

Abstract
Agonist-induced glucocorticoid receptor [GR] transport from the cytoplasm to the nucleus was used as a model to identify dynein-mediated cargo transport inhibitors. Cell-based screening of the library of pharmacologically active compound (LOPAC)-1280 collection identified several small molecules that stalled the agonist-induced transport of GR-green fluorescent protein (GFP) in a concentration-dependent manner. Fluorescent images of microtubule organization, nuclear DNA staining, expression of GR-GFP, and its subcellular distribution were inspected and quantified by image analysis to evaluate the impact of compounds on cell morphology, toxicity, and GR transport. Given the complexity of the multi-protein complex involved in dynein-mediated cargo transport and the variety of potential mechanisms for interruption of that process, we therefore developed and validated a panel of biochemical assays to investigate some of the more likely intracellular target(s) of the GR transport inhibitors. Although the apomorphine enantiomers exhibited the most potency toward the ATPase activities of cytoplasmic dynein, myosin, and the heat-shock proteins (HSPs), their apparent lack of specificity made them unattractive for further study in our quest. Other molecules appeared to be nonspecific inhibitors that targeted reactive cysteines of proteins. Ideally, specific retrograde transport inhibitors would either target dynein itself or one of the other important proteins associated with the transport process. Although the hits from the cell-based screen of the LOPAC-1280 collection did not exhibit this desired profile, this screening platform provided a promising phenotypic system for the discovery of dynein/HSP modulators.
Introduction
To promote cell survival, essential cellular components must be organized and transported to specific locations within the cell when needed. When some steroid nuclear receptors (glucocorticoid receptor [GR], estrogen receptor [ER], and androgen receptor [AR]) become activated by agonist binding, they translocate from the cytoplasm to the nucleus, where they initiate target gene transcription leading to a cascade of important cellular events necessary for cell function and survival. For example, the GR is a nuclear receptor that, when bound to a glucocorticoid, is transported to the nucleus to activate transcription of target genes involved in the regulation of inflammatory and immune responses.1
The cell's cytoskeleton provides a highly sophisticated network of railways for motor proteins to transport macromolecules to their desired locations, including the retrograde translocation of cytoplasmic GR to the nucleus by the motor protein cytoplasmic dynein.2 Cytoplasmic dynein is a large multi-protein complex that consists of homodimers each of heavy, intermediate, light-intermediate, and light chains.3 The dynein heavy chain is a member of the adenosine triphosphatases (ATPases) associated with diverse activities (AAA+) family and the motor domain contains six AAA domains, although only the first four are capable of binding ATP.4 The first AAA domain is largely responsible for providing the force needed for cytoplasmic dynein movement, whereas the remaining AAA domains are believed to have regulatory functions (e.g., increasing the rate of microtubule [MT] movement driven by dynein).5 A coiled-coil stalk that protrudes from the dynein heavy chain contains an MT-binding domain, allowing cytoplasmic dynein to interact with and bind to MTs.6 The remaining chains connect the motor domain to a multi-protein complex that connects with the cargo. Protein cargos transported by cytoplasmic dynein are often linked through dynactin and immunophilin-heat-shock protein complexes.7 In addition to transporting GR, cytoplasmic dynein carries cargos that vary from other proteins, including ER8 and p53,9 to much larger viruses10,11 and Golgi vesicles.12 Cytoplasmic dynein has also been shown to provide force in nuclear envelope breakdown,13 chromosome segregation,14 mitotic spindle formation,15 and cytokinesis.16
Because cytoplasmic dynein contributes to many important cellular processes, small molecule modulators with specificity for dynein would provide useful chemical and cell biology tools to further investigate the critical roles of cytoplasmic dynein in the cell. To date, there are only a handful of small molecules that target cytoplasmic dynein, most of which are either ATP/transition state mimics (e.g., erythro-9-(2-hydroxy-3-nonyl)adenine, adenylyl imidodiphosphate, and orthovanadate),17,18 or sulfhydryl-reactive agents (e.g., thiourea and N-ethylmaleimide [NEM]).18,19 We recently examined purealin, a natural product derived from the sea sponge Psammaplysilla purea, that has been shown to enhance the ATPase activity of myosin20 while also inhibiting the ATPase activity of axonemal dynein.21 Synthetic analogs of purealin inhibited the ATPase activity of rat cytoplasmic dynein heavy chain 1.22 Although purealin itself was a potent inhibitor of cytoplasmic dynein heavy chain in vitro, the compound was unable to do so in cells.
We therefore chose a new method to screen for dynein modulators, one that would include cellular activity. Using a cell-based high-content screen (HCS) assay to quantify the agonist-induced translocation of GR from the cytoplasm to the nucleus as a cellular model of dynein-mediated cargo transport, we identified several concentration-dependent inhibitors in a screen of the 1280-member library of pharmacologically active compound (LOPAC) collection. In the current report, we present these results as well as those from a panel of secondary assays that we developed to characterize the cell-based screen hits and to investigate the potential mechanisms of action of these inhibitors of cytoplasmic dynein-mediated cargo transport. The distribution of GR-green fluorescent protein (GFP) present in the cytoplasm and nuclei of cells was first examined and compared to determine which compounds inhibited agonist-induced GR translocation. From the same cell images, the cellular toxicity of compounds, defined by a loss of cells, was evaluated, as were the compound-induced changes in the morphology of α-tubulin antibody staining along with the intensity and the overall MT organization of treated cells. Competitive displacement GR binding assays and in vitro MT polymerization assays were performed to identify potential GR binding antagonists and MT perturbing agents, respectively. Compounds were then tested biochemically against the ATPase activity of heat-shock protein (Hsp)90 and Hsp70 molecular chaperones, which are both important components of the GR cargo transport process.7 The ability of the GR translocation inhibitors to inhibit both the basal and the MT-stimulated ATPase activity of the recombinant dynein motor domain was then tested. Finally, to evaluate the ATPase selectivity of the hits, compounds were screened against the ATPase activity of the myosin molecular motor since the only known inhibitor of dynein (purealin) has also been shown to perturb myosin activity.
Materials and Methods
Materials
ATP, guanosine triphosphate (GTP), 2-(N-morpholino)ethanesulfonic acid (MES), monosodium glutamate (MSG), and piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) were from USB, Affymetrix, Inc. The LOPAC®1280 library, CaCl2, dimethylsulfoxide (DMSO), ethylene glycol tetraacetic acid (EGTA), HEPES, Hoechst 33342, imidazole, KCl, 2-mercaptoethanol, MgCl2, NEM, phosphate-buffered saline (PBS), Protease Inhibitor Cocktail, and tris(hydroxymethyl)aminomethane hydrochloric acid (TRIS-HCl), were from Sigma-Aldrich. Ethylenediaminetetraacetic acid (EDTA) was from Boehringer Mannheim. Alexa-595, Dulbecco's modified Eagle's medium (DMEM), G418, L-glutamine, Hi5™ Cells, Express Five® Serum Free Medium, nonessential amino acids, penicillin and streptomycin, Green Polar Screen™ Glucocorticoid Receptor Competitor Assay Kit, sodium pyruvate, and trypan blue stain were from Invitrogen, Inc. Fetal bovine serum (FBS) was from Gemini Bio-Products. The BCA™ Protein Assay Kit and bovine serum albumin (BSA) were from Pierce. PiColorLock™ Gold was from Innova Biosciences. Myosin II from rabbit skeletal muscle was from Cytoskeleton, Inc. Nickel nitrilotriacetic acid (Ni-NTA) Superflow was from Qiagen. PD-10 Desalting and Q-Sepharose™ columns were from GE Healthcare Life Sciences. The baculovirus expression vector pVL1393 was from BD Biosciences. The 3617.4 mouse mammary adenocarcinoma cell line stably expressing GR-GFP under the control of a tetracycline (Tet) regulated promoter was kindly provided by Dr. Gordon Hager from the Laboratory of Receptor Biology and Gene Expression, NCI, Bethesda, MD. Triton X-100 and Tween 20 were from BioRad. Mouse monoclonal anti-α-tubulin antibody was obtained from NeoMarkers/Lab Vision Inc. Cy3-labeled donkey anti-mouse was obtained from Jackson ImmunoResearch Laboratories, Inc. Paclitaxel (PTX) was obtained from the NIH Drug Synthesis Branch. Formaldehyde was obtained from Fisher Scientific.
Methods
Dexamethasone-induced GR translocation HCS assay protocol
The 3617.4 cell line23 was maintained in complete culture medium containing 10 μg/mL Tet (Tet-on) in a humidified incubator at 37°C, 5% CO2, and 95% humidity to keep the expression of GR-GFP repressed; DMEM with 2 mM L-glutamine supplemented with 10% FBS, 100 μM nonessential amino acids, 100 μM sodium pyruvate, 100 U/mL penicillin and streptomycin, and containing 0.96 mg/mL G418. To induce GR-GFP expression in the 3617.4 cell line, cells were cultured for 48 h in the same tissue culture medium without the Tet (Tet-off). 3617.4 cells that had been cultured in Tet-containing complete culture medium were detached from tissue culture flasks by trypsinization; cells were centrifuged at 500 g for 5 min and re-suspended in 10 mL of Tet-free induction medium; and viable cells that excluded trypan blue were counted in a haemocytometer. 3617.4 cells were adjusted to 4.2×104 cells/mL in Tet-free induction medium and then 60 μL of cell suspension per well was dispensed into the wells of 384-well black-walled clear-bottom plates using the Zoom liquid handler (Titertek) to give a final seeding density of 2,500 cells/well. Assay plates were incubated under Tet-off conditions for 48 h at 37°C, 5% CO2 in a humidified incubator and then diluted compounds (20 μL) were added to wells in columns 3 through 22 using an Evolution P3 (Perkin-Elmer) outfitted with a 384-well transfer head for a final screening concentration after dexamethasone (DEX) addition of 20 μM. Compound-treated plates were incubated at 37°C, 5% CO2 in a humidified incubator for 60 min and then 20 μL of 5.0 μM DEX (1.0 μM final in well) was transferred to assay plates using the Evolution P3 liquid handler outfitted with a 384-well transfer head. The plate control wells were located in columns 1, 2, 23, and 24 and the minimum controls (n=24) were treated with 0.5% DMSO and the maximum control wells (n=32) were exposed to 1.0 μM DEX and 0.5% DMSO. Assay plates were incubated for 30 min at 37°C, 5% CO2, and 95% humidity, and then the contents of the wells were aspirated and replaced with 50 μL of 3.7% formaldehyde containing 2 μg/mL Hoechst 33342 in PBS without Ca2+ and Mg2+, prewarmed to 37°C, using a BioTek ELx405 (BioTek) plate washer, and cells were fixed for 10–30 min at ambient temperature. The fixative was then aspirated and plates were then washed twice with 50 μL PBS using the BioTek ELx405 (BioTek) plate washer and sealed with adhesive aluminum plate seals using the Abgene Seal-IT 100 plate sealer (Abgene) with the last 50 μL wash of PBS in place.
Image acquisition
Images of two fluorescent channels (Hoechst and fluorescein isothiocyanate [FITC]) were sequentially acquired on the ArrayScan VTI (AS-VTI) automated imaging platform (Thermo Fisher Scientific) using a 10×0.3NA objective and the XF100 excitation and emission filter set to obtain images of stained nuclei and GR-GFP. Excitation was provided by an X-CITEÒ 120 W high-pressure metal halide arc lamp with intelli-lamp™ technology (Photonic Solutions Inc.). Typically with the 10×0.3NA objective, the AS-VTI was set up to acquire 500 selected objects (nuclei) or two fields of view, whichever occurred first.
Image analysis
The nucleic acid dye Hoechst 33342 was used to stain and identify the nucleus, and this fluorescent signal was used to focus the instrument and to define a nuclear mask for the molecular translocation (MTL) image analysis algorithm. To quantify the DEX-induced translocation of GR-GFP from the cytoplasm to the nucleus, images were analyzed using the MTL image analysis algorithm as described previously.24 Hoechst stained objects in channel 1 (Ch1) that exhibited the appropriate fluorescent intensities above background and morphological size characteristics (width, length, and area) were identified and classified by the image segmentation as nuclei. The nuclear mask derived from Ch1 was then used to segment the GR-GFP images from Ch2 into nuclear (Circ) and cytoplasmic (Ring) regions. The nuclear mask was eroded by 1 pixel to reduce cytoplasm contamination within the nuclear area, and the reduced mask was used to quantify the amount of target Ch GR-GFP fluorescence within the nuclear region. The nuclear mask was then dilated to cover as much of the cytoplasm region as possible without going outside the cell boundary. Removal of the original nuclear region from this dilated mask created a ring mask in the cytoplasm region outside the nuclear envelope. The number of pixels away from the nuclear mask (1 pixel) and the number of pixels (width=3 pixels) between the inner and outer ring masks were selectable within the MTL bio-application software. The ring masks were then used to quantify the amount of target Ch GR-GFP fluorescence within the cytoplasm region. The MTL image analysis algorithm outputs quantitative data including the total selected object or cell counts from Ch1, and the total and average fluorescent intensities of the GR-GFP in the nuclear (Circ) or cytoplasm (Ring) compartments of the cell. To quantify the relative distribution of the GR-GFP within the nucleus and the cytoplasm regions of the 3617.4 cells, the MTL image analysis algorithm provides a mean average intensity difference calculated by subtracting the average GR-GFP intensity in the Cytoplasm (Ring) region from the average GR-GFP intensity in the Nuclear (Circ) region of Ch2 to produce a mean circ-ring average intensity difference.
Indirect immunofluorescent staining of 3617.4 cell MTs
To observe the organization of MTs in images of 3617.4 cells acquired on the AS-VTI, we adapted previously described indirect immunofluorescence sample preparation methods.24 Briefly, fixed, Hoechst-stained 3617.4 cells prepared as described above for the GR-GFP translocation HCS assay were permeabilized in 0.5% (v/v) Triton X-100 in PBS (without Ca2+ and Mg2+) for 5 min at ambient temperature; cells were washed once in PBS (without Ca2+ and Mg2+) and then incubated for 15 min in 0.1% (v/v) Tween 20 in PBS (without Ca2+ and Mg2+) blocking buffer; cells were incubated for 1 h with a 1:2,000 dilution of a mouse monoclonal anti-α-tubulin antibody in PBS (without Ca2+ and Mg2+). Cells were then washed once in PBS (without Ca2+ and Mg2+) and then incubated for 15 min in Tween 20 blocking buffer and then incubated for 45 min with a 1:1,000 dilution of goat anti-mouse immunoglobulin G antibody conjugated to Alexa-595 in PBS (without Ca2+ and Mg2+). Cells were then washed twice with 50 μL PBS (without Ca2+ and Mg2+) and sealed with the last 50 μL wash of PBS in place. Fluorescent images from three fluorescent Chs were then sequentially acquired on the AS-VTI using a 20 × 0.4NA objective with the XF53 (Hoechst and FITC) and the XF32 (tetramethylrhodamine-5-(and 6)-isothiocyanate) excitation and emission filter sets to obtain images of stained nuclei, GR-GFP, and anti-α-tubulin antibody-stained MTs. The protocol used for this HCS assay has been summarized in Table 1.
Table 1.
Summary of Dexamethasone-Induced Glucocorticoid Receptor Translocation High-Content Screen Assay and Microtubule Staining Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Plate cells | 60 μL | 2,500 3617.4 GR-GFP cells |
| 2 | Incubate cells | 48 h | Induction medium at 37°C, 5% CO2, and 95% humidity |
| 3 | Library compounds/DMSO to controls wells | 20 μL | 20 μM final concentration in well, 0.5% DMSO |
| 4 | Incubate plates | 1 h | At 37°C, 5% CO2, and 95% humidity |
| 5 | Add DEX/media | 20 μL | DEX 1.0 μM final in well, media to min controls |
| 6 | Incubate plates | 30 min | at 37°C, 5% CO2, and 95% humidity |
| 7 | Aspirate media & fix cells | 50 μL | 3.7% formaldehyde containing 2 μg/mL Hoechest 33342 in PBS without Ca2+ and Mg2+, prewarmed to 37°C |
| 8 | Incubate plates | 10–30 min | Ambient temperature |
| 9 | Aspirate fixative & wash 2×with PBS | 50 μL | Fixative was aspirated and plates were then washed twice with 50 μL PBS without Ca2+ and Mg2+, 50 μL PBS in well |
| 10 | Cell permeabilization | 50 μL | 0.5% (v/v) Triton X-100 in PBS (without Ca2+ and Mg2+) for 5 min at ambient temperature |
| 11 | PBS wash | 50 μL | PBS (without Ca2+ and Mg2+) |
| 12 | Blocking buffer incubation | 50 μL | 15 min in 0.1% (v/v) Tween 20 in PBS (without Ca2+ and Mg2+) at ambient temperature |
| 13 | PBS wash | 50 μL | PBS (without Ca2+ and Mg2+) |
| 14 | Primary antibody incubation | 25 μL | 1 h with a 1:2000 dilution of a mouse monoclonal anti-α-tubulin antibody in PBS (without Ca2+ and Mg2+) |
| 15 | PBS wash | 50 μL | PBS (without Ca2+ and Mg2+) |
| 16 | Blocking buffer incubation | 50 μL | 15 min in 0.1% (v/v) Tween 20 in PBS (without Ca2+ and Mg2+) at ambient temperature |
| 17 | Secondary antibody incubation | 25 μL | 45 min incubation with a 1:1000 dilution of goat anti-mouse immunoglobulin G antibody conjugated to Alexa-595 in PBS (without Ca2+ and Mg2+) |
| 18 | PBS wash 2× | 50 μL | PBS (without Ca2+ and Mg2+), 50 μL PBS in well |
| 19 | Seal plates | 1× | Sealed with adhesive aluminum plate seals |
| 20 | Acquire images | 20×, 0.4NA objective | Images of the Hoechst (Ch1), GR-GFP (Ch2) and microtubules (Ch3) were sequentially acquired on the ArrayScan VTI using the XF53 (Hoechst and FITC) and the XF32 (TRITC) excitation and emission filter sets |
| 21 | Assay readout | MCRAID | Images were analyzed using the Compartmental Analysis image analysis Algorithm using the Mean Circ (Nucleus)–Ring (Cytoplasm) Average Intensity Difference to quantify the GR-GFP translocation |
1. 384-well black-walled clear-bottom plates, Greiner Bio-one Cat # 781091, Zoom liquid handler (Titertek, Huntsville, AL).
2. DMEM+2 mM L-glutamine+10% charcoal-stripped FBS+100 μM nonessential amino acids+100 μM sodium pyruvate+100 U/mL penicillin and streptomycin +0.96 mg/mL G418.
3. Compounds added to wells in columns 3–22. Controls in columns 1, 2, 23, and 24. VPrep (Velocity 11, Menlo Park, CA) or an Evolution P3 (Perkin-Elmer, Waltham, MA) outfitted with a 384-well transfer head.
4. Pre-exposure of cells to 20 μM compound before DEX agonist addition.
5. 5.0 μM DEX (1.0 μM final in well) to max controls and compound wells, media to min control wells.
6. DEX-induced translocation of GR-GFP from the cytoplasm to the nucleus.
7. Aspiration of media and fixative addition automated on BioTek ELx405 (BioTek) plate washer.
8. 10–30 min incubation at ambient temperature to fix cells and stain nuclei with Hoechst.
9. Aspiration of fixative and PBS wash steps automated on BioTek ELx405 (BioTek) plate washer.
10. Permeabilization buffer added with the Microflo bulk reagent dispenser (BioTek).
11. Aspiration of antibody and PBS wash steps automated on BioTek ELx405 (BioTek) plate washer.
12. Blocking buffer added with the Microflo bulk reagent dispenser (BioTek).
13. PBS wash step automated on BioTek ELx405 (BioTek) plate washer.
14. Primary antibody added with the Microflo bulk reagent dispenser (BioTek).
15. PBS wash step automated on BioTek ELx405 (BioTek) plate washer.
16. Secondary antibody added with the Microflo bulk reagent dispenser (BioTek).
17. PBS wash steps automated on BioTek ELx405 (BioTek) plate washer.
18. Plates sealed with adhesive aluminum plate seals using the Abgene Seal-IT 100 plate sealer (Abgene).
19. Plates loaded into the ArrayScan VTI for scanning using a Twister II robotic plate handler (Thermo Fisher Scientific).
20. Compartmental Analysis bio-application (Thermo Fisher Scientific).
GR, glucocorticoid receptor; Ch, channel; DMEM, Dulbecco's modified Eagle's medium; DMSO, dimethylsulfoxide; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; GFP, green fluorescent protein; MCRAID, mean circ-ring average intensity difference; PBS, phosphate-buffered saline; TRITC, tetramethylrhodamine-5-(and 6)-isothiocyanate; DEX, dexamethasone.
Protein expression and isolation
Tubulin: Tubulin was isolated from three freshly obtained bovine brains according to a modified procedure of that reported by Hamel and Lin.25 Brains were homogenized in a blender containing 0.75 mL/g of 0.1 M MES, pH 6.4, containing 1 mM EGTA, 4 M glycerol, 1 mM MgCl2, 1 mM 2-mercaptoethanol, 0.1 mM EDTA, and 0.1 mM GTP (Solution A). The homogenate was clarified by centrifuging at 6,400 g for 15 min at 0°C with a JA-10 Beckman-Coulter rotor. All remaining centrifugations listed were performed using a Ti-45 Beckman-Coulter rotor. The supernatant was centrifuged at 90,000 g for 44 min at 0°C. ATP and GTP were added to the supernatant to give final concentrations of 1 and 0.3 mM, respectively, and the solution was incubated for 40 min at 37°C. The solution was centrifuged at 90,000 g for 44 min at 37°C. The supernatant was discarded and the pellet was resuspended in 10 mL/brain of the above-described buffer without glycerol (Solution B) and incubated on ice for 30 min. For each 1 mL of solution, 0.553 g of glycerol was added to give a final concentration of 4 M. ATP and GTP were then added to give final concentrations of 1 and 0.3 mM, respectively, and the mixture was incubated for 1 h at 37°C. The solution was centrifuged at 90,000 g for 44 min at 37°C and the pellet was resuspended in 6.25 mL/brain of Solution B. The resuspended pellet was incubated on ice for 30 min and centrifuged at 67,000 g for 30 min at 0°C. Lyophilized MES (2M, pH 6.9) was added to the solution to give a final concentration of 1.6 M followed by the addition of 1 mM GTP and 2 mM dithiothreitol and the mixture was incubated for 45 min at 37°C. The mixture was centrifuged at 90,000 g for 44 min at 37°C and the pellet was resuspended in 10 mL cold 1 M MSG, pH 6.6. The mixture was incubated on ice for 2 h and then centrifuged at 90,000 g for 30 min at 0°C. To the supernatant, 1 mM GTP was added and the solution was incubated for 1 h at 37°C and the mixture was then centrifuged at 90,000 g for 1.5 h at 37°C. The pellet was resuspended in 10 mL of cold 1 M MSG, pH 6.6 and incubated on ice for 1 h. The solution was centrifuged at 90,000 g for 1 h at 0°C and the supernatant contained soluble tubulin. The electrophoretic homogeneity of the protein was determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and the concentration determined by a bicinchoninic acid (BCA) Protein Assay kit using BSA as a standard.
Hsp70: Yeast Hsp70 (Ssa1p) was purified as described by McClellan and Brodsky.26 In brief, yeast expressing an active, hexahistidine-tagged form of Ssa1p on a galactose-regulated promoter were grown to log-phase and harvested, and the cell walls were digested. The resulting spheroplasts were lysed with glass beads and the lysate was chromatographed on an ATP-affinity column. The bound protein was released with ATP, and then analyzed on a Q-Sepharose column. Ssa1p was eluted through the use of a KCl gradient and then further purified by using nickel ion affinity chromatography. The protein, which was eluted from this column with imidazole, was dialyzed, and aliquots were retained at −80°C until use.
Hsp90: Yeast Hsp90 (Hsc82p) was purified according to the protocol outlined by Youker et al.27 Yeast containing a vector engineered for the overexpression of Hsc82p were propagated, harvested, and then lysed with glass beads, and a crude Hsc82p-containing fraction was obtained by running a KCl gradient on a DE52 anion exchange column. After dialysis to reduce the salt concentration, the chaperone was highly enriched by chromatography on a Q-Sepharose column that was similarly treated with a KCl gradient. Peak fractions containing Hsc82p were pooled and again dialyzed, and the protein was aliquoted and stored at −80°C.
Cytoplasmic dynein: The 380 kDa motor domain fragment was expressed as described by Hook et al.28 Briefly, the gene encoding rat cytoplasmic dynein motor domain (Gly1286-Glu4644) with a C-terminal in-frame hexahistidine tag was inserted into the baculovirus expression vector pVL1393. Hi5™ cells plated with Express Five™ Serum Free Medium were infected with the virus for 40 h. The cells were then washed and resuspended in PBS buffer, and the recombinant motor domain fragments were extracted from the cells by homogenization. Extracts were centrifuged at 5,000 g for 10 min and then 100,000 g for 30 min. The supernatant was applied to an Ni-NTA Superflow column pre-equilibrated with 50 mM PIPES, 50 mM HEPES, pH 7.2, containing 2 mM MgCl2 and 2 mM EGTA. The column was washed with 16 volumes of buffer containing 20 mM imidazole before eluting the protein with buffer supplemented with 125 mM imidazole. The imidazole was removed using a PD-10 Desalting Column and sample was resuspended in 10 mM PIPES, 10 mM HEPES, pH 7.2, containing 0.4 mM MgCl2 and 0.4 mM EGTA. Protein concentrations and purity were then determined by a BCA assay with albumin as the standard and gel electrophoresis, respectively. ATPase activity of the motor domain was determined using a modified malachite kit (PiColorlock™ Gold Assay) according to the manufacturer's protocol. Samples were then read with a SpectraMax® M5 Microplate Reader (MDS Analytical Devices Inc.) at 630 nm.
Activity assays
GR competitor assay: The Green Polar Screen™ Glucocorticoid Receptor Competitor Assay Kit was used according to the manufacturer's recommendations with the final sample volume reduced to 40 μL and is summarized in Table 2. Briefly, human recombinant GR was used at the recommended concentration of 15 nM and the Fluormone™ GS1 was used at a concentration of 1 nM. An initial screen was performed with aliquots (20 μL) of the mixture of test compounds in DMSO screening buffer with final concentrations of 12.5 and 50 μM were distributed in 384-well, black flat-bottom plates. 4×Fluormone (10 μL) followed by 4×GR (10 μL) was then added to the wells. Each compound was tested in triplicate, and 50 μM DEX was used as a positive control. The DMSO concentration was kept at 1% (v/v) throughout the experiment. After 2 h, the fluorescence polarization (FP) (485 nm excitation and 530 nm emission) was measured using a SpectraMax M5 reader. Data were then fitted to a one-site competition curve using KaleidaGraph Software V.4.03. The fit was constrained by the high polarization control, which was the GR/GS1 complex with no competition, and the low polarization control, which was the GR/GS1 complex with 50 μM DEX for 100% competition.
Table 2.
Summary of Glucocorticoid Receptor Competitor Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 0.5 μL | 1 or 4 mM compound in DMSO added to black 384-well plates |
| 2 | Buffer addition | 19.5 μL | 1×GR screening buffer, 1×Stabilizing peptide, 5 mM DTT |
| 3 | Fluorescent ligand addition | 10 μL | 4×Fluormone™ in assay buffer |
| 4 | Protein addition | 10 μL | 4× GR in assay buffer |
| 5 | Incubation time | 2 h | Covered with aluminum foil at ambient temperature |
| 6 | Assay readout | 485 and 513 nm | Fluorescence polarization, 100 readings/well |
1. 1 or 4 mM compound used for 12.5 and 50 μM final concentrations, respectively.
2. Room temperature assay performed.
4. Prepare GR on ice.
DTT, dithiothreitol.
MT polymerization assay: Test agents at 10 μM or DMSO (vehicle control) with 10 μM bovine tubulin in 1 M MSG, pH 6.6, and 400 μM GTP were combined to give a total volume of 100 μL per well and incubated on ice for 15 min to screen for MT destabilizers. The same mixture was used without GTP to identify MT stabilizers. Positive and negative controls were performed using 10 μM PTX and colchicine (CHC), respectively. Sample mixtures were transferred to a 96-well plate preheated to 37°C and absorbance readings were taken immediately every 1 min for 30 min. MT polymerization was measured by the change in absorbance of the solution at 340 nm with a SpectraMax M5 Microplate Reader. The relative MT stabilization or destabilization was measured from the absolute change in absorbance after 20 min normalized to the PTX or CHC absorbance, respectively, also at the 20 min time point. Table 3 provides a summary of the MT polymerization assay.
Table 3.
Summary of Microtubule Polymerization Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 1 μL | 1 mM compound in DMSO added to 96-well plates |
| 2 | Protein addition | 80 μL | 1.25 mg/mL tubulin in 1 M MSG, pH 6.6 |
| 3 | Incubation time | 15 min | On ice |
| 4 | GTP addition | 19 μL | 2.1 mM in assay buffer |
| 5 | Assay readout | 340 nm | Absorbance–kinetic mode, 1 reading/min for 30 min at 37°C |
4. GTP only added for MT destabilizer screen. MT stabilizer screen replaced with buffer only.
GTP, guanosine triphosphate; MSG, monosodium glutamate; MT, microtubule.
Hsp70 ATPase assay: Hsp70 (1300 nM) was incubated on ice with test agents at 10 and 50 μM or DMSO for 15 min in buffer consisting of 50 mM HEPES, pH 7.2, containing 2 mM MgCl2. Sodium orthovanadate (100 μM) was used as the positive control. Then, 1 mM ATP was added to the sample mixtures and were incubated at 32°C for 1 h in a final volume of 30 μL per well of a 384-well clear, flat-bottom plate. The amount of free phosphate was detected using the modified malachite kit according to the manufacturer's protocol. Samples were then read with a SpectraMax M5 Microplate Reader at 630 nm. The absorbance of an ATP blank was subtracted from all sample readings. Amount of ATPase activity inhibition was calculated by the amount of absorbance normalized to DMSO control. Table 4 provides a summary of this ATPase assay.
Table 4.
Summary of Hsp70 Adenosine Triphosphatase Activity Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 0.6 μL | 0.5 or 2.5 mM compound in DMSO added to 384-well plates |
| 2 | Protein addition | 24.4 μL | 1600 nM Hsp70 in 50 mM HEPES, pH 7.2, containing 2 mM MgCl2 |
| 3 | Incubation time | 15 min | On ice |
| 4 | ATP addition | 5 μL | 6 mM ATP in assay buffer |
| 5 | Incubation time | 1 h | Lidded, 32°C |
| 6 | Malachite reagent addition | 7.5 μL | PiColorLock™ Gold mixture |
| 7 | Incubation time | 5 min | Lidded, ambient temperature |
| 8 | Stabilizer addition | 3 μL | |
| 9 | Assay Readout | 630 nm | Absorbance |
1. 0.5 or 2.5 mM compound used for 10 and 50 μM final concentrations, respectively.
8. Do not directly mix PiColorLock™ Gold with stabilizer.
ATP, adenosine triphosphate.
Hsp90 ATPase assay: Hsp90 (1,300 nM) was incubated on ice with test agents at 10 and 50 μM or DMSO for 15 min in buffer consisting of 20 mM Tris-HCl, pH 7.4, containing 2 mM MgCl2. 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) (50 μM) was used as the positive control. Then, 1 mM ATP was added to the sample mixtures and were incubated at 32°C for 1 h in a final volume of 30 μL per well of a 384-well clear, flat-bottom plate. The amount of free phosphate was detected and ATPase activity inhibition was determined as described above and the protocol summarized in Table 5.
Table 5.
Summary of Hsp90 Adenosine Triphosphatase Activity Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 0.6 μL | 0.5 or 2.5 mM compound in DMSO added to 384-well plates |
| 2 | Protein addition | 24.4 μL | 1600 nM Hsp90 in 20 mM Tris-HCl, pH 7.4, containing 2 mM MgCl2 |
| 3 | Incubation time | 15 min | On ice |
| 4 | ATP addition | 5 μL | 6 mM ATP in assay buffer |
| 5 | Incubation time | 1 h | Lidded, 32°C |
| 6 | Malachite reagent addition | 7.5 μL | PiColorLock™ Gold mixture |
| 7 | Incubation time | 5 min | Lidded, ambient temperature |
| 8 | Stabilizer addition | 3 μL | |
| 9 | Assay Readout | 630 nm | Absorbance |
1. 0.5 or 2.5 mM compound used for 10 and 50 μM final concentrations, respectively.
8. Do not directly mix PiColorLock™ Gold with stabilizer.
Tris-HCl, tris(hydroxymethyl)aminomethane hydrochloric acid.
Cytoplasmic dynein ATPase assay: Test agents at 10 and 50 μM or DMSO were added to buffer consisting of 20 mM Tris-HCl, pH 7.6, containing 50 mM KCl, 5 mM MgCl2, and 28 nM dynein and were incubated for 15 min on ice. A positive control was performed using 100 μM sodium orthovanadate. Separately, MTs were performed by incubating 10 μM bovine tubulin with 10 μM PTX in 1 M MSG, pH 6.6, at 37°C for 30 min. The preformed MTs were then centrifuged at 16,000 g for 30 min at 37°C. The pellet was resuspended in the same buffer used for the cytoplasmic dynein with 10 μM PTX and incubated for an additional 20 min while maintaining samples at 37°C. Then, 1 mM ATP was added to the sample mixtures and were incubated at 37°C for 30 min with or without preformed MTs for a final volume of 50 μL per well of a 384-well clear, flat-bottom plate. The amount of free phosphate was detected and ATPase activity inhibition was determined as described above and the protocol summarized in Table 6.
Table 6.
Summary of Dynein Adenosine Triphosphatase Activity Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 1 μL | 0.5 or 2.5 mM compound in DMSO added to 384-well plates |
| 2 | Protein addition | 42 μL | 33.3 nM dynein in 20 mM Tris-HCl, pH 7.6, containing 50 mM KCl, 5 mM MgCl2 |
| 3 | Incubation time | 15 min | On ice |
| 4 | Warm samples | 10 min | Lidded, 37°C |
| 5 | Preformed MT addition | 2 μL | 10 μM MTs stabilized by 10 μM PTX in assay buffer |
| 6 | ATP addition | 5 μL | 10 mM ATP in assay buffer |
| 7 | Incubation time | 30 min | Lidded, 37°C |
| 8 | Malachite reagent addition | 12.5 μL | PiColorLock™ Gold mixture |
| 9 | Incubation time | 5 min | Lidded, ambient temperature |
| 10 | Stabilizer addition | 5 μL | |
| 11 | Assay Readout | 630 nm | Absorbance |
1. 0.5 or 2.5 mM compound used for 10 and 50 μM final concentrations, respectively.
5. For basal dynein assays, 2 μL buffer was used to replace MTs.
6. ATP prewarmed to 37°C before addition to plate.
10. Do not directly mix PiColorLock™ Gold with stabilizer.
PTX, paclitaxel.
Myosin Ca2+-ATPase assay: Rabbit skeletal muscle myosin II (125 nM) and the test agents at 10 and 50 μM were premixed in 50 mM Tris-HCl, pH 7.9, containing 0.23 M KCl and 2.5 mM CaCl2 and incubated on ice for 15 min. Then, 1 mM ATP was added to give a final volume of 50 μL per well of a 384-well clear, flat-bottom plate. NEM (100 μM) was used as the positive control. The reaction mixture was incubated at room temperature for 30 min. The amount of free phosphate was detected and activity inhibition was determined as described above and the protocol summarized in Table 7.
Table 7.
Summary of Myosin Ca2+-Adenosine Triphosphatase Assay
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Compound addition to plate | 1 μL | 0.5 or 2.5 mM compound in DMSO added to 384-well plates |
| 2 | Protein addition | 44 μL | 142 nM Tris-HCl, pH7.9, containing 0.23 M KCl, 2.5 mM CaCl2 |
| 3 | Incubation time | 15 min | On ice |
| 4 | ATP addition | 5 μL | 10 mM ATP in assay buffer |
| 5 | Incubation time | 30 min | Lidded, ambient temperature |
| 6 | Malachite reagent addition | 7.5 μL | PiColorLock™ Gold mixture |
| 7 | Incubation time | 5 min | Lidded, ambient temperature |
| 8 | Stabilizer addition | 3 μL | |
| 9 | Assay Readout | 630 nm | Absorbance |
1. 0.5 or 2.5 mM compound used for 10 and 50 μM final concentrations, respectively.
8. Do not directly mix PiColorLock™ Gold with stabilizer.
Statistics
The Z′-factor statistical coefficient of the cell-based GR-GFP translocation assay was calculated defined by 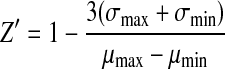 ,29 where σ is the standard deviation of the max and min fluorescence signals, and μ is the mean value of max and min signals, respectively. The Z-factor over a 3-day window to assess signal variability was reported as the mean±standard deviation (SD). Quantitation of cell images, percent GR ligand competition, MT polymerization assay, and percent ATPase inhibition were also reported as the mean±SD. The coefficient of determination (R2) was reported for the concentration-dependent curve fits.
,29 where σ is the standard deviation of the max and min fluorescence signals, and μ is the mean value of max and min signals, respectively. The Z-factor over a 3-day window to assess signal variability was reported as the mean±standard deviation (SD). Quantitation of cell images, percent GR ligand competition, MT polymerization assay, and percent ATPase inhibition were also reported as the mean±SD. The coefficient of determination (R2) was reported for the concentration-dependent curve fits.
Results and Discussion
In an effort to identify compounds that inhibited the retrograde transport of GR via cytoplasmic dynein, a HCS cell-based assay was used to screen the LOPAC-1,280 collection from Sigma. Based on these studies, we were able to identify a handful of compounds (shown in Fig. 1) that inhibited the DEX-induced translocation of GR-GFP to the nucleus mediated by cytoplasmic dynein. We further investigated the molecular targets of these compounds via a series of biochemical assays.
Fig. 1.

Chemical Structures of library of pharmacologically active compound-1280 hits. (A) 4-Phenyl-3-furoxancarbonitrile (3-cyano-4-phenyl-1,2,5-oxadiazole 2-oxide), (B) Parthenolide ((1aR,7aS,10aS,10bR,E)-1a,5-dimethyl-8-methylene-2,3,6,7,7a,8,10a,10b-octahydrooxireno[2′,3′:9,10]cyclodeca[1,2-b]furan-9(1aH)-one), (C) Apomorphine (rac-6-methyl-5,6,6a,7-tetrahydro-4H-dibenzo[de,g]quinoline-10,11-diol, (D) 6-Nitroso-1,2-benzopyrone (6-nitroso-2H-chromen-2-one), (E) Bay 11–7085 ((E)-3-(4-tert-butylphenylsulfonyl)prop-2-eneitrile).
Multi-parameter automated immunofluorescence microscopy was first utilized to provide high-content information on the cellular effects of the compounds. Such an analysis provided information on the location of GR-GFP within the cell, as well as effects on MT organization and mass, nuclear morphology, and cytotoxicity by surveying different components of the cell with specific labels: GR-GFP, tubulin antibodies, and Hoechst (Fig. 2A). The cellular phenotype of the DMSO vehicle control demonstrated well-organized cells with a few cells undergoing mitosis and GR-GFP located predominantly in the cytoplasm. The cells were also uniformly rounded with numerous sizeable cellular projections. Upon the addition of the GR agonist DEX, the cell morphology appeared very similar to the DMSO vehicle control with the exception that all of the GR-GFP had translocated into the nuclei as expected (Fig. 2A, B). The addition of several compounds in the LOPAC set 60 min before DEX treatment noticeably altered the GR-GFP distribution phenotype and decreased the DEX-induced nuclear GR-GFP content relative to the DEX control (Fig. 2). Compounds that caused a ≥50% decrease in GR-GFP accumulation in the nuclei were considered hits. Pretreatment with parthenolide, apomorphine, Bay 11–7085, and 4-phenyl-3-furoxancarbonitrile all inhibited DEX-induced GR nuclear translocation and led to the retention of GR-GFP in the cytoplasm (Fig. 2A, B). These five compounds also appeared to alter cell morphology in that cells exposed to these molecules exhibited fewer and shorter cellular projections protruding from the cell body when compared to the images of the DMSO- and DEX-treated control cells. With respect to the MT mass, however, only exposure to Bay 11–7085 altered the α-tubulin staining intensities of the treated cells relative to the DMSO and DEX control cells, and significantly reduced the cytoplasmic (mean ring average intensity) signal (Fig. 2C). In addition, only exposure to Bay 11–7085 significantly altered the number of cell nuclei detected in treated cells relative to the DMSO and DEX control cells (Fig. 2D). At concentrations >12.5 μM, Bay 11–7085 treatment was cytotoxic as indicated by the apparent reductions in cell counts and MT mass (Fig. 2C, D). The apparent reduction in MT mass produced in cells treated with 50 μM Bay 11–7085 (Fig. 2C) was probably secondary to its cytotoxic effects on cell number (Fig. 2D). Even though Bay 11–7085 at 12.5 μM was noncytotoxic, inhibition of GR-GFP translocation was still observed at this concentration. In contrast, the other compounds that inhibited DEX-induced GR-GFP translocation did not significantly affect either cell counts or MT mass relative to the DMSO and DEX controls. The Z′-factor for the GR-GFP translocation assay was determined to be 0.52±0.11 by measuring the max and min fluorescence signal of GR-GFP from fixed cells over a 3-day period, suggesting that the assay is well-suited and reliable for high-throughput screening.29
Fig. 2.

Multi-parameter immunofluorescence visualization and quantitation of treated cells. (A) GR-GFP (green) was used to track cytoplasmic dynein translocation, a fluorescent tubulin antibody (red) to label MTs, and Hoechst 33342 was used to label DNA (blue) to observe the nuclei. (B) Mean circ-ring average intensity difference of GR-GFP content was measured to determine extent of GR translocation inhibition. (C) Mean mass of stained MTs by tubulin antibody provided information on cell morphology. (D) Mean number of cells was counted to evaluate cell viability after treatment with compounds. SD was calculated for DMSO and DEX from 14 and 16 independent experiments, respectively. SD for test agents was determined from three independent experiments. The statistical effect size for the assay was determined to be Z′=0.52±0.11. GR, glucocorticoid receptor; DEX, dexamethasone; DMSO, dimethylsulfoxide; GFP, green fluorescent protein; MT, microtubule; SD, standard deviation.
Since ligand binding to GR is required to induce translocation from the cytoplasm to the nucleus,30 we investigated the ability of the five inhibitors to disrupt the binding of a fluorescent-labeled ligand to GR in a FP competitive binding assay. The GR agonist DEX inhibited ligand binding in the FP assay in a concentration-dependent manner (Fig. 3A) and exhibited an half maximal inhibitory concentration (IC50) value of 10.1±1.5 nM, corresponding well to previously published values.31 At both of the concentrations tested, 12.5 and 50 μM, neither parthenolide nor 4-phenyl-3-furoxancarbonitrile disrupted agonist binding to GR (Fig. 3B). In contrast, at 50 μM the other GR-translocation inhibitors appeared to compete for the GR ligand binding domain by an average of >60% (Fig. 2B). At 12.5 μM, however, the percent competition of GR-binding exhibited by these compounds was considerably lower and much more variable (Fig. 3B). Given the relatively large standard errors in the compound binding data, it is unclear whether the apparent competition of GR binding at high concentrations contributes to the inhibition of DEX-induced GR translocation observed at compound concentrations <12.5 μM. This was perhaps an indication that these compounds inhibited GR translocation through interactions with other targets within the cell.
Fig. 3.

Fluorescence polarization GR competitive binding assay. (A) Concentration-dependent evaluation of the competitive binding of the GR agonist DEX fitted by a one-site competition curve with R2=0.9976 and an half maximal inhibitory concentration value of 10.1±1.5 nM. (B) Percent competition of test compounds against binding to GR normalized to DEX control. Data represented as mean values with SD from two independent experiments done in triplicate.
Since retrograde transport requires the existence of functionally structured MTs,30 alterations in MTs can stall GR translocation by cytoplasmic dynein. Many compounds have been previously demonstrated to alter MT dynamics by either acting as MT stabilizers (PTX, discodermolide)32,33 or destabilizers (CHC, vincristine).34,35 MT perturbing agents have been shown to inhibit transport of p53 via cytoplasmic dynein to the nucleus.36 Compounds identified as MT perturbing agents may also serve as cytoplasmic dynein enhancers when used at low concentrations.37 Although only exposure to Bay 11–7085 was shown to alter MT mass in treated cells (Fig. 2C), we investigated the ability of the selected inhibitors to perturb MT dynamics in vitro relative to the MT stabilizer PTX and the MT destabilizer CHC, respectively (Table 8). Of the six compounds tested, 4-phenyl-3-furoxancarbonitrile and Bay 11–7085 both exhibited MT destabilization characteristics. 4-Phenyl-3-furoxancarbonitrile is a nitric oxide donor,38 whereas Bay 11–7085 is an nuclear factor kappa B inhibitor that induces apoptosis,39 and neither compound has been reported to alter MT dynamics. In contrast, parthenolide has been previously reported to exhibit MT stabilizing activity,40 and was the only compound tested that was found to induce MT polymerization in vitro. MT perturbation measurements by turbidity are effective for a small number of samples, but are not robust enough for high-throughput assays. We have recently examined the ability to polymerize MTs on biosensor surfaces and distinguish MT stabilizers from destabilizers,41 an assay format that may be applied to screening MT-perturbing agents at greater throughput in the future.
Table 8.
Inhibition of Guanosine Triphosphate-Induced Assembly and Hypernucleation/Polymer Stabilization of Bovine Brain Tubulin by Hits from the High-Content Screen Analysis of the Library of Pharmacologically Active Compound-1280 Collection
| Compound (10 μM) | Percent destabilization relative to 10 μM CHC | Percent stabilization relative to 10 μM PTX |
|---|---|---|
| 4-Phenyl-3-furoxancarbonitrile | 50.71±10.7 | NP |
| Parthenolide | −26.41±11.7 | 80.6±7.9 |
| (R)-Apomorphine HCl hydrate | −2.51±54.8 | NP |
| (S)-Apomorphine HCl hydrate | 23.81±6.8 | NP |
| 6-Nitroso-1,2-benzopyrone | 42.21±44.8 | NP |
| Bay 11–7085 | 64.5±27.4 | NP |
Data are mean values±SD (n=3).
NP, no polymerization; CHC, colchicines; SD, standard deviation.
Other factors, including the stability of the dynein-cargo complex, play an important role in the transport of cytoplasmic dynein. In the case of the GR, complex formation begins with the ATP-dependent binding of Hsp70 to the GR along with the nonessential co-chaperone Hsp40 creating a “primed” GR/Hsp70 complex.42 The GR/Hsp70 complex then binds to Hsp90 followed by the binding of p23 to stabilize this newly formed complex.43 GR translocation is dependent on its association with Hsp90 and it has been suggested that the dynamics of this complex is dependent on ligand binding to the receptor.7 A number of Hsp-binding immunophilins then link the cargo complex to the light and intermediate chains of cytoplasmic dynein.2 Inhibition of Hsp90 has been demonstrated to halt GR translocation.44 Hsp90 has a unique ATP binding pocket near its N-terminus, where specific inhibitors such as geldanamycin bind.45 The use of 17-AAG, which is an ansamycin antibiotic that binds Hsp90,46 as a control (Fig. 4) exhibited expected inhibition of Hsp90 activity.47 Given the importance of HSP in stabilizing the cargo complex with cytoplasmic dynein, we investigated whether or not the targeting of these proteins inhibited GR translocation. Ssa1p and Hsc82p, the yeast versions of Hsp70 and 90, respectively, were used in this study because of the shared homology with mammalian versions48 along with the fact that the formation of the cargo complex is conserved among different species.49 Therefore, screening for compounds that alter the ATPase activity of HSPs can indicate potential targets of compounds inhibiting GR translocation. In addition, the identification of HSP inhibitors/activators could prove to be useful since Hsps are a therapeutic target in oncology50 and against neurodegenerative diseases because of their involvement in protein folding.51 Through the use of a colorimetric assay, the percent inhibition of ATPase activity was calculated by comparing the amount of inorganic phosphate released in the presence of the compounds relative to a DMSO control. At the highest concentration tested, 50 μM, none of the compounds had any significant effect on Hsc82p (Hsp90) (Fig. 4). Concentrations of test agents >10 μM caused Ssa1p to precipitate, rendering measurement of ATPase activity inaccurate.
Fig. 4.

HSP ATPase inhibition assays. (A) Percent inhibition of ATPase activity of yeast Hsp70 (Ssa1p) with test compounds compared to DMSO control and sodium orthovanadate as a control. (B) Percent inhibition of ATPase activity yeast Hsp90 (Hsc82p) with test compounds compared to DMSO control and 17-(allylamino)-17-demethoxygeldanamycin as a control. Data represented as mean values with SD from two independent experiments done in triplicate. ATP, adenosine triphosphate; HSP, heat-shock protein.
The ability of the compounds to inhibit cytoplasmic dynein's ATPase activity was then investigated. Only four of the compounds exhibited a significant concentration-dependent inhibition of the ATPase activity of both basal and MT-enhanced activity of cytoplasmic dynein (Fig. 5). Both enantiomers of apomorphine inhibited cytoplasmic dynein with IC50 values of ∼1.5 μM, whether in the basal or MT-stimulated state (Table 9). 6-Nitroso-1,2-benzopyrone and Bay 11–7085 also inhibited cytoplasmic dynein, but there was a more substantial change in their IC50 values between basal and MT-stimulated states. Although there was no indication that any of the compounds significantly altered the interaction of cytoplasmic dynein's MT-binding domain with the MTs, this type of comparison can be important in identifying compounds that interfere with the dynein-MT interaction in future screens. The decreased IC50 value of 6-nitroso-1,2-benzopyrone in the MT-stimulated state could indicate the targeting of the sulfhydryls on tubulin heterodimers52 since nitroso derivatives have been shown bind to protein sulfhydryls,53 subsequently destabilizing the MTs as suggested in Table 8, rather than binding to a defined active site. The double sigmoidal behavior of 6-nitroso-1,2-benzopyrone also suggested its nonspecific binding to cytoplasmic dynein (Fig. 5B, D).
Fig. 5.

Cytoplasmic dynein ATPase inhibition assay. (A) Percent inhibition of ATPase activity of basal dynein with test compounds compared to DMSO control and sodium orthovanadate as a control. (B) Concentration-dependent inhibition of basal cytoplasmic dynein ATPase activities of (R)-apomorphine (R2=0.9943), (S)-apomorphine (R2=0.9918), 6-nitroso-1,2-benzopyrone (R2=0.9866), Bay 11–7805 (R2=0.9929). (C) Percent inhibition of ATPase activity of MT-stimulated dynein with test compounds compared to DMSO control and sodium orthovanadate as a control. (D) Concentration-dependent inhibition of MT-stimulated cytoplasmic dynein ATPase activities of (R)-apomorphine (R2=0.9973), (S)-apomorphine (R2=0.9937), 6-nitroso-1,2-benzopyrone (R2=0.9379), Bay 11–7805 (R2=0.9916). Data represented as mean values with SD from two independent experiments done in quadruplet.
Table 9.
Biochemical Half Maximal Inhibitory Concentration Values of Test Agents Against Both Basal and Microtubule-Stimulated Cytoplasmic Dynein
| |
IC50 (μM) |
|
|---|---|---|
| Compound | Basal | MT Stimulated |
| (R)-Apomorphine HCl hydrate | 1.5±0.5 | 1.7±0.3 |
| (S)-Apomorphine HCl hydrate | 1.5±0.3 | 1.3±0.2 |
| 6-Nitroso-1,2-benzopyrone | 1.8±1.1 | 0.7±1.2 |
| Bay 11–7085 | 4.6±0.5 | 8.9±0.5 |
Data are mean values±SD (n=3).
IC50, half maximal inhibitory concentration.
Finally, to rule out any compounds that might be general ATPase motor protein inhibitors, we tested the ability of compounds to inhibit the ATPase activity of myosin. At the two concentrations tested, 10 and 50 μM, only the apomorphines exhibited any significant inhibition of the ATPase activity of myosin (Fig. 6A) with concentration-dependent behavior and IC50 values of 1.4±0.4 and 3.2±0.7 μM for the R and S enantiomers, respectively (Fig. 6B). Despite being the most potent cytoplasmic dynein ATPase inhibitors, the broad spectrum of target activities that the apomorphine enantiomers inhibited in our panel of assays suggests that they were promiscuous hits.
Fig. 6.

Myosin ATPase inhibition assay. (A) Percent inhibition of ATPase activity of myosin with test compounds compared to DMSO control and NEM as a control. (B) Concentration-dependent inhibition of myosin ATPase activity by (R)-apomorphine (R2=0.9661) and (S)-apomorphine (R2=0.9287). Data represented as mean values with SD from two independent experiments done in triplicate. NEM, N-ethylmaleimide.
Conclusions
Cell-based screening reliably revealed a number of small molecules from a LOPAC collection that stalled the agonist-induced transport of GR from the cytoplasm to the nucleus. Since there are a number of potential reasons for interruption of this dynein-mediated transport, we sought to identify the target(s) of these molecules through a number of biochemical assays applicable to the translocation process. From the five compounds tested, the apomorphine enantiomers exhibited the most potency toward the ATPase activities of cytoplasmic dynein and myosin, but also appeared to inhibit GR binding. The lack of specificity these mirror image molecules exhibit, however, made them unattractive for further study in our quest. Parthenolide is a previously recognized MT stabilizer that most likely altered the tracks needed for dynein to transport GR-GFP to the nuclei. Based on the chemical structures of the remaining cell-based screening hits, it is likely that the compounds targeted reactive sulfhydryls of dynein and MTs, both contributing to inhibition of dynein-mediated transport. The reliability of the hits from the cell-based screen was further supplemented by the concentration-dependent inhibition of activity determined from the biochemical assays and with good R2 values. Ideally, specific dynein-mediated transport inhibitors would either target dynein itself or one of the other important proteins associated with the transport process. Although the hits from the cell-based screen of the LOPAC-1280 collection did not exhibit this desired profile, these assays demonstrated the ability to examine cell permeable inhibitors of GR translocation and attempt to identify the target(s) of these molecules through a series of readily implemented biochemical assays. We envision that screening of a larger library of compounds for inhibition of cell-based cytoplasmic dynein transport will reveal specific cytoplasmic dynein and/or HSP inhibitors. We also believe that this route of identifyifollowed by target validation is an improvement over conventional biochemical screening that suffers when hits eventually are cell impermeable and/or severely cytotoxic.
Abbreviations
- AAA
ATPases associated with diverse activities
- AR
androgen receptor
- ATP
adenosine triphosphate
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- Ch
channel
- CHC
colchicine
- DEX
dexamethasone
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethylsulfoxide
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol tetraacetic acid
- ER
estrogen receptor
- FBS
fetal bovine serum
- FP
fluorescence polarization
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- GR
glucocorticoid receptor
- GTP
guanosine triphosphate
- HCS
high-content screen
- HEPES
-4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HSC
heat-shock cognate
- HSP
heat-shock protein
- IC50
half maximal inhibitory concentration
- LOPAC
library of pharmacologically active compounds
- MCRAID
mean circ-ring average intensity difference
- MES
2-(N-morpholino)ethanesulfonic acid
- MSG
monosodium glutamate
- MT
microtubule
- MTL
molecular translocation
- NEM
N-ethylmaleimide
- Ni-NTA
nickel nitrilotriacetic acid
- PBS
phosphate-buffered saline
- PIPES
piperazine-N,N0-bis(2-ethanesulfonic acid)
- PTX
paclitaxel
- SD
standard deviation
- 17-AAG
17-(allylamino)-17-demethoxygeldanamycin
- Tet
tetracycline
- TRIS-HCl
tris(hydroxymethyl)aminomethane hydrochloric acid
- TRITC
tetramethylrhodamine-5-(and 6)-isothiocyanate.
Acknowledgments
We thank Prof. Richard Vallee along with Drs. Peter Hook and Richard McKenney from Columbia University for generously providing the baculovirus required for dynein purification as well as their continued support and discussions throughout the project. We also thank Robert Youker and Christine Wright for supplying some of the purified Hsc82p and Ssa1p, respectively, that was used in this study and Dr. Annette Chiang for her technical assistance. This work was funded by National Institutes of Health grants NS057026 to B.W.D. and DK079307 (the University of Pittsburgh George O'Brien Kidney Research Core Center) to J.L.B.
Disclosure Statement
No competing financial interests exist.
References
- 1.Glass CK. Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 2.Harrell JM. Murphy PJM. Morishima Y. Chen H. Mansfield JF. Galigniana MD. Pratt WB. Evidence for glucocorticoid receptor transport on microtubules by dynein. J Biol Chem. 2004;279:54647–54654. doi: 10.1074/jbc.M406863200. [DOI] [PubMed] [Google Scholar]
- 3.Samso M. Radermacher M. Frank J. Koonce MP. Structural characterization of a dynein motor domain. J Mol Biol. 1998;276:927–937. doi: 10.1006/jmbi.1997.1584. [DOI] [PubMed] [Google Scholar]
- 4.Vale RD. AAA proteins: lords of the ring. J Cell Biol. 2000;150:F13–F20. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reck-Peterson SL. Vale RD. Molecular dissection of the roles of nucleotide binding and hydrolysis in dynein's AAA domain in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2004;101:1491–1495. doi: 10.1073/pnas.2637011100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Carter AP. Garbarino JE. Wilson-Kubalek EM. Shipley WE. Cho C. Milligan RA. Vale RD. Gibbons IR. Structure and functional role of dynein's microtubule-binding domain. Science. 2008;322:1691–1695. doi: 10.1126/science.1164424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pratt WB. Galigniana MD. Harrell JM. DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signaling protein movement. Cell Signal. 2004;16:857–872. doi: 10.1016/j.cellsig.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Rayala SK. der Hollander P. Balasenthil S. Yang Z. Broaddus RR. Kumar R. Function regulation of oestrogen receptor pathway by the dynein light chain 1. EMBO Rep. 2005;6:538–545. doi: 10.1038/sj.embor.7400417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galigniana MD. Harrell JM. O'Hagen HM. Ljungman M. Pratt WB. Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J Biol Chem. 2004;279:22483–22489. doi: 10.1074/jbc.M402223200. [DOI] [PubMed] [Google Scholar]
- 10.McDonald D. Vodicka MA. Lucero G. Svitkina TM. Borisy GG. Emerman M. Hope TJ. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bremner KH. Scherer J. Yi J. Vershinin M. Gross SP. Vallee RB. Adenovirus transport via direct interaction of cytoplasmic dynein with viral capsid hexon subunit. Cell Host Microbe. 2009;6:523–535. doi: 10.1016/j.chom.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corthésy-Theulaz I. Pauloin A. Pfeffer SR. Cytoplasmic dynein participates in the centrosomal localization of the golgi complex. J Cell Biol. 1992;118:1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salina D. Bodoor K. Eckley DM. Schroer TA. Rattner JB. Burke B. Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell. 2002;108:97–107. doi: 10.1016/s0092-8674(01)00628-6. [DOI] [PubMed] [Google Scholar]
- 14.Sharp DJ. Rogers GC. Scholey JM. Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat Cell Biol. 2000;2:922–930. doi: 10.1038/35046574. [DOI] [PubMed] [Google Scholar]
- 15.Vaisberg EA. Koonce MP. McIntosh JR. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J Cell Biol. 1993;123:849–858. doi: 10.1083/jcb.123.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh E. Skibbens RV. Cheng JW. Salmom ED. Bloom K. Spindle dynamics and cell cycle regulation of dynein in the budding yeast, Saccharaomyces cerevisiae. J Cell Biol. 1995;130:687–700. doi: 10.1083/jcb.130.3.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouchard P. Penningroth SM. Cheung A. Gagnon C. Bardin CW. Erythro-9-[3-(2-Hydroxynonyl)]adenine is an inhibitor of sperm motility that blocks dynein ATPase and protein carboxylmethylase activities. Proc Natl Acad Sci USA. 1981;78:1033–1036. doi: 10.1073/pnas.78.2.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallee RB. Shpetner HS. Motor proteins of cytoplasmic microtubules. Annu Rev Biochem. 1990;59:909–932. doi: 10.1146/annurev.bi.59.070190.004401. [DOI] [PubMed] [Google Scholar]
- 19.Blum JJ. Hayes A. Effect of thiourea and substituted thioureas on dynein ATPase and on the turbidity response of Tetrahymena cilia. J Supramol Struct. 1979;12:23–34. doi: 10.1002/jss.400120104. [DOI] [PubMed] [Google Scholar]
- 20.Takito J. Nakamura H. Kobayashi J. Ohizumi Y. Ebisawa K. Nonomura Y. Purealin, a novel stabilizer of smooth muscle myosin filaments that modulates ATPase activity of dephosphorylated myosin. J Biol Chem. 1986;261:13861–13865. [PubMed] [Google Scholar]
- 21.Fang Y. Yokota E. Mabuchi I. Nakamura H. Ohizumi Y. Purealin blocks the sliding movement of sea urchin flagellar axonemes by selective inhibition of half the ATPase activity of axonemal dyneins. Biochemistry. 1997;36:15561–15567. doi: 10.1021/bi971363n. [DOI] [PubMed] [Google Scholar]
- 22.Zhu G. Yang F. Balachandran R. Höök P. Vallee RB. Curran DP. Day BW. Synthesis and biological evaluation of purealin and analogues as cytoplasmic dynein heavy chain inhibitors. J Med Chem. 2006;49:2063–2076. doi: 10.1021/jm051030l. [DOI] [PubMed] [Google Scholar]
- 23.Walker D. Htun H. Hager GL. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods. 1999;19:386–393. doi: 10.1006/meth.1999.0874. [DOI] [PubMed] [Google Scholar]
- 24.Trask OJ. Nickischer D. Burton A. Williams RG. Kandasamy RA. Johnston PA. Johnston PA. High-throughput automated confocal microscopy imaging screen of a kinase-focused library to identify p38 mitogen-activated protein kinase inhibitors using the GE InCell 3000 Analyzer. Methods Mol Biol. 2009;565:159–186. doi: 10.1007/978-1-60327-258-2_8. [DOI] [PubMed] [Google Scholar]
- 25.Hamel E. Lin CM. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry. 1984;23:4173–4184. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 26.McClellan AJ. Brodsky JL. Mutation of the ATP-binding pocket of SSA1 indicates that a functional interaction between ssa1p and ydj1p is required for post-translational translocation into the yeast endoplasmic reticulum. Genetics. 2000;156:501–512. doi: 10.1093/genetics/156.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youker RT. Walsh P. Beilharz T. Lithgow T. Brodsky JL. Distinct roles for the hsp40 and hsp90 molecular chaperones during cystic fibrosis transmembrane conductance regulator degradation in yeast. Mol Biol Cell. 2004;15:4787–4797. doi: 10.1091/mbc.E04-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hook P. Mikami A. Shafer B. Chait BT. Rosenfeld SS. Vallee RB. Long range allosteric control of cytoplasmic dynein ATPase activity by the stalk and C-terminal domains. J Biol Chem. 2005;280:33045–33054. doi: 10.1074/jbc.M504693200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J. Chung TDY. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 30.Galigniana MD. Scruggs JL. Herrington J. Welsh MJ. Carter-Su C. Housley PR. Pratt WB. Heat shock protein 90-dependent (geldanamycin-inhibited) movement of glucocorticoid receptor through the cytoplasm to the nucleus requires intact cytoskeleton. Mol Endocrinol. 1998;12:1903–1913. doi: 10.1210/mend.12.12.0204. [DOI] [PubMed] [Google Scholar]
- 31.Reynolds PD. Pittler SJ. Scammell JG. Cloning and expression of the glucocorticoid receptor from the squirrel monkey (Saimiri boliviensis boliviensis), a glucocorticoid resistant primate. J Clin Endocrinol Metab. 1997;82:465–472. doi: 10.1210/jcem.82.2.3755. [DOI] [PubMed] [Google Scholar]
- 32.Schiff PB. Fant J. Horwitz SB. Promotion of microtubule assembly in vitro by taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 33.Ter Haar E. Kowalski RJ. Hamel E. Lin CM. Longley RE. Gunasekera SP. Rosenkranz HS. Day BW. Discodermolide, a cytotoxic marine agent that stabilizes microtubules more potently than taxol. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 34.Bhattacharyya B. Panda D. Gupta S. Banerjee M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097. [DOI] [PubMed] [Google Scholar]
- 35.Jordan MA. Himes RH. Wilson L. Comparison of the effects of vinblastine, vincristine, vindesine, and vinepidine on microubule dynamics and cell proliferation in vitro. Cancer Res. 1985;45:2741–2747. [PubMed] [Google Scholar]
- 36.Giannakakou P. Sackett DL. Ward Y. Webster KR. Blagosklonny MV. Fojo T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol. 2000;2:709–717. doi: 10.1038/35036335. [DOI] [PubMed] [Google Scholar]
- 37.Giannakakou P. Nakano M. Nicolaou KC. O'Brate A. Yu J. Blagosklonny MV. Greber UF. Fojo T. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc Natl Acad Sci USA. 2002;99:10855–10860. doi: 10.1073/pnas.132275599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Medana C. Ermondi G. Fruttero R. Di Stilo A. Ferretti C. Gasco A. Furoxans as nitric oxide donors. 4-Phenyl-3-furoxancarbonitrile: thiol-mediated nitric oxide release and biological evaluation. J Med Chem. 1994;37:4412–4416. doi: 10.1021/jm00051a020. [DOI] [PubMed] [Google Scholar]
- 39.Pierce JW. Schoenleber R. Jesmok G. Best J. Moore SA. Collins T. Gerritsen ME. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 40.Miglietta A. Bozzo F. Gabriel L. Bocca C. Microtubule-interfering activity of parthenolide. Chem Biol Interact. 2004;149:165–173. doi: 10.1016/j.cbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 41.Daghestani HN. Fernig DG. Day BW. Evaluation of biosensor surfaces for the detection of microtubule perturbation. Bionsens Bioelectron. 2009;25:136–141. doi: 10.1016/j.bios.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy PJM. Morishima Y. Chen H. Galigniana MD. Mansfield JF. Simons SS. Pratt WB. Visualization and mechanism of assembly of a glucocorticoid receptor·hsp70 complex that is primed for subsequent hsp90-dependent opening of the steroid binding cleft. J Biol Chem. 2003;278:34764–34773. doi: 10.1074/jbc.M304469200. [DOI] [PubMed] [Google Scholar]
- 43.Dittmar KD. Demady DR. Stancato LF. Krishna P. Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery—the role of p23 is to stabilize receptor·hsp90 heterocomplexes formed by hsp90·p60·hsp70. J Biol Chem. 1997;272:21213–21220. doi: 10.1074/jbc.272.34.21213. [DOI] [PubMed] [Google Scholar]
- 44.Czar MJ. Galigniana MD. Silverstein AM. Pratt WB. Geldanamycin, a heat shock protein 90-binding benzoquinone ansamycin, inhibits steroid-dependent translocation of the glucocorticoid receptor from the cytoplasm to the nucleus. Biochemistry. 1997;36:7776–7785. doi: 10.1021/bi970648x. [DOI] [PubMed] [Google Scholar]
- 45.Grenert JP. Sullivan WP. Fadden P. Haystead TA. Clark J. Mimnaugh E. Krutzsch H. Ochel HJ. Schulte TW. Sausville E. Neckers LM. Toft DO. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem. 1997;272:23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- 46.Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- 47.Rowlands MG. Newbatt YM. Prodromou C. Pearl LH. Workman P. Aherne W. High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Anal Biochem. 2004;327:176–183. doi: 10.1016/j.ab.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 48.Schlesinger MJ. Heat shock proteins: the search for functions. J Cell Biol. 1986;103:321–325. doi: 10.1083/jcb.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrell JM. Kurek I. Breiman A. Radanyi C. Renoir JM. Pratt WB. Galigniana MD. All of the protein interactions that link steroid receptor·hsp90·immunophilin heterocomplexes to cytoplasmic dynein are common to plant and animal cells. Biochemistry. 2002;41:5581–5587. doi: 10.1021/bi020073q. [DOI] [PubMed] [Google Scholar]
- 50.Calderwood SK. Khaleque MA. Sawyer DB. Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–172. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 51.Luo W. Sun W. Taldone T. Rodina A. Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5:24–31. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikeda Y. Steiner M. Sulfhydryls of platelet tubulin: their role in polymerization and colchicine binding. Biochemistry. 1978;17:3454–3459. doi: 10.1021/bi00610a005. [DOI] [PubMed] [Google Scholar]
- 53.Ji Y. Akerboom TPM. Sies H. Thomas JA. S-Nitrosylation and S-glutathiolation of protein sulfhydryls by S-nitroso glutathione. Arch Biochem Biophys. 1999;362:67–78. doi: 10.1006/abbi.1998.1013. [DOI] [PubMed] [Google Scholar]