
Table 1. Overview on publications on mAC inhibitors.
| Inhibitor name and structure | Pharmacological key data | Comments | |||
|---|---|---|---|---|---|
MANT-GTP
|
Ki values in AC/GC assay (+ Mn2+): AC1: 90 nM; AC2: 610 nM; AC5: 53 nM; AC6: 91 nM; sAC: > 100 μM; sGC: 710 nM; EF: 1.7 μM; CyaA: 6.5 μM; mouse heart AC: 21 nM. [22,28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-GTP is the reference AC inhibitor for the group of (M)ANT-NTPs. MANT-GTPγS and MANT-GppNHp are hydrolysis-resistant versions of MANT-GTP. MANT-GTPγS and MANT-GppNHp were originally used as G-protein probes, but they possess higher affinities for mACs than for G-proteins [9,22]. MANT-GTPγS has been used as AC5 inhibitor in electrophysiological experiments [60]. MANT-GTP has been widely used for fluorescence studies with purified VC1:IIC2 to characterize both the catalytic and the diterpene site [23-25,28,40]. MANT-GTP has been used in crystallographic studies (PDB:1TL7) [23]. Note the lack of selectivity of MANT-GTP for AC5 relative to AC6. MANT-GTP is commercially available as an experimental tool. | |||
MANT-ITP
|
Ki values in AC assay (+ Mn2+): AC1: 2.8 nM; AC2: 14 nM; AC5: 1.2 nM; mouse heart AC: 4 nM. [28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-ITP is the most potent competitive mAC inhibitor known so far. Based on previous data obtained with ITPγS and MANT-ITPγS [22], the exceptional potency of MANT-ITP at mACs was predicted. MANT-ITPγS has a higher mAC-selectivity relative to G-proteins than MANT-GTPγS [22]. The high affinity of MANT-ITP for mACs is explained by a tight interaction of the triphosphate chain with the protein [25]. However, in electrophysiological experiments, MANT-ITP exhibits off-target effects that are independent of AC inhibition (PDB:3G82) [30], despite its high affinity. MANT-ITP has been used in crystallographic and fluorescence spectroscopy studies [25,28]. The base hypoxanthine is generic and can bind to both adenine- and guanine nucleotide-binding proteins with substantial affinity. MANT-ITP should also be a potent sGC inhibitor. | |||
MANT-ATP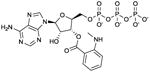
|
Ki values in AC/GC assay (+ Mn2+): AC1: 150 nM; AC2: 330 nM; AC5: 100 nM; AC6: 280 nM; sAC: 5.6 μM; sGC: 430 nM; EF: 230 nM; CyaA: 5.4 μM; mouse heart AC: 64 nM. [22,28,29]. Competitive AC inhibition. Binds to the catalytic site. | On first glance, it was quite unexpected to find that the adenine nucleotide MANT-ATP was not a more potent mAC inhibitor than the guanine nucleotide MANT-GTP [22]. Along the same line, sGC has no preference for guanine nucleotide-based inhibitors. However, with only few mutations substrate- and inhibitor-specificities of ACs and GCs can be switched [36,61]. MANT-ATP gives rise to smaller fluorescence signals with VC1:IIC2 than MANT-GTP, probably due to less favorable positioning of the MANT-group in the hydrophobic pocket [24]. MANT-ATP has been used in crystallographic studies (PDB:2GVZ) [24]. Note the lack of selectivity of MANT-ATP for AC5 relative to AC6. MANT-ATP is commercially available as experimental tool. In electrophysiological studies, MANT-ATP exerts paradoxical stimulatory effects that cannot be explained by AC5 inhibition, but must be due to hitherto unexplained off-target effects [30]. | |||
MANT-UTP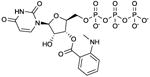
|
Ki values in AC assay (+ Mn2+): AC1: 46 nM; AC2: 460 nM; AC5: 32 nM; mouse heart AC: 12 nM [28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-UTP is a prototypical representative of the pyrimidine-based mAC inhibitors, surpassing the affinities of MANT-ATP. The high-affinity inhibition of mACs by MANT-UTP reflects the broad base-specificity of the catalytic site of these enzymes [24]. MANT-UTP yields distinct fluorescence responses with VC1:IIC2 compared to other MANT-NTPs, reflecting unique positioning of the nucleotide in the catalytic site [28]. MANT-UTP could also be a potent sGC inhibitor. The broad base-specificity of mACs and sGC with a slight preference for the pyrimidine uracil relative to adenine is puzzling. The situation is reminiscent to the identification of UTP-binding P2Y-receptors two decades ago [62]. These UTP-binding GPCRs are now being firmly established. | |||
TNP-ATP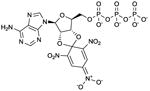
|
Ki values in AC/GC assay (+ Mn2+): AC1: 9.0 nM; AC2: 99 nM; AC5: 3.7 nM; sAC: 710 nM; sGC: 7.3 nM [27]. Competitive AC inhibition. Binds to the catalytic site. | TNP-ATP is the reference AC inhibitor for the group of TNP-NTPs [27].Overall, TNP-NTPs have been less extensively studied than (M)ANT-NTP-based AC inhibitors. TNP-AMP is effectively phosphorylated to TNP-ATP in AC reaction mixtures by the myokinase/pyruvate kinase-based NTP-regenerating system. Phosphorylation can also occur with MANT-NDPs [22]. This must be considered when studying mono- and diphosphates. TNP-NTPs give rise to large basal fluorescence signals upon interaction with purified VC1:IIC2. In contrast to the data obtained with (M)ANT-NTPs, FS reduces fluorescence signals with TNP-NTPs [27,28]. TNP-ATP has been used in crystallographic studies (PDB:2GVD) [24]. Notably, TNP-NTPs are also very potent sGC inhibitors [27]. One could envisage that TNP-NTPs (and possibly MANT-NTPs) are useful tools for achieving an elusive goal in nucleotidyl cyclase research, namely the crystallization of sGC. TNP-ATP binds to numerous types of nucleotide-binding proteins and is not specific for mACs or sGC [27,31]. TNP-ATP is commercially available as experimental tool. | |||
Bis-Cl-ANT-ATP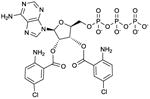
|
Ki values in AC assay (+ Mn2+): AC1: 1.7 μM; AC2: 2.4 μM; AC5: 1.6 μM; CyaA: 16 nM; EF: 220 nM [26,63]. Competitive AC inhibition. Binds to the catalytic site. | Bis-Cl-ANT-ATP serves as reference AC inhibitor for the group of bis-(M)ANT-NTPs. Bis-Cl-ANT-ATP constitutes a very potent CyaA inhibitor with substantial selectivity relative to mACs, indicating that in principle, by targeting the catalytic site, AC isoform-selectivity can be obtained. The catalytic site of EF is less spacious than the catalytic site of CyaA. Hence Bis-Cl-ANT-ATP is a less potent inhibitor of EF than of CyaA [63]. Bis-(M)ANT-nucleotides are also characterized by low basal fluorescence and high signal-to noise ratio in studies with purified CyaA. At mACs, these nucleotides have not yet been examined in fluorescence studied [26,63]. The affinity of mACs for bis-(M)ANT-nucleotides may be too low for fluorescence spectroscopy studies, but the high signal-to-noise ratio of the nucleotides may compensate for this disadvantage The introduction of two (M)ANT groups into an inhibitor increases substantially the number of possible chemical substitutions. This property may facilitate identification of mAC-selective inhibitors. However, spatial constraints in mACs are of concern. | |||
|
UTPγS |
Ki values in AC assay (+ Mn2+): VC1:IIC2: 8.5 μM; AC1: 53 μM; AC2: > 100 μM; AC5: 18 μM; sGC: 4.1 μM; sAC: > 100 μM [22]. Competitive AC inhibition. Binds to the catalytic site. | Originally, UTPγS was described as relatively potent Gs activator, giving rise to AC stimulation [22]. However, at concentrations above 1 μM, UTPγS (like ITPγS) causes strong AC inhibition. UTPγS and ITPγS have been very important tools for unmasking the broad base-specificity of mACs [22,64]. Notably, UTPγS is also a quite potent sGC inhibitor [22]. Based thereon, we assume that uracil nucleotide-based nucleotides can be developed into highly potent sGC inhibitors. For sGC crystallography, such compounds may be most useful. UTPγS is commercially available as experimental tool. | |||
|
2′,5′-dd-3′-ATP |
IC50 values in AC assay (+ Mn2+): AC1: 170 nM; AC2: 280 nM; AC6: 150 nM; AC7: 90 nM; AC8: 150 nM [15]. VC1:IIC2: 38 nM; AC1: 37 nM; AC2: 220 nM; AC5: 37 nM; sAC: 690 nM [22]. Non-competitive/un-competitive AC inhibition. Binds to the catalytic site. | 2′,5′-dd-3′-ATP is a prototypical polyphosphate-containing potent P-site inhibitor of mAC that has also been used in crystallographic studies (PDB:1CUL) [38]. The compound illustrates the difficulties in obtaining AC isoform-selective P-site inhibitors [15]. Notably, the compound is a relatively potent sAC inhibitor [22]. 2′,5′-dd-3′-ATP is commercially available as experimental tool. | |||
AraAde
|
IC50 values in AC assay (+ Mg2+): AC2: 700 μM; AC2: 380 μM; AC5: 9.8 μM [18]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound is a virustatic drug [17] and constitutes a prototypical low-affinity P-site inhibitor. Clinically, potential cancerogenic, embryotoxic and antiproliferative effects of AraAde and structurally related compounds must be taken into consideration. Compounds structurally related to AraAde are supposed to act as selective AC5 inhibitors [17], but AC isoform has not yet been studied in sufficient detail. In addition, the low potency is of concern, increasing the probability of interactions with other targets and toxic effects when used in animals or humans. Also, it is difficult to obtain fully saturated concentration/response curves even in in vitro experiments. AraAde is commercially available as experimental tool. | |||
PMC6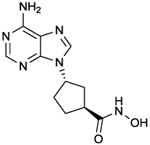
|
IC50 values in AC assay (+ Mg2+): AC2: 85 μM; AC2: 11 μM; AC5: 320 nM [18]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound exhibits substantial selectivity for AC5 relative to ACs 2 and 3, but from molecular modelling studies (Fig. 3C), it remained unclear what the molecular basis for this selectivity may be. Formally, it cannot be excluded that PMC6 also binds to another site than the catalytic site. Similar considerations also apply to AraAde, NKY80 and NB001. Crystallographic studies would be required to answer the question although identification of a binding site beyond the crystallized VC1:IIC2 domains, e.g. in the transmembrane domains, is very challenging. The selectivity of PMC6 for AC6 and other targets (except for ACs 2 and 3) has not yet been examined. In cardiomyocytes, PMC6 exhibits beneficial effects on apoptosis, supposedly mediated via AC5 inhibition [18]. | |||
SQ 22,536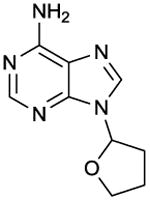
|
IC50 values in AC assay (+ Mg2+): AC1: 120 μM; AC2: 670 μM; AC6: 360 μM; AC8: 120 μM [15]. AC2: 290 μM; AC3: 100 μM; AC5: 2.2 μM [14]. Note the differences in IC50 values between ACs 5 and 6 between the two studies. The difference is difficult to interpret because a direct comparison of both AC isoforms has not yet been performed. Non-competitive AC inhibition. Probably binds to the catalytic site. | SQ 22,536 is one of the first mAC inhibitors developed and was introduced into experimental pharmacology [13]. Considering the structural similarity between ACs 5 and 6, the supposed AC5-selectivity of SQ 22,536 is quite amazing, but a direct side-by-side comparison of SQ 22,526 at both ACs 5 and 6 has not yet been presented. A problem in the analysis of SQ 22,536 is its rather low potency so that it is difficult to obtain fully saturated inhibition curves for all mAC isoforms. SQ 22,536 is probably the most-widely used mAC inhibitor, specifically with respect to intact cell studies. A pubmed research on September 15, 2011, revealed 377 entries with the key word “SQ 22,536”. In many studies the specificity of SQ 22,536 for mACs is taken for granted without considering off-target effects. However, considering the rather low potency of SQ 22,536, such effects cannot be excluded. Conversely, in some studies, SQ 22,536 was used at rather low concentrations (1 μM) [65] that may be insufficient to inhibit AC and prevent cAMP formation. SQ 22,536 is commercially available as experimental tool. | |||
NKY80
|
IC50 values in AC assay (+ Mg2+): AC2: 2.6 mM; AC3: 230 μM; AC5: 15 μM [18]. AC2: 1.7 mM; AC3: 130 μM; AC5: 8.3 μM [14]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound constitutes a prototypical low-affinity P-site inhibitor and was identified in a virtual screen of > 850,000 compounds [14]. The compound is marketed as “selective AC5 inhibitor” for experimental purposes, but AC isoform has not yet been studied in sufficient detail. In fact, there are doubts whether NKY80 is selective for AC5 relative to AC6 [20]. In addition, the low potency of NKY80 for AC5 is of concern, increasing the probability of interactions with other targets. The lack of effects of NKY80 in a typical AC5 system has also been noted [18]. Due to the low affinity of NKY80 for mACs, it is difficult to obtain fully saturated concentration/response curves for precise calculation of IC50 values [14]. NKY80 is commercially available as experimental tool. | |||
NB001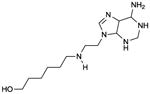
|
IC50 values in cAMP accumulation assay in intact transfected HEK293 cells: AC1: 10 μM; AC5: 210 μM; AC6: 170 μM; AC7: 190 μM; AC8: 140 μM [8]. Molecular mechanism undefined. Probably binds to the catalytic site. | The molecular mechanism of AC inhibition by NB001 has not yet been demonstrated because AC activity studies with membranes have not yet been performed. Only studies with intact cells have been performed. It cannot be taken for granted that cAMP accumulation assays in intact cells reflect AC activity even if phosphodiesterases are blocked. Specifically, intracellularly formed cAMP may be exported from cells via multidrug resistance proteins [66], introducing bias into the experimental setting. This specific experimental design renders comparison with other AC inhibitors that are routinely tested in the broken cell AC assay difficult. Based on our modelling (Fig. 3C), NB001 presumably acts as a non-competitive P-site inhibitor. However, our modelling failed to reveal the molecular basis for the AC1-selectivity. NB001 is a low-affinity mAC inhibitor, raising questions whether in addition to AC1, the compound also interacts with other targets in mammalian cells. The paper by Wang et al. [8] does not provide information on structure/activity relationships for AC inhibition by NB001 and related compounds. | |||
MDL 12330A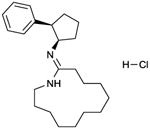
|
MDL 12330 inhibits histamine-stimulated AC activity in the guines pig ventricle according to a biphasic concentration/response curve. IC50-1 ∼20 μM; IC50-2 ∼ 300 μM [67]. MDL 12330 (100 μM) shows modest inhibition of ACs 2 and 3, but not of AC5 [14]. However, complete concentration-response curves have not yet been presented. | MDL 12330A, like SQ 22,536, is one of the most widely used mAC inhibitor for intact cell studies. A pubmed research on September 15, 2011, revealed 166 entries with the key word “MDL 12330A”.MDL 12330A is a non-nucleoside-based mAC inhibitor. It was introduced into experimental pharmacology into the early 1980s [67]. It is very well documented that in addition to AC inhibition, the compound exhibits numerous pleiotropic effects including inhibition of Na+/K+-ATPase and phosphodiesterases [16,67,68]. Nonetheless, in several studies using MDL 12330A, specificity for mAC inhibition is assumed although exceedingly high concentrations (up to 10 mM) are used [69]. Overall, MDL 12330A has not yet been thoroughly examined at mAC isoforms. In some studies, MDL 12330A exerts also stimulatory effects on AC [70], a possible indication for isoform-specific effects like with BODIPY-FS [11,34]. MDL 12330A is commercially available as experimental tool. | |||
BODIPY-FS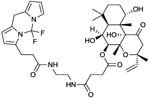
|
EC50 values in AC assay (+ Mn2+): AC1: 2.9 μM; AC5: 2.3 μM. IC50 value in AC assay (+ Mn2+): AC2: 1.2 μM [34]. The effects of BODIPY-FS strongly depend on the experimental conditions, the type of divalent cation being a critical determinant. In a follow-up study [11], the following data were obtained: EC50 values in AC assay (+ Mn2+): AC1: 1.2 μM; AC5: 2.7 μM. IC50 value in AC assay (+ Mn2+): AC2: 170 nM. EC50 values in AC assay (+ Mg2+): AC1: 900 nM; AC5: 24 μM. IC50 value in AC assay (+ Mg2+): AC2: 500 nM. Binds to the diterpene site. | BODIPY-FS is a partial agonist (compared to FS) in terms of AC1- and AC5 activation, but reduces the catalytic activity of AC2 in the presence of GTPγS [11,34]. The potencies and efficacies of BODIPY-FS are strongly determined by the divalent cation. Most notably, in the presence of Mg2+, the inhibitory effect of BODIPY-FS is much more pronounced than in the presence of Mn2+ [11]. BODIPY-FS is only weakly effective at activating VC1:IIC2 but inhibits the stimulatory effect of FS (3 μM) with an IC50 of 800 nM [34]. BODIPY-FS is a fluorescent molecule and has been studied to a very limited extent to localize mACs in cells [41]. BODIPY-FS could also be used as fluorescence probe for VC1:IIC2, but so far, no data have been published. BODIPY-FS was withdrawn from the market because of lack of demand, but because of the renewed interest in the compound, it is now again commercially available as experimental tool. Other fluorescent groups than BODIPY have not yet been assessed, but linkage of the FS core to other fluorophores is technically feasible. Thus, BODIPY-FS serves as a promising starting point for extensive structure/activity relationship studies. Based on the experimental data [11,34] and molecular modelling (Figs. 3A and B), the development of mAC isoform-selective diterpenes is anticipated. The BODIPY substituent increases the affinity of FS for mACs substantially. As control reagent for BODIPY-FS, the free dye, BODIPY was used [11,34]. This compound is devoid of effects on AC activity. | |||
1,9dd-FS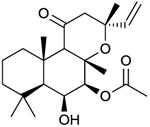
|
No activation of VC1:IIC2, AC1, AC2 or AC5 but apparently non-competitive inhibition of FS-stimulated catalysis of mACs [34,40]. Binds to the diterpene site. A recent study [41] suggests 1,9dd-FS can also exert stimulatory effects on as yet unidentified mAC isoforms | Historically, the compound was considered not to interact with mACs [4,39]. However, fluorescence studies with MANT-GTP at VC1:IIC2 clearly demonstrated that 1,9dd-FS (and other “inactive” 1d-FS derivatives) bind to the diterpene site without activating catalysis [34,40]. The non-competitive inhibition is probably due to the slow exchange of FS and 1,9dd-FS. Concentration-response curves for the inhibitory effects of 1,9dd-FS have not yet been performed. Only a fixed 1,9-dd-FS concentration of 100 μM was studied. 1,9dd-FS is very lipophilic so that very high concentrations of dimethyl sulfoxide (up to 6%, v/v) have to be used in experiments. Fortunately, mACs expressed in Sf9 cells and VC1:IIC2 are very dimethyl sulfoxide-resistant. In fact, we recommend to use high dimethyl sulfoxide concentrations in experiments with 1,9dd-FS in particular and with diterpenes in general because otherwise, incomplete dissolution of compounds introduces substantial experimental errors. The poor water-solubility of 1,9dd-FS limits the use of this compound in intact cell studies. 1,9dd-FS is commercially available as experimental tool. | |||
6A7DA-FS
|
AC assay in the presence of Mg2+:AC1: EC50, 6.5 μM. AC2: IC50, 1.8 μM; EC50, 61 μM. AC5: EC50, 52 μM [11]. The inhibitory effect of 6A7DA on AC2 in the presence of Mg2+ is relatively small and is not observed in the presence of Mn2+. Binds to the diterpene site. | 6A7DA is also referred to as iso-forskolin because the acetyl group is switched from the 7-position to the 6-position of the diterpene ring. In the presence of Mn2+, FS and 6A7DA-FS are similarly potent activators of ACs 1, 2 and 5, but there are differences in efficacy. In the presence of Mg2+, FS and 6A7DA-FS are similarly potent activators of AC1, but at AC5, 6A7DA is about ten-fold less potent than FS [11]. Most notably, in the presence of Mg2+, 6A7DA-FS exerts high-potency inhibitory effects and low-potency stimulatory effects on AC2 [11]. These data corroborate the unique position of AC2 among mACs in terms of inhibition. Biphasic effects on mACs were also observed for calmidazolium [12]. | |||
Calmidazolium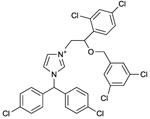
|
IC50 values in AC assay (+ Mg2+): mouse AC9: ∼6 μM; human AC9: ∼15 μM; rat AC2, ∼30 μM; soluble C1-C2 fusion protein from AC9: 8 μM [12]. Binds to a still undefined site. The IC50 of calmidazolium for calmodulin in a fluorescence binding assay is 2-3 nM [71]. | Calmidazolium is a well-known high-affinity calmodulin antagonist [71]. However, calmidazolium also binds to other proteins exhibiting hydrophobic sites, i.e. many effects of calmidazolium are actually calmodulin-independent [72]. The precise molecular mechanism by which calmidazolim inhibits mAC activity has not yet been determined [12], but it is not dependent on calmodulin. Notably, at a concentration of ∼5 μM, calmidazolium stimulates human AC9 by up to ∼60%. At a concentration of ∼2-3 μM, calmidazolium stimulates mouse AC9 by up to ∼70%. At a concentration of 10 μM, calmidazolium increases AC2 activity even by ∼220%. At higher concentrations, very steep inhibition isotherms are observed for all mACs studied so far, indicative for cooperative inhibitor binding. The peculiar (biphasic and steep) concentration-response curves for calmidazolium could point to the existence of multiple ligand binding sites or to a single binding site adopting two affinity states and displaying positive cooperativity. To our knowledge, this is the first study in which species-dependent sensitivity of a mAC to activators and inhibitors has been observed. Recently, we observed biphasic stimulatory and inhibitory effects of 6A7DA-FS on AC2 [11]. Binding of calmidazolium to hydrophobic sites in proteins other than calmodulin has been repeatedly reported [72]. Hence, again, target-specificity of a mAC inhibitor is of concern. The concentrations required to inhibit mACs are considerably higher than these needed to block calmodulin function [71]. Calmidazolim is commercially available as experimental tool. Calmidazolium may represent an archetype of allosteric mAC inhibitor targeting as yet unexplored sites that may encompass the poorly studied transmembrane domains. | |||
Tyrphostin A25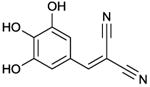
|
IC50 value in AC/GC assay (+ Mn2+): GC-C: 5.8 μM; sGC: 34 μM; AC-HEK cells: 120 μM; C1-C2 fusion protein (C1 from AC1 and C2 from AC2): 16 μM [59]. Binds to a still undefined site. | The precise molecular mechanism by which tyrphostin A25 inhibits mAC and GC activity has not yet been determined [59]. Tyrphostin A25 has been suggested to bind to hydrophobic residues close to the catalytic site [59]. An interesting aspect of [59] concerns the fact that this is, to our knowledge, the only report reporting on inhibition of a particulate GC in comparison to mACs. The area of particulate GC inhibition needs to be developed much more intensively. Tyrphostin A25 is commercially available as experimental tool. Tyrphostin may represent an archetype of allosteric mAC inhibitor targeting as yet unexplored sites that may encompass the poorly studies transmembrane domains. | |||
In the Table, representative mAC inhibitors from various chemical classes are listed. Structures and pharmacological key data are shown, and specific compound properties and problems are discussed. Please, note that due to space limitations, the list of inhibitors is not comprehensive. This table focuses only on these inhibitors that have been examined at various ACs (and GCs). Inhibition data of (M)ANT- and TNP-nucleotides for VC1:IIC2 are listed in Fig. 2F [22,24,28]. It should be noted that the potencies of mAC inhibitors differ vastly from each other. For example, MANT-ITP inhibits AC5 with a Ki of a bout 1 nM, whereas the “AC5-selective inhibitor” NKY80 inhibits AC5 with an IC50 of about 10 μM, i.e. the potency difference amounts to about 10,000-fold. Even the highly potent MANT-ITP exhibits effects unrelated to AC5 [30], rendering it likely that much less potent mAC inhibitors such as NKY80 and NB001 also exhibit off-target effects. The possibility of off-target effects of mAC inhibitors has not yet been comprehensively studied and must be carefully examined in future studies. For competitive inhibitors, Ki values are given; for non-competitive inhibitors, IC50 values are given.