, Fujimoto K, Kornfeld S. 1987. The interaction of phosphorylated oligosaccharides and lysosomal enzymes with bovine liver cation-dependent mannose 6-phosphate receptor. J. Biol. Chem. 262:123–129 [PubMed] [Google Scholar]
- 23. Infante RE, et al. 2008. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. U. S. A. 105:15287–15292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon HJ, et al. 2009. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 137:1213–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lazzarino DA, Gabel CA. 1988. Biosynthesis of the mannose 6-phosphate recognition marker in transport-impaired mouse lymphoma cells. Demonstration of a two-step phosphorylation. J. Biol. Chem. 263:10118–10126 [PubMed] [Google Scholar]
- 26. Lazzarino DA, Gabel CA. 1989. Mannose processing is an important determinant in the assembly of phosphorylated high mannose-type oligosaccharides. J. Biol. Chem. 264:5015–5023 [PubMed] [Google Scholar]
- 27. Lemansky P, Gieselmann V, Hasilik A, von Figura K. 1985. Synthesis and transport of lysosomal acid phosphatase in normal and I-cell fibroblasts. J. Biol. Chem. 260:9023–9030 [PubMed] [Google Scholar]
- 28. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 29. Lübke T, Lobel P, Sleat DE. 2009. Proteomics of the lysosome. Biochim. Biophys. Acta 1793:625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S. 2011. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science 333:87–90 [DOI] [PubMed] [Google Scholar]
- 31. Meredith MJ. 1988. Rat hepatocytes prepared without collagenase: prolonged retention of differentiated characteristics in culture. Cell Biol. Toxicol. 4:405–425 [DOI] [PubMed] [Google Scholar]
- 32. Möbius W, et al. 2003. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4:222–231 [DOI] [PubMed] [Google Scholar]
- 33. Müller-Loennies S, Galliciotti G, Kollmann K, Glatzel M, Braulke T. 2010. A novel single-chain antibody fragment for detection of mannose 6-phosphate-containing proteins: application in mucolipidosis type II patients and mice. Am. J. Pathol. 177:240–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neuhoff V, Arold N, Taube D, Ehrhardt W. 1988. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9:255–262 [DOI] [PubMed] [Google Scholar]
- 35. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567 [DOI] [PubMed] [Google Scholar]
- 36. Pohl S, et al. 2007. Increased expression of lysosomal acid phosphatase in CLN3-defective cells and mouse brain tissue. J. Neurochem. 103:2177–2188 [DOI] [PubMed] [Google Scholar]
- 37. Pohl S, Marschner K, Storch S, Braulke T. 2009. Glycosylation- and phosphorylation-dependent intracellular transport of lysosomal hydrolases. Biol. Chem. 390:521–527 [DOI] [PubMed] [Google Scholar]
- 38. Rohrbach M, Clarke JTR. 2007. Treatment of lysosomal storage disorders: progress with enzyme replacement therapy. Drugs 67:2697–2716 [DOI] [PubMed] [Google Scholar]
- 39. Ruivo R, Anne C, Sagne C, Gasnier B. 2009. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta 1793:636–649 [DOI] [PubMed] [Google Scholar]
- 40. Saftig P, et al. 1997. Mice deficient in lysosomal acid phosphatase develop lysosomal storage in the kidney and central nervous system. J. Biol. Chem. 272:18628–18635 [DOI] [PubMed] [Google Scholar]
- 41. Saftig P, Klumperman J. 2009. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10:623–635 [DOI] [PubMed] [Google Scholar]
- 42. Schröder B, et al. 2007. Integral and associated lysosomal membrane proteins. Traffic 8:1676–1686 [DOI] [PubMed] [Google Scholar]
- 43. Schröder BA, Wrocklage C, Hasilik A, Saftig P. 2010. The proteome of lysosomes. Proteomics 10:4053–4076 [DOI] [PubMed] [Google Scholar]
- 44. Schulze H, Kolter T, Sandhoff K. 2009. Principles of lysosomal membrane degradation: cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 1793:674–683 [DOI] [PubMed] [Google Scholar]
- 45. Shevchenko A, et al. 1996. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. U. S. A. 93:14440–14445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sleat DE, Zheng H, Qian M, Lobel P. 2006. Identification of sites of mannose 6-phosphorylation on lysosomal proteins. Mol. Cell Proteomics 5:686–701 [DOI] [PubMed] [Google Scholar]
- 47. Sun P, et al. 2008. Acid phosphatase 5 is responsible for removing the mannose 6-phosphate recognition marker from lysosomal proteins. Proc. Natl. Acad. Sci. U. S. A. 105:16590–16595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suter A, et al. 2001. Overlapping functions of lysosomal acid phosphatase (LAP) and tartrate-resistant acid phosphatase (Acp5) revealed by doubly deficient mice. Development 128:4899–4910 [DOI] [PubMed] [Google Scholar]
- 49. Tanaka Y, Tanaka R, Kawabata T, Noguchi Y, Himeno M. 2000. Lysosomal cysteine protease, cathepsin B, is targeted to lysosomes by the mannose 6-phosphate-independent pathway in rat hepatocytes: site-specific phosphorylation in oligosaccharides of the proregion. J. Biochem. 128:39–48 [DOI] [PubMed] [Google Scholar]
- 50. Tiede S, et al. 2005. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat. Med. 11:1109–1112 [DOI] [PubMed] [Google Scholar]
- 51. Varki A, Kornfeld S. 1983. The spectrum of anionic oligosaccharides released by endo-beta-N-acetylglucosaminidase H from glycoproteins. Structural studies and interactions with the phosphomannosyl receptor. J. Biol. Chem. 258:2808–2818 [PubMed] [Google Scholar]
- 52. Varki A, Kornfeld S. 1980. Structural studies of phosphorylated high mannose-type oligosaccharides. J. Biol. Chem. 255:10847–10858 [PubMed] [Google Scholar]
- 53. Wattiaux R, Wibo M, Baudhuin P. 1963. Effect of the injection of Triton WR 1339 on the hepatic lysosomes of the rat. Arch. Int. Physiol. Biochim. 71:140–142 [PubMed] [Google Scholar]
- 54. Winchester B. 2005. Lysosomal metabolism of glycoproteins. Glycobiology 15:1R–15R [DOI] [PubMed] [Google Scholar]
- 55. Xu Z, Farver W, Kodukula S, Storch J. 2008. Regulation of sterol transport between membranes and NPC2. Biochemistry 47:11134–11143 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.