

Abstract
Sindbis virus (SBV) has been shown to possess oncolytic potential in many human xenograft tumor models in immunocompromised mice. However, the mechanism underlying the tumor selectivity of SBV remains undetermined. In this study, we provide evidence that the tumor tropism of SBV infection is not determined by the levels of SBV receptor but by the status of the type I interferon (IFN) response in the tumors. Our results demonstrate that cells with defects in the IFN response (in either IFN-β production or IFN signaling) were highly susceptible to SBV infection in vitro. The results of oncolysis experiments conducted in immunocompetent animals further confirmed that the success of SBV-mediated oncolysis is greatly dependent on the presence of defects in IFN signaling in tumors. In all cases, viral titers rapidly declined in tumors due to host immune responses in immunocompetent animals. Interestingly, however, tumor-specific immune responses were concomitantly elicited, which might contribute to the sustained antitumor effect observed after the clearance of SBV. These findings indicate that SBV-mediated virotherapy is a promising therapeutic strategy for cancers defective in the IFN response and underscore the importance of bystander antitumor immunity in the efficacy of this virotherapy.
Introduction
One major obstacle to the development of gene therapy for solid tumors is the low transduction efficiency of replication-defective viral vectors or nonviral vectors. Oncolytic viruses, which undergo tumor-selective replication and lead to tumor cell destruction, provide an attractive new tool because they are capable of replicating and spreading efficiently within tumors.
Sindbis virus (SBV) has been shown to have great oncolytic potential of eradicating several types of human and mouse tumors in immunocompromised mice.1,2,3,4,5 SBV belongs to the Alphavirus genus of the Togaviridae family and has a positive sense, single-stranded RNA genome. SBV infects many animal species such as mosquitoes and birds and causes very mild symptoms, if any, in humans.6 The underlying mechanism of the tumor selectivity of SBV is not clear; however, it has been hypothesized to be caused by the higher levels of laminin receptor, the receptor for SBV, observed in numerous tumors as compared to normal cells.7 In fact, there are two cellular receptors for SBV, the laminin receptor and the heparin sulfate proteoglycan.8,9 Thus, the fact that normal cells have lower levels of laminin receptor than tumor cells does not explain why SBV replication fails in normal cells.
Many RNA viruses, such as Newcastle disease virus and vesicular stomatitis virus (VSV), are interferon (IFN)-sensitive and have been shown to exert significant oncolytic effects in IFN-defective tumor cells.10,11,12 Therefore, we hypothesized that a defective type I IFN response may be the major determinant of the tumor tropism of SBV because it is also sensitive to IFN.13,14,15,16 Type I IFNs, including IFN-α and IFN-β, can activate hundreds of IFN-stimulated genes (ISGs),17 which constitute the first line of host defense against viral infection. The IFN response consists of the early phase of IFN-β production and the late phase of IFN signaling. IFN-β production is triggered via the recognition of viral nucleic acids by host Toll-like receptors, retinoic acid inducible gene-I (RIG-I), or melanoma differentiation-associated gene 5 (MDA5),18,19,20 which then transduce these signals to activate the IFN-β promoter. The IFN-β produced binds to the IFN receptor (IFNAR) in an autocrine or a paracrine manner and activates the JAK-STAT signaling pathway, leading to the expression of ISGs.
In this study, we investigated the mechanism underlying the tumor tropism of SBV. Our results do not support the assumption that the levels of SBV receptor determine tumor selectivity but indicate that the status of the IFN response, particularly in terms of IFN signaling, is critical to the success of Sindbis virotherapy in vivo. Our data also demonstrate that apart from its oncolytic activity, SBV-mediated oncolysis induces a bystander antitumor immunity that continuously constrains tumor growth after SBV clearance.
Results
Cellular susceptibility to SBV infection is not determined by the levels of SBV receptor
We first determined the tumor selectivity of SBV. The susceptibilities of several cell lines or primary cells to SBV infection were examined using a recombinant SBV that expresses enhanced green fluorescence protein (EGFP). As shown in Figure 1a, the cell lines were roughly divided into two groups, one with high susceptibility (>50% infectivity) and the other with low susceptibility (<50% infectivity). Of the primary cells, all but STKO, a STAT1-knockout mouse embryonic fibroblast cell line, had low susceptibilities to SBV infection. The EGFP expression was strongly correlated with SBV RNA replication, revealed by the northern blot which showed that the SBV RNA species were expressed in abundant levels in representative cell lines with high susceptibilities, but were low in those with low susceptibilities (Figure 1d). Moreover, robust SBV replication in cell lines with high susceptibilities resulted in significant cell death (Supplementary Figure S1).
Figure 1.
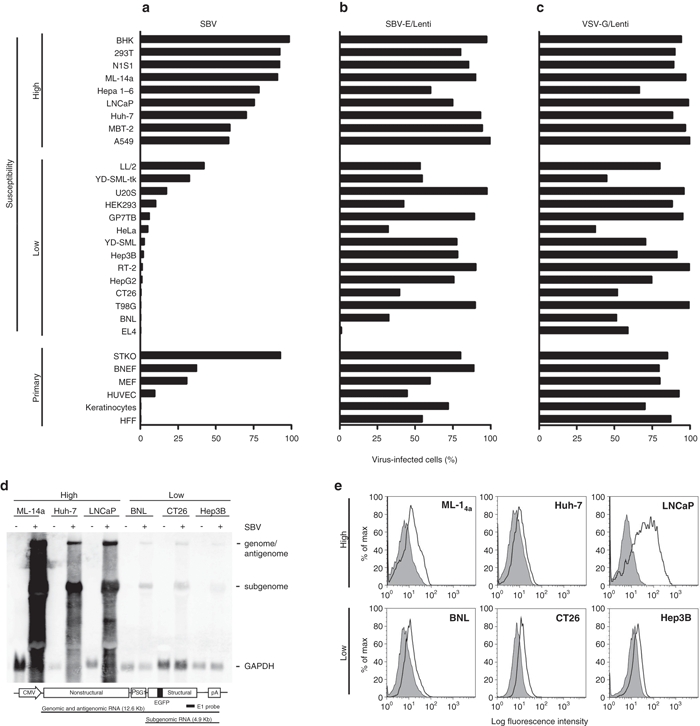
Cellular susceptibility to SBV infection. (a) Various cell lines were infected with EGFP-expressing SBV, (b) EGFP-expressing lentivirus pseudotyped with SBV envelope proteins (SBV-E/Lenti), or (c) EGFP-expressing lentivirus pseudotyped with VSV glycoprotein-G (VSV-G/Lenti), at a multiplicity of infection of 5. The percentages of virus-infected cells were determined by flow cytometry. Cell lines with SBV-infectivity >50% were classified as having high susceptibility to infection, whereas those with SBV-infectivity <50% were classified as having low susceptibility. Some primary cells were also used for infectivity assays, including STAT1-knockout mouse embryonic fibroblasts (STKO), Brown Norway rat embryonic fibroblasts (BNEF), mouse embryonic fibroblasts (MEF), human umbilical vein endothelial cells (HUVECs), human keratinocytes, and human foreskin fibroblasts (HFF). Data presented are representatives of three independent experiments with similar results. (d) Northern blot showing the SBV replicative RNA species. Five micrograms of cellular RNA from the indicated cell lines with (+) or without (−) SBV infection were analyzed by northern blot using the SBV E1 and the GAPDH probes. The region of the E1 probe is indicated at the bottom of the figure. (e) Surface expression of laminin receptor. Representative cell lines with extremely high or low susceptibilities to SBV infection were analyzed for their surface expression of laminin receptor using the monoclonal antibody MLuC5. The black lines show the expression levels of laminin receptor and the gray areas show the background levels. GAPDH, glyceraldehydes-3-phosphate dehydrogenase; SBV, Sindbis virus; VSV, vesicular stomatitis virus.
Based on these cell lines, we examined whether the various susceptibilities to SBV infection were caused by various levels of SBV receptor expression on the cells, as previously suggested.7 An EGFP-expressing lentiviral vector pseudotyped with the SBV envelope protein (SBV-E/Lenti) was used to infect these cell lines. The results showed that the SBV-E-pseudotyped lentiviruses could enter and express EGFP in most cells, regardless of their susceptibilities to SBV infection (Figure 1b); these results are reminiscent of the infection pattern of VSV-G/Lenti, which are lentiviruses pseudotyped with VSV glycoprotein-G (Figure 1c). We further examined the surface expression of laminin receptor in some representative cell lines with extremely high or low susceptibilities to SBV infection. As shown in Figure 1e, the receptor was expressed in all these cell lines; however, the expression levels were not correlated with their susceptibilities. These results thus demonstrate that SBV receptor is widely expressed on the surface of many cells and exclude the possibility that various levels of SBV receptor expression account for the various susceptibilities of these cell lines to SBV infection.
Cell lines with defects in IFN-β production or IFN signaling are all susceptible to SBV infection
Since SBV is highly sensitive to IFN activity,13,14,15,16 we hypothesized that cells that are susceptible to SBV infection may have defects in eliciting the IFN response, either in IFN-β production or in IFN signaling, thus allowing for robust SBV replication. To test this hypothesis, we chose several mouse and human cell lines with extremely low or high susceptibilities to SBV infection and examined their abilities to synthesize IFN-β upon poly(I:C) stimulation. Cells were transfected with poly(I:C), and IFN-β mRNA expression was analyzed using quantitative RT-PCR (qRT-PCR). As shown in Figure 2a, all of the cell lines with low susceptibilities to SBV infection experienced a more than 100-fold increase in IFN-β expression upon stimulation, whereas all those with high susceptibilities to SBV infection (except LNCaP) had tenfold lower inductions of IFN-β. Conditioned medium from the poly(I:C)-treated cells had inhibitory effects on SBV replication (which was strongly correlated with IFN-β production levels) in mouse ML-14a and human 293T cells, both of which have intact IFN signaling (vide infra) (Figure 2b). Interestingly, although LNCaP cells express high levels of IFN-β, and although the conditioned medium also strongly inhibited SBV replication in 293T cells, LNCaP themselves are highly susceptible to SBV infection (Figure 1a). The results suggest that LNCaP cells may have defects in IFN signaling rather than IFN-β production.
Figure 2.
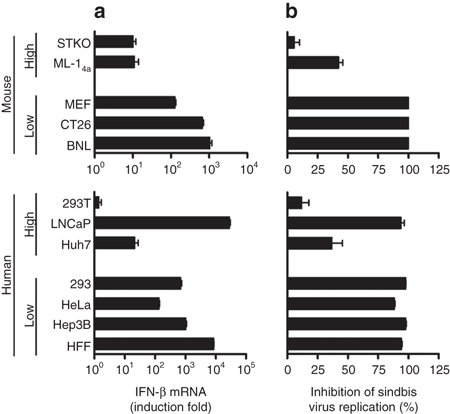
Correlation of low IFN-β production with high susceptibility to SBV infection. Representative mouse and human cell lines with extremely low or high susceptibility to SBV infection were examined with respect to their abilities to synthesize IFN-β in response to poly(I:C) stimulation. (a) IFN-β production. Cells were left untreated or transfected with poly(I:C) (1 µg/ml) for 6 hours. Total RNA was isolated from the control or poly(I:C)-transfected cells, and the levels of IFN-β mRNA were determined by qRT-PCR. The results are expressed as fold induction, with the IFN-β mRNA levels of poly(I:C)-treated cells divided by those of the control cells. (b) Inhibition of SBV replication by conditioned medium. Representative cell lines were transfected with poly(I:C) for 6 hours. Fresh medium was then placed on the cells, and they were incubated for another 18 hours. The conditioned medium from the poly(I:C)-treated mouse or human cells was collected, diluted with fresh medium at a 1:1 ratio, and then applied to ML-14a or 293T cells, respectively. After an overnight incubation, the cells were challenged with EGFP-expressing SBV for 24 hours. EGFP(+) cells were analyzed by flow cytometry. Data were presented as percentage of inhibition of SBV infection, calculated using the formula: [(A − B)/A] × 100%, where A is the percentage of EGFP(+) cells from cells treated with the control medium, and B is the percentage of EGFP(+) cells from cells treated with the conditioned medium. EGFP, enhanced green fluorescence protein; IFN, interferon; SBV, Sindbis virus.
Therefore, we further examined the intactness of IFN signaling in these cells. Cells were treated with IFN-α, and the mRNA expression of two ISGs, ISG56 and ZAP, was analyzed using qRT-PCR (Figure 3a and Supplementary Figure S2, respectively). The IFN-α-treated cells were also challenged with SBV to determine whether IFN signaling was induced to inhibit SBV replication (Figure 3b). In mouse STKO cells, significantly lower levels of ISG56 and ZAP mRNA were induced, and the cells had lower ability to inhibit SBV replication upon IFN stimulation, which is in agreement with their lack of STAT1. The human LNCaP cells had a similar phenotype to STKO cells, supporting our aforementioned hypothesis. All the other cell lines expressed reasonable levels of ISG56 or ZAP and significantly inhibited SBV replication, suggesting that they have normal IFN signaling. Collectively, these data indicate that cell lines with high susceptibilities to SBV infection do indeed possess defects in the IFN response. ML-14a, Huh-7, 293T, and STKO cells have partial defects in IFN-β production, whereas STKO and LNCaP cells have defects in IFN signaling.
Figure 3.
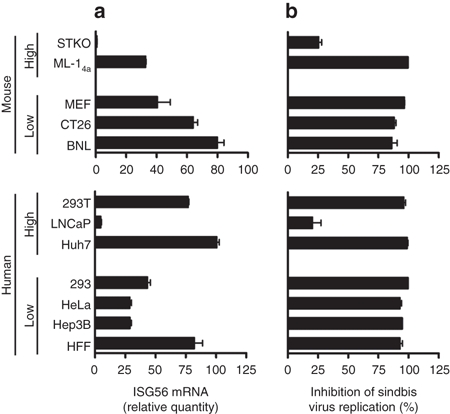
Correlation of poor IFN signaling with high susceptibility to SBV infection. Representative mouse and human cell lines with extremely low or high susceptibilities to SBV infection were examined for the presence of intact IFN signaling upon IFN-α stimulation. Cells were left untreated or treated with IFN-α (100 IU/ml) for 24 hours. (a) ISG56 mRNA expression, reflecting the status of IFN signaling, was determined in various cell lines. Total RNA was isolated from the IFN-α-treated cells, and the levels of ISG56 mRNA were determined by qRT-PCR. (b) Control or IFN-α-treated cells were challenged with luciferase-expressing SBV for 24 hours, and cell lysates were analyzed for luciferase activity. The results were presented as percentage of inhibition of SBV infection, calculated based on the formula: [(A − B)/A] × 100%, where A is the luciferase activity from cells without IFN-α treatment, and B is the luciferase activity from cells treated with IFN-α. IFN, interferon; SBV, Sindbis virus.
Modulation of the IFN response alters susceptibility to SBV infection
To confirm that the IFN response plays a crucial role in determining the cell tropism of SBV infection, we used genetic approaches to modulate the IFN response in cells. Hep3B cells have a low susceptibility to SBV infection (Figure 1a). However, when the expression of MDA5 was knocked down in Hep3B (Figure 4a), their ability to express IFN-β upon poly(I:C) stimulation was significantly attenuated (Figure 4b), leading to an increased susceptibility to SBV infection (Figure 4c). Similarly, when STAT1 was knocked down (Figure 4d), the ability of Hep3B cells to induce ISG56 mRNA expression upon IFN-α stimulation was also reduced (Figure 4e). As a result, SBV infection significantly increased (Figure 4f).
Figure 4.
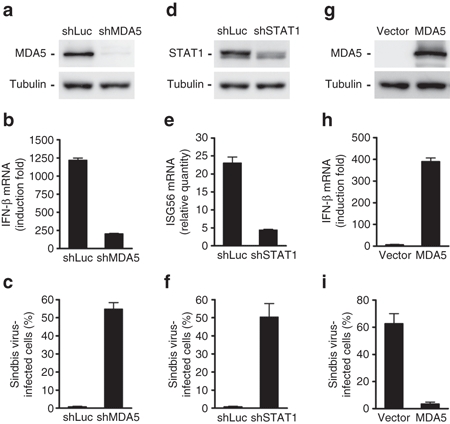
Modulation of the IFN response can change cellular susceptibility to SBV infection. Hep3B cells, which are not susceptible to SBV infection, were converted to be susceptible by knocking down the expression of MDA5 or STAT1. Conversely, 293T cells, which are susceptible to SBV infection, were converted to be more resistant to SBV infection by overexpressing MDA5. (a,d) The protein levels of MDA5 or STAT1, respectively, in Hep3B cells knocked down by shRNA. (b) Reduced IFN-β mRNA expression in response to poly(I:C) stimulation in the Hep3B cells with MDA5 knockdown. (e) Reduced ISG56 mRNA expression in response to IFN-α stimulation in Hep3B cells under STAT1 knockdown. (c,f) Increased susceptibility to SBV infection in Hep3B cells under MDA5 or STAT1 knockdown, respectively. (g) Protein levels of ectopic MDA5 expression in 293T cells. (h) Increased IFN-β mRNA synthesis in response to poly(I:C) stimulation in 293T cells overexpressing MDA5. (i) Reduced susceptibility to SBV infection in 293T cells overexpressing MDA5. IFN, interferon; SBV, Sindbis virus.
Contrarily, 293T cells are highly susceptible to SBV infection due to partial defects in IFN-β production (Figure 2a). We later discovered that 293T cells expressed extremely low levels of MDA5. Therefore, when MDA5 was overexpressed in 293T cells (Figure 4g), the cells regained the ability to express IFN-β upon poly(I:C) stimulation (Figure 4h) and became resistant to SBV infection (Figure 4i). Taken together, these data confirm that the IFN response plays a major influential role in determining the cell tropism of SBV infection.
Defects in IFN signaling are important to the success of Sindbis virotherapy in vivo
Animal experiments were performed to investigate how the IFN response of tumor cells might influence the antitumor effects of Sindbis virotherapy in vivo. ML-14a is a mouse hepatoma cell line that is partially defective in IFN-β production (Figure 2a) but intact in IFN signaling (Figure 3a). As a result, when implanted in vivo and treated by Sindbis virotherapy, ML-14a cells may produce low levels of IFN-β but can respond well to the IFNs secreted by the surrounding normal cells. As a result, these tumor cells may still be capable of mounting an antiviral response and reducing the efficacy of virotherapy. To test this possibility and to demonstrate that cells defective in IFN signaling are better targets for virotherapy in vivo, we established a stable IFN receptor 1 (IFNAR1)-knockdown cell line, ARKD, based on the ML-14a cell line. ARKD retained only ~20% of the original level of IFNAR1 expression (Supplementary Figure S3a). Thus, upon IFN-α stimulation, IFN signaling and ISG56 expression were reduced in ARKD as compared to parental ML-14a cells; these cells were also less able to inhibit SBV replication (Supplementary Figure S3b,c).
Both ML-14a and ARKD cells have same genetic background and similar growth rates in vivo (Figure 5a,b, the phosphate-buffered saline (PBS)-treated groups), thus enabling easier interpretation of the role of IFN signaling in Sindbis virotherapy. BALB/c mice bearing subcutaneous ML-14a or ARKD tumors were treated with a single dose of SBV (1 × 107 PFU/mouse) or PBS intratumorally on day 7 after tumor implantation, and tumor growth was followed for up to 41 days. The results showed that Sindbis virotherapy significantly retarded tumor growth in both tumor models as compared to the PBS control (Figure 5a,b). In contrast, it had no effect on the BNL tumor model (Figure 5c), a mouse hepatoma cell line with normal IFN response (Figures 2a and 3a). A lower dose of SBV (1 × 105 PFU/mouse) or an UV-inactivated SBV (1 × 107 PFU/mouse) exerted no therapeutic effect on the ML-14a or ARKD tumors (Figure 5a,b). Notably, the volumes of ARKD tumors were significantly smaller than those of ML-14a tumors (P < 0.001, Figure 5d). The SBV-induced oncolysis was demonstrated by the staining of apoptotic cells in the tumor region. Both SBV-treated ML-14a and ARKD tumors exhibited higher levels of apoptosis than did the PBS-treated tumors; the apoptotic cells in the ARKD tumors were also significantly higher than those in the ML-14a tumors (Figure 5e and Supplementary Figure S4).
Figure 5.
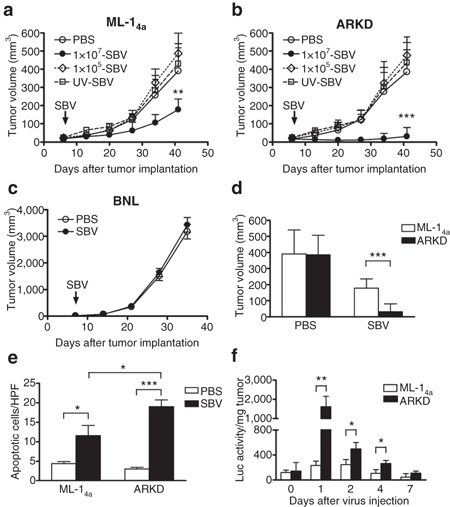
Defective IFN signaling in tumors facilitates Sindbis virotherapy in immunocompetent mouse models. Sindbis virotherapy in (a) the ML-14a tumor mode, (b) the ARKD tumor model, and (c) the BNL tumor model. Tumor cells (5 × 105/mouse for ML-14a and ARKD, or 3 × 105/mouse for BNL) were subcutaneously inoculated in syngenic BALB/c mice to generate tumors. A single dose of SBV (1 × 107 PFU/mouse) or PBS was intratumorally injected on day 7 after tumor implantation. In panels a and b, a lower dose of SBV (1 × 105 PFU/mouse) and an UV-inactivated SBV (1 × 107 PFU/mouse) were also injected as controls. Tumor growth was monitored weekly. (d) Tumor sizes at day 41 were compared between the ML-14a and the ARKD tumor models. (e) SBV induced apoptosis in tumors. Animals were killed seven days after SBV virotherapy and tumor cell apoptosis was analyzed by TUNEL assay. Data presented are the quantification of apoptotic cells from three high power fields (HPFs) per tumor and three tumors per group. (f) Viral loads in the tumors. BALB/c mice bearing ML-14a or ARKD tumors were intratumorally treated with SBV expressing a luciferase reporter. Animals were killed at the time indicated, and the luciferase activity was measured in the tumor lysates to assess SBV replication. The results are expressed as relative luciferase activity per mg tumor tissue. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test. PBS, phosphate-buffered saline; SBV, Sindbis virus.
To demonstrate the relationship between viral loads and tumor sizes, we used a recombinant SBV expressing luciferase to treat the tumors, in which system viral replication is required for the expression of luciferase. At the time indicated, tumor tissues were harvested, and luciferase activity in tumor lysates was measured to determine viral loads. As shown in Figure 5f, the luciferase activity in ARKD tumors was significantly higher than that in ML-14a tumors on days 1, 2, and 4 after virus injection. It is worth mentioning that viral replication in ML-14a or in ARKD tumor cells was comparably high in vitro (data not shown). These results imply that tumors defective only in IFN-β production may not be sufficiently susceptible to SBV replication in vivo and that additional defects in IFN signaling would make tumors more vulnerable to SBV-mediated oncolysis.
Sindbis virotherapy induces bystander antitumor immune responses
We noted that the SBV viral loads rapidly (in about a week) declined in both tumor models (Figure 5f), implying that immune mechanisms rapidly clear SBV in immunocompetent animals. However, the tumor volumes remained small for a period of time even after SBV had disappeared; this effect was particularly evident in the ARKD tumor model (Figure 5b). We interpreted that a bystander antitumor immunity might have been induced by SBV-mediated oncolysis. Therefore, we examined immune cell infiltration in tumors treated with Sindbis virotherapy. Both ML-14a and ARKD tumors treated with SBV displayed higher levels of CD8+ and CD4+ cell infiltration compared with the PBS-treated tumors (P < 0.05, Figure 6a and Supplementary Figure S5), suggesting that immune responses were induced. To demonstrate whether these immune responses involve antitumor immunity or not, the splenocytes of the animals bearing ML-14a or ARKD tumors were isolated on day 7 after virotherapy, and ML-14a- or ARKD-specific cytotoxic T lymphocyte activity was assayed. In both ML-14a and ARKD tumors treated with SBV, an enhanced cytotoxic T lymphocyte response was induced compared to the control (Figure 6b). The cytotoxic T lymphocyte response was specific to ML-14a/ARKD tumors because it had no effect on the syngenic BNL hepatoma cells (Supplementary Figure S6). To further confirm that T cell immune responses were involved in the SBV-induced antitumor effect, we depleted CD8+ T cells in the mice bearing ARKD tumors which were treated with SBV virotherapy. The depletion efficiency of CD8+ T cells was >97% as determined by flow cytometric analysis (Figure 6c). The results showed that CD8+ T cells depletion significantly impaired the antitumor effect of SBV virotherapy, compared with the mice treated with an IgG2a isotype control antibody (P < 0.001, Figure 6d). These results strongly suggested that Sindbis virotherapy induces the development of tumor-specific immune responses that may continuously restrict tumor growth even after the cessation of SBV-mediated oncolysis.
Figure 6.
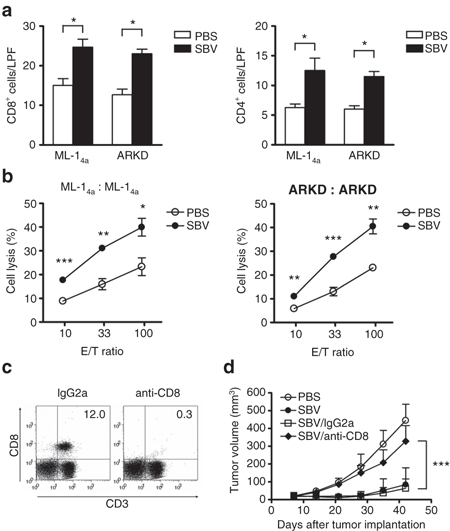
Sindbis virotherapy generates tumor-specific immune response. Tumor cells (5 × 105/mouse) were subcutaneously inoculated in syngenic BALB/c mice on day 0. A single dose of SBV (1 × 107 PFU/mouse) or PBS was intratumorally injected on day 7. Animals were killed on day 14 for the analysis of the Sindbis virotherapy-induced immune response. (a) Increased numbers of tumor-infiltrating CD8+ and CD4+ lymphocytes in both ML-14a and ARKD tumors treated with Sindbis virotherapy. Data presented are the quantification of IHC staining results from three low power fields (LPFs) per tumor and three tumors per group. (b) Enhancement of tumor-specific CTL response in the Sindbis virotherapy-treated animals. Mouse splenocytes were prepared from the mice bearing ML-14a or ARKD tumors. They were stimulated with mitomycin C-treated respective tumor cells for 5 days. The stimulated splenocytes were recovered and incubated with fresh ML-14a or ARKD cells, respectively, for 4 hours at the indicated E/T ratios. CTL activity was determined by lactate dehydrogenase release assays. (c) The efficiency of CD8+ T cells depletion. Animals were depleted of CD8+ T cells following a protocol as described in the Supplementary Materials and Methods by the anti-CD8 monoclonal antibody (53-6.72). An irrelevant IgG2a antibody was used as an isotype control. One day after the last antibody injection, the percentages of CD8+ T cells remained in the spleens were examined by flow cytometry. (d) The roles of CD8+ T cells in the Sindbis virotherapy-mediated antitumor activity. The growth of ARKD tumor was examined as described in the legend to Figure 5 in the SBV-treated animals which were depleted of CD8+ T cells or treated with an irrelevant IgG2a control antibody. *P < 0.05, **P < 0.01, ***P < 0.001, Student's t-test. CTL, cytotoxic T lymphocyte; PBS, phosphate-buffered saline; SBV, Sindbis virus.
Discussion
This study demonstrates that cellular susceptibility to SBV infection is not determined by SBV receptor levels, because the lentivirus pseudotyped with the SBV envelope glycoproteins could infect diverse tumor cell lines and primary cells as efficiently as the lentivirus pseudotyped with the VSV-G glycoproteins. The results imply that SBV can attach to tumor cells and primary cells comparably well in vitro. Thus, the laminin receptor may not be a major determinant for the tumor selectivity of SBV. However, given that the membrane and matrix components of the in vitro cell lines may not completely mimic those of the in vivo tissues, this study can not rule out the possibility that the attachment properties of SBV to tumor tissues and to normal tissues are highly variable in vivo, which may partly attribute to the tumor selectivity in vivo. Despite this possibility, herein we provide strong evidence that the status of IFN response in tumor cells plays critical roles in determining cellular susceptibility to SBV virotherapy.
By dissecting the IFN response into IFN-β production and IFN signaling phases, we showed that tumors defective in either phase had an attenuated IFN response and were therefore more susceptible to SBV infection in vitro (Figures 2 and 3). However, tumors defective only in IFN-β production did not sufficiently allow SBV-mediated oncolysis in vivo (Figure 5a), probably because of IFN production by neighboring normal cells, which limits SBV replication in the tumor cells intact in IFN signaling. The results of animal experiments support this scenario, showing that only about a fifth of the total SBV load was produced in the parental ML-14a tumors as compared to the IFN-signaling-defective ARKD tumors on day 1 after virotherapy (Figure 5f). Nevertheless, SBV was still quickly cleared in both the ML-14a and the ARKD tumor models.
We provide the following possible explanations for this phenomenon. First, because IFNAR1 knockdown was not 100% in ARKD cells, it is possible that the remaining IFN receptors may still be able to mount an antiviral response in ARKD tumors in vivo. Second, in immunocompetent animals, other forms of immunity that can limit SBV replication (e.g., innate or antiviral adaptive immunity) could have been induced. We have evidence showing that the titers of anti-SBV neutralizing antibodies rose significantly starting on day 3 after virotherapy (Supplementary Figure S7), indicating that virus-specific adaptive immunity was quickly induced by one dose of virotherapy. Notably, previous studies used xenograft tumor models in immunocompromised mice and did not observe clearance of SBV,1,2,3,4,5 which could be due to the lack of adaptive immunity in immunocompromised mice and to the lack of cross reactivity of IFN across different species.
Though host immune responses strongly limit SBV replication and thus reduce the efficacy of oncolysis, they may not be detrimental in terms of overall antitumor effects. As shown in our study and in many others,21,22,23 virotherapy actually induced a bystander antitumor immune response, for several reasons. First, oncolytic viral infection increases the release of tumor-associated antigens; second, it triggers the production of inflammatory cytokines. The inflammatory response acts as a danger signal and enhances the phagocytosis of tumor-associated antigens by antigen-presenting cells.24 Consequently, oncolytic virotherapy can lead to the priming and development of adaptive antitumor immunity. An increasing body of evidence has demonstrated that bystander immune responses play significant roles in the efficacy of oncolytic virotherapy.21,22,23 In our tumor model, antitumor immune responses play important roles in continuously restricting tumor cell growth after the termination of oncolysis, thus prolonging the therapeutic effects of virotherapy.
In conclusion, virotherapy can induce antiviral and antitumor immunities in immunocompetent hosts. Host immune responses act as a double-edged sword for the success of oncolytic virotherapy, depending on the balance between the antiviral and antitumor immune responses.24 Therefore, strategies to alter this balance in favor of a therapeutic benefit may increase the therapeutic efficacy of oncolytic virotherapy.
Materials and Methods
Cell culture. Primary human foreskin fibroblasts were isolated from human foreskin tissue obtained with patients' informed consent. Mouse embryonic fibroblasts were isolated from a 13.5-day-old mouse embryo. STKO, a mouse embryonic fibroblast cell line derived from STAT1-knockout mice, was kindly provided by Dr Lee (National Taiwan University, Taipei, Taiwan). The mouse hepatoma cell line ML-14a was kindly provided by Dr Lei (National Cheng Kung University, Tainan, Taiwan). The ARKD cell line, an IFNR knockdown cell line, was generated from ML-14a by lentiviral transduction of a shRNA targeting IFNR and was adapted in BALB/c mice for one generation. The remaining cell lines used in Figure 1 are described in Supplementary Materials and Methods.
Preparation of SBVs and lentivirus pseudotyped with the Sindbis viral envelope proteins. The DNA-based Sindbis viral vector, dSinF, was modified from dsTE12Q plasmid (kindly provided by Dr Hardwick).25 To express viral RNA in mammalian cells, the SP6 promoter in the dsTE12Q was replaced with a cytomegaloviral immediate early gene promoter via recombinant PCR, in a way that enabled the transcription to be initiated at the first nucleotide of the SBV sequence. In addition, a ribozyme sequence derived from hepatitis D virus and the polyadenylation signal sequences from bovine growth hormone were also inserted downstream of the poly A sequences in dsTE12Q, enabling the transcript to be terminated exactly at the 3′ end of the SBV sequences. An in-frame fusion of the 2A protease of the foot-and-mouth disease virus was cloned downstream of the capsid gene as previously described.26 Two restriction sites, MluI and NotI, were introduced at the 5′ end of the 2A protease sequences. Therefore, the cDNAs of transgenes, including EGFP and luciferease, that were PCR amplified included an MluI and a NotI restriction site at their 5′ and 3′ ends, respectively. The cDNA fragments were then inserted into the SB viral vector via the MluI and NotI sites. SBVs were produced by transfection of the viral genomic DNAs into BHK cells using a calcium phosphate precipitation method. Sixteen hours after transfection, cells were replaced with Opti-MEM I medium (Invitrogen, Carlsbad, CA) and cultured for another 32 hours. The virus-containing culture medium was collected, filtered through a 0.22 µm filter, and then stored at −80 °C. Sindbis viral titer was determined by standard plaque assay on monolayers of BHK cells.
To produce lentiviruses pseudotyped with Sindbis viral envelope protein, the lentiviral vector expressing EGFP, the gag-pol expressing plasmid p8.91, and the plasmid encoding the SBV envelope protein, pIntron-SB,27 were co-transfected into 293FT cells using a calcium phosphate precipitation method. Viruses were collected 2 days after DNA transfection.
Assays measuring IFN-β production or IFN signaling in various cell lines. To examine the IFN-β production capabilities of various cell lines, cells were transfected with 1 µg/ml of poly(I:C) using Lipofectamine 2000 (Invitrogen). Six hours after transfection, the cultures were replaced with fresh medium and incubated for another 18 hours. The conditioned medium was collected, diluted with fresh medium at 1:1 ratio, and then used to prime ML-14a or 293T cells for 24 hours. The ML-14a and 293T cells were further challenged with an EGFP-expressing SBV at a multiplicity of infection of 5 for 24 hours. The percentages of EGFP(+) cells were analyzed by flow cytometry. To examine the status (intact or defective) of IFN signaling in various cell lines, cells were first treated with 100 IU/ml of IFN-α (Sigma, St Louis, MO) for 24 hours and then challenged with a luciferase-expressing SBV. Luciferase activity was analyzed 24 hours after SBV infection.
Animal studies. Male BALB/c mice aged 7–8 weeks were obtained from the National Laboratory Animal Center, Taipei, Taiwan. The mice were housed in standard conditions, and all the experiments were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals” prepared by the Institutional Animal Care and Use Committee of National Yang-Ming University. Subcutaneous tumors were generated by inoculating 5 × 105 ML-14a or ARKD cells in the posterior flanks of the mice on day 0. Seven days after tumor implantation, a single dose of SBV (1 × 107 PFU/mouse) was intratumorally injected. A 100-fold lower dose of SBV (1 × 105 PFU/mouse) and an UV-inactivated SBV (exposed to UV at 2000 µWatts/cm2 for 20 minutes on ice) were used as controls. The efficiency of UV irradiation on virus was confirmed by plaque assay. The growth of tumors was monitored weekly, and tumor volumes were calculated using the following formula: volume = 0.52 × A × B2, where A and B are the longest and the shortest diameters of the tumors, respectively. To quantify the viral loads in tumor tissues, the SBV expressing a luciferase reporter was used. Three mice from each group were killed on days 0 (6 hours after injection), 1, 2, 4, and 7 after SBV injection. Tumors were explanted, weighed, and homogenized in 0.2 ml cell culture lysis reagent (Promega, Madison, WI). Ten microliters of the lysate was subjected to a luciferase activity assay using the Luciferase Assay System (Promega). The luciferase activity results were further normalized to the tumor weights.
Statistical analysis. GraphPad Prism 4 was used for all statistical analysis. Data are presented as the means ± SD. The statistical analysis was performed using an unpaired, two-tailed Student's t-test. Differences with a P value <0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Images of SBV-induced cytopathology of representative cell lines. Figure S2. Induction folds of ZAP mRNA in various cell lines upon IFN-α stimulation. Figure S3. Establishment of the IFN signaling-defective ML-14a tumor cell line ARKD. Figure S4. TUNEL analysis showing apoptotic cells in tumor tissues. Figure S5. Immunohistochemistry of CD8+ and CD4+ cells showing the lymphocyte infiltration in tumor tissues. Figure S6. Syngenic BNL hepatoma cells were used as a control for CTL specificity. Figure S7. Production of SBV neutralizing antibodies in tumor-bearing mice after virotherapy. Table S1. Sequences of the primers used for quantitative RT-PCR analysis. Materials and Methods.
Acknowledgments
This work was supported by a grant from the National Health Research Institutes (NHRI-EX100–9831BI). We thank the RNAi Core Facility, Academia Sinica, Taiwan for preparing the lentiviral shRNA.
Supplementary Material
Images of SBV-induced cytopathology of representative cell lines.
Induction folds of ZAP mRNA in various cell lines upon IFN-α stimulation.
Establishment of the IFN signaling-defective ML-14a tumor cell line ARKD.
TUNEL analysis showing apoptotic cells in tumor tissues.
Immunohistochemistry of CD8+ and CD4+ cells showing the lymphocyte infiltration in tumor tissues.
Syngenic BNL hepatoma cells were used as a control for CTL specificity.
Production of SBV neutralizing antibodies in tumor-bearing mice after virotherapy.
Sequences of the primers used for quantitative RT-PCR analysis.
REFERENCES
- Tseng JC, Levin B, Hirano T, Yee H, Pampeno C., and, Meruelo D. In vivo antitumor activity of Sindbis viral vectors. J Natl Cancer Inst. 2002;94:1790–1802. doi: 10.1093/jnci/94.23.1790. [DOI] [PubMed] [Google Scholar]
- Tseng JC, Hurtado A, Yee H, Levin B, Boivin C, Benet M.et al. (2004Using sindbis viral vectors for specific detection and suppression of advanced ovarian cancer in animal models Cancer Res 646684–6692. [DOI] [PubMed] [Google Scholar]
- Tseng JC, Levin B, Hurtado A, Yee H, Perez de Castro I, Jimenez M.et al. (2004Systemic tumor targeting and killing by Sindbis viral vectors Nat Biotechnol 2270–77. [DOI] [PubMed] [Google Scholar]
- Unno Y, Shino Y, Kondo F, Igarashi N, Wang G, Shimura R.et al. (2005Oncolytic viral therapy for cervical and ovarian cancer cells by Sindbis virus AR339 strain Clin Cancer Res 114553–4560. [DOI] [PubMed] [Google Scholar]
- Wollmann G, Tattersall P., and, van den Pol AN. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J Virol. 2005;79:6005–6022. doi: 10.1128/JVI.79.10.6005-6022.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH., and, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montuori N., and, Sobel ME. The 67-kDa laminin receptor and tumor progression. Curr Top Microbiol Immunol. 1996;213 (Pt 1):205–214. doi: 10.1007/978-3-642-61107-0_13. [DOI] [PubMed] [Google Scholar]
- Wang KS, Kuhn RJ, Strauss EG, Ou S., and, Strauss JH. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J Virol. 1992;66:4992–5001. doi: 10.1128/jvi.66.8.4992-5001.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes AP., and, Griffin DE. Binding of Sindbis virus to cell surface heparan sulfate. J Virol. 1998;72:7349–7356. doi: 10.1128/jvi.72.9.7349-7356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S, Takimoto T, Scroggs RA., and, Portner A. Differentially regulated interferon response determines the outcome of Newcastle disease virus infection in normal and tumor cell lines. J Virol. 2006;80:5145–5155. doi: 10.1128/JVI.02618-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N.et al. (2000Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus Nat Med 6821–825. [DOI] [PubMed] [Google Scholar]
- Parato KA, Senger D, Forsyth PA., and, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- Ryman KD, Klimstra WB, Nguyen KB, Biron CA., and, Johnston RE. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74:3366–3378. doi: 10.1128/jvi.74.7.3366-3378.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Williams BR, Rice CM., and, Frolov I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J Virol. 2004;78:8455–8467. doi: 10.1128/JVI.78.16.8455-8467.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman KD, Meier KC, Nangle EM, Ragsdale SL, Korneeva NL, Rhoads RE.et al. (2005Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells J Virol 791487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Burke CW, Ryman KD., and, Klimstra WB. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR., and, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Kawai T., and, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M.et al. (2004The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses Nat Immunol 5730–737. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K.et al. (2005Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity J Immunol 1752851–2858. [DOI] [PubMed] [Google Scholar]
- Zamarin D, Martínez-Sobrido L, Kelly K, Mansour M, Sheng G, Vigil A.et al. (2009Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses Mol Ther 17697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD., and, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther. 2011;19:335–344. doi: 10.1038/mt.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujar SA, Pan DA, Marcato P, Garant KA., and, Lee PW. Oncolytic virus-initiated protective immunity against prostate cancer. Mol Ther. 2011;19:797–804. doi: 10.1038/mt.2010.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA., and, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8:1581–1588. doi: 10.1586/14737140.8.10.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Levine B, Boise LH, Thompson CB., and, Hardwick JM. Bax-independent inhibition of apoptosis by Bcl-XL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- Thomas JM, Klimstra WB, Ryman KD., and, Heidner HW. Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. J Virol. 2003;77:5598–5606. doi: 10.1128/JVI.77.10.5598-5606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizono K, Bristol G, Xie YM, Kung SK., and, Chen IS. Antibody-directed targeting of retroviral vectors via cell surface antigens. J Virol. 2001;75:8016–8020. doi: 10.1128/JVI.75.17.8016-8020.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Images of SBV-induced cytopathology of representative cell lines.
Induction folds of ZAP mRNA in various cell lines upon IFN-α stimulation.
Establishment of the IFN signaling-defective ML-14a tumor cell line ARKD.
TUNEL analysis showing apoptotic cells in tumor tissues.
Immunohistochemistry of CD8+ and CD4+ cells showing the lymphocyte infiltration in tumor tissues.
Syngenic BNL hepatoma cells were used as a control for CTL specificity.
Production of SBV neutralizing antibodies in tumor-bearing mice after virotherapy.
Sequences of the primers used for quantitative RT-PCR analysis.