

Abstract
Recent expansion of immunocompromised population has led to significant rise in zygomycosis caused by filamentous fungus Rhizopus oryzae. Due to emergence of fungal resistance and side-effects of antifungal drugs, there is increased demand for novel drug targets. The current study elucidates molecular interactions of peptide drugs with G-6-P synthase (catalyzing the rate-limiting step of fungal cell wall biosynthetic pathway) of R.oryzae by molecular docking studies. The PDB structures of enzyme in R.oryzae are not known which were predicted using I-TASSER server and validated with PROCHECK. Peptide inhibitors, FMDP and ADGP previously used against enzyme of E.coli (PDBid: 1XFF), were used for docking studies of enzyme in R.oryzae by SchrödingerMaestro v9.1. To investigate binding between enzyme and inhibitors, Glide and Induced Fit docking were performed. IFD results of 1XFF with FMDP yielded C1, R73, W74, T76, G99 and D123 as the binding sites. C379 and Q427 appear to be vital for binding of R.oryzae enzymes to inhibitors. The comparison results of IFD scores of enzyme in R.oryzae and E.coli (PDBid: 2BPL) yield appreciable score, hinting at the probable effectiveness of inhibitors FMDP and ADGP against R.oryzae, with ADGP showing an improved enzyme affinity. Moreover, the two copies of gene G-6-P synthase due to extensive fungal gene duplication, in R. oryzae eliminating the problem of drug ineffectiveness could act as a potential antifungal drug target in R. oryzae with the application of peptide ligands.
Keywords: 2-amino-2-deoxy-D-glucitol-6-phosphate (ADGP), Docking, N3-(4-methoxyfumaroyl)-L-2, 3-diaminopropanoic acid (FMDP), Glucosamine-6-phosphate (G-6-P) synthase, Phylogenetics, Zygomycosis
Background:
A significant increase in the immunocompromised populations suffering from AIDS, cancer, diabetes mellitus or undergoing organ transplants and immunosuppressive therapy have been encountered in the past two decades [1–3]. Such patients are prone to be infected with opportunistic invasive fungal infections such as zygomycosis caused by the filamentous fungus Rhizopus oryzae belonging to the phylum Zygomycetes. Zygomycosis is characterized by nodular lesions and inflammation leading to tissues undergoing extensive necrosis, formation of ulcers and finally dissemination. Number of recent cases of zygomycosis has been observed globally with an overall mortality rate of approximately 80% [2]. Its treatment requires parallel intervention of surgery and antifungal drugs. The antifungals comprise Amphotericin B, Posaconazole and Echinocandins that are nephrotoxic and hepatotoxic [3]. The drugs are mostly used in combinations for synergistic effects, but owing to the emergence of drug-resistant fungal strains, their varied susceptibility towards antifungal agents [4] and early diagnoses inadequacies of mycoses, further research is critical for the mining of new antifungal targets leading to the development of next generation antifungal drugs. The enzyme G-6-P synthase [EC 2.6.1.16] catalyzes the rate-limiting step of the fungal cell wall biosynthetic pathway (Figure 1) and forms chitin, a major cell wall component [5, 6]. Its potentiality as an antifungal target has been studied in E. coli [6]. G-6-P synthase has been cloned, purified and biochemically characterized in E. coli [6], S. cerevisiae [7], Candida albicans [8], Volvariella volvacea [9] and humans [10]. Further, the enzyme kinetics and IC50 values of inhibitors like FMDP have been determined. In R. oryzae, the enzyme has not been cloned and characterized so far to the best of our knowledge, subsequently, its X-ray crystallographic structure is not yet known. Rapid progress in genomic studies has accelerated the drug discovery process. Genome sequencing of R. oryzae was carried out by Broad Institute of MIT and Harvard [ http://www.broadinstitute.org/] and it was found that G-6-P synthase in R. oryzae is encoded by two genes viz. RO3G_04247.3 and RO3G_14807.3 located at chromosomes 7 and 15, respectively. Both the genes consist of 12.4% introns, share a nucleotide sequence similarity of 89% and amino acid sequence similarity of 97% (Table 1 see supplementary material). Two chemically synthesized peptide inhibitors, FMDP and ADGP have been previously studied against G-6-P synthase of E. coli [6], S. cerevisiae [7], C. albicans [11], V. volvacea [9] and humans [12]. FMDP is a highly selective irreversible inhibitor of G-6-P synthase which is effectively transported into the cells and have shown promising in-vitro activity against S.cerevisiae and in-vivo activity to mammalian cell lines or to mice without toxicity. Therefore, in the current study, these inhibitors have been chosen for docking studies with the predicted structures of G-6-P synthase of R. oryzae using the software Schrödinger Maestro v9.1 in an attempt to discuss its potentiality as an antifungal drug target in R. oryzae and to provide insights into the use of inhibitors against the target such that they can be exploited further for the treatment of zygomycosis in the near future.
Figure 1.

Fungal cell wall biosynthetic pathway eventually leading to the formation of chitin. Glucosamine-6-phosphate synthase [EC 2.6.1.16] is the drug target selected in R. oryzae which forms chitin, a major component of fungal cell wall. (Source: KEGG)
Methodology:
Prediction, validation and preparation of protein (receptor):
The 3-dimensional structures of G-6-P synthase of two genes of R. oryzae (RO3G_04247.3 and RO3G_14807.3) were predicted using the online protein structure and function prediction server I-TASSER [13– 15]. The stereochemical quality of the predicted structures was measured employing PROCHECK which yielded Ramachandran Plots displaying favourable conformations obtained after an analysis of 163 structures at resolution 2.0 Å or better [16]. Subsequently, the proteins were analyzed for their potential binding pockets with the help of CASTp server [17]. The preparation of predicted proteins was carried out with the help of docking software SchrödingerMaestro v9.1. Foremost, the missing side-chain atoms within the protein residues were predicted by Prime (Schrödinger). The crystallographic water molecules, ions and cofactors were deleted, hydrogen atoms were added and formal charges along with bond orders were assigned to the structures.
Sketching and preparation of ligand:
The ligands FMDP and ADGP were sketched in ACD/ChemSketch (Freeware) and saved in MDL molfiles [V2000\. Subsequently, they were prepared using LigPrep (Schrödinger) by modifying the torsions of the ligands and assigning them appropriate protonation states. In Glide (Schrödinger), 32 stereochemical structures were generated per ligand with possible states at target pH 7.0 ± 2.0 using Ionizer, tautomerized, desalted and optimized by producing lowenergy 3D structure for the ligand under the OPLS 2005 force field while retaining the specified chiralities of the input Maestro file.
Receptor grid generation:
Receptor grids were calculated for prepared proteins such that various ligand poses bind within the predicted active site during docking. In Glide, grids were generated keeping the default parameters of van der Waals scaling factor 1.00 and charge cutoff 0.25 subjected to OPLS 2001 force field. A cubic box of specific dimensions centred around the centroid of the active site residues (predicted by CASTp) was generated for each receptor. The bounding box was set to 14 Å × 14 Å × 14 Å for docking experiments.
Glide Standard Precision (SP) ligand docking:
SP flexible ligand docking was carried out in Glide of Schrödinger-Maestro v8.5 [18– 20] within which penalties were applied to non-cis/trans amide bonds. Van der Waals scaling factor and partial charge cutoff was selected to be 0.80 and 0.15, respectively for ligand atoms. Final scoring was performed on energy-minimized poses and displayed as Glide score. The best docked pose with lowest Glide score value was recorded for each ligand.
Induced fit docking (IFD) Extra Precision (XP):
IFD XP was performed using the module Induced Fit Docking of Schrödinger-Maestro v9.1 [21]. The entire receptor molecule constrained minimized with an RMSD cutoff of 0.18 Å was selected for generation of centroid of the residues and the box size was generated automatically. The initial Glide docking for each ligand was carried out. Side chains were trimmed automatically based on B-factor, with receptor and ligand van der Waals scaling of 0.70 and 0.50, respectively; and the number of poses generated were set to be 20. Prime side chain prediction and minimization was carried out in which residues were refined within 5.0 Å of ligand poses and side chains were optimized. This leads to a ligand structure and conformation that is induced fit to each pose of the receptor structure. Finally, Glide XP redocking was carried out into structures within 30.0 kcal/mol of the best structure, and within the top 20 structures overall. The ligand was rigorously docked into the induced-fit receptor structure and the results yielded an IFD score for each output pose.
Results and Discussion:
Prediction and validation of protein:
The 3D structures of the two genes encoding G-6-P synthase in R. oryzae have been predicted by I-TASSER (Figure 2) which yield the Confidence score (C-score) ranging from -5 to 2, for estimating the quality of the predicted models and higher score indicates better predicted structure. TM-score and Root Mean Square Deviation (RMSD) are standard parameters used to indicate the closeness of the predicted model to the native structure. TM-score > 0.15 signifies a model of correct topology. On comparison of the C-scores and TM-scores of predicted 3D structures, RO3G_04247.3 seems to be slightly better predicted than RO3G_14807.3. In context with the proteins' secondary structure, the N-terminal domain of the enzyme is made up of anti-parallel β-sheets surrounded by α-helices (αββα- core structure) as is observed in other members of the superfamily of the N-terminal nucleophile (Ntn) hydrolases. PROCHECK sketches Ramachandran plots (Figure 3) between the Phi (Φ) and Psi (Ψ) torsion angles of all residues of the predicted proteins, except the amino acids present at the chain termini. Glycine residues are shown in black triangles. The residues in the core (red coloured) region represent the most favoured combinations of the torsion values indicating low-energy regions and the additional/generously allowed regions are in yellow. On evaluation of the Ramachandran plots, it is observed that the torsion angles in the plot of RO3G_14807.3 slightly lack real clustering in the core regions and residues are more dispersed as compared to the plot of RO3G_04247.3. Hence, there is a trivial difference between the residues falling in the core region for both the genes of R. oryzae. The validated structures were further used to predict active sites by CASTp out of which the binding pocket with deepest cleft was selected for our study (results not shown).
Figure 2.

Secondary structure prediction of G-6-P synthase by ITASSER. a. RO3G_04247.3 C-score: 0.35, Exp. TM-score: 0.67±0.13, Exp. RMSD: 8.8±4.6 Å b. RO3G_14807.3 C-score: - 0.53, Exp. TM-score: 0.65±0.13, Exp. RMSD: 9.3±4.6 Å. It consists of α-helices (in red), β-sheets (in cyan) and loops (in gray).
Figure 3.

Ramachandran plots of predicted structures of G-6-P synthase estimated by PROCHECK. 67.3% residues of a. RO3G_04247.3 and 64.0% of b. RO3G_14807.3 fall in the most favoured regions (red coloured). Yellow shaded areas display the residues in additional/generously allowed regions. The labeling is as follows: A- Core alpha, a- Allowed alpha, ˜a- Generous alpha, B- Core beta, b- Allowed beta, ˜b- Generous beta, L- Core left-handed alpha, l- Allowed left-handed alpha, ˜l- Generous left-handed alpha, p- Allowed epsilon, ˜p- Generous epsilon
Docking studies:
The best ligand binding pose with the least Glide/IFD score or energy was chosen. Previous docking studies on G-6-P synthase of E. coli [6] were carried out with PDB ID: 1gdo which is currently superseded by 1XFF in the Protein Data Bank (PDB). The Glide SP docking calculations for Glutaminase domain of G-6-P synthase of E. coli (PDB ID: 1XFF) with FMDP were not reproduced as per literature. In view of the fact that, Glide docking uses the basic assumption of a rigid receptor; it may not be adequate if the ligands (Figure 4) induce significant conformational changes in the receptor. Hence, flexible receptors were generated by carrying out induced fit XP docking which uses Glide with Prime to allow for receptor flexibility. The protein-ligand binding interactions of 1XFF with FMDP were consistent with literature after performing IFD (Table 2 see supplementary material). The functions of few of the amino acids have already been elucidated in previous studies [6]. Cys1 is known to play a major catalytic role in the substrate-induced activation of the enzyme by acting as a nucleophile and Thr76, Gly99 and Asp123 are responsible to form stable hydrogen bonds with the amido moiety of the inhibitor. Docking studies in R. oryzae have not been conducted till date, to the best of our knowledge; hence, molecular docking was performed for the organism (Figure 5). Cys379 and Gln427 emerged out to be crucial for binding of R.oryzae enzymes to inhibitors and they could be part of coevolving residues that were fixed over time [22]. The docking calculations reveal that the two genes of R. oryzae encoding G-6-P synthase interact slightly differently to both the inhibitors as is apparent from their binding energies (Table 2, see supplementary material).
Figure 4.
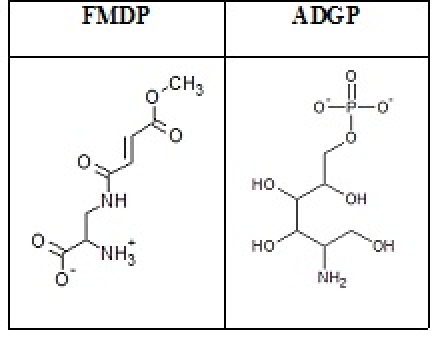
Peptide inhibitors. FMDP and ADGP drawn in ACD/ChemSketch (Freeware).
Figure 5.

Screenshots of docking calculations performed using IFD of Schrödinger-Maestro v9.1. a. RO3G_04247.3 docked with FMDP b. RO3G_04247.3 docked with ADGP c. RO3G_14807.3 docked with FMDP d. RO3G_14807.3 docked with ADGP e. 1XFF docked with FMDP f. 2BPL docked with ADGP. 1XFF (PDB ID: Glutaminase domain of G-6-P synthase of E. coli), 2BPL (PDB ID: G-6-P synthase of E. coli). Figures represent ligand docking of the enzyme within its active site. Amino acid residues are labelled with which ligand interactions have taken place. Yellow dotted lines represent hydrogen bonds. Wireframe structure of the enzyme is coloured by atoms: gray, white, blue, red and yellow represent carbon, hydrogen, nitrogen, and oxygen and sulphur atoms, respectively.
The scores also illustrate that ADGP has a much improved enzyme affinity than FMDP. Comparative study of the IFD scores for R.oryzae and 2BPL (G-6-P synthase of E. coli) reveal that inhibitors show appreciable binding with R.oryzae, implying that they could be used to delimit the growth of the fungus. The result needs to be validated by wet lab experimentation in the future by determining the IC50 values for FMDP and ADGP in R.oryzae. It is common knowledge that in fungi, gene duplication is a frequent event. Several enzymes in fungi exist in multiple copies such as 16 genes of chitin synthase in R.oryzae. Hence, presence of different isoforms of the enzyme may be one of the reasons that the antifungal drugs are not very effective against them. However, an additional advantage which has been observed for G-6-P synthase in R.oryzae is that it has only two copies and both the protein molecules have high similarity as observed through molecular docking studies, making it a highly lucrative antifungal target in R. oryzae. Moreover, existing literature [6] on the enzyme revealed that the enzyme is of vital importance to both fungal and mammalian cells, but slight enzyme inactivation in fungi is fatal while the same is not true in the case of humans because of rapidly expressing enzyme. Therefore, G-6-P synthase shows promise as a potential antifungal drug target in R.oryzae.
Conclusion:
Rapid progress made in genome sequencing studies of zygomycetes has greatly helped in identifying the drug target. Due to varied antifungal susceptibility, further research is crucial in the field of drug discovery to unearth new compounds, especially peptides as drug molecules. The present study yielded few significant amino acids involved in inhibition of the drug target G-6-P synthase in R. oryzae and ligands FMDP or ADGP could be used in the near future to inhibit its growth. This study facilitates initiation of the drug discovery process for zygomycosis to present the scientific community with better inhibitors and/or drugs.
Supplementary material
Acknowledgments
We are thankful to Jaypee Institute of Information Technology, Noida for providing the necessary facility to conduct the study and Prof. G.B.K.S Prasad, SOS Biotechnology, Jiwaji University, Gwalior, M.P. for his visionary support.
Footnotes
Citation:Banerjee et al, Bioinformation 7(6): 285-290 (2011)
References
- 1.V Krcmery. Jr. J Hosp Infect. 1996;33:109. [Google Scholar]
- 2.JA Ribes, et al. Clin Microbiol Rev. 2000;13:236. doi: 10.1128/cmr.13.2.236-301.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.M Chayakulkeeree, et al. Eur J Clin Microbiol Infect Dis. 2006;25:215. doi: 10.1007/s10096-006-0107-1. [DOI] [PubMed] [Google Scholar]
- 4.U Gupta, et al. Bioinformation. 2011;6:196. [Google Scholar]
- 5.H Komatsuzawa, et al. Mol Microbiol. 2004;53:1221. doi: 10.1111/j.1365-2958.2004.04200.x. [DOI] [PubMed] [Google Scholar]
- 6.M Wojciechowski, et al. Acta Biochim Pol. 2005;52:647. [PubMed] [Google Scholar]
- 7.G Watzele, W Tanner. J Biol Chem. 1989;264:8753. [PubMed] [Google Scholar]
- 8.P Sachadyn, et al. Protein Expr Puri. 2000;3:343. [Google Scholar]
- 9.C Luo, et al. Protein J. 2009;28:34. [Google Scholar]
- 10.GL McKnight, et al. J Biol Chem. 1992;267:25208. [PubMed] [Google Scholar]
- 11.RJ Smith, et al. J Bacterio. 1996;178:2320. doi: 10.1128/jb.178.8.2320-2327.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.KO Broschat, et al. J Biol Chem. 2002;277:14764. doi: 10.1074/jbc.M201056200. [DOI] [PubMed] [Google Scholar]
- 13.A Roy, et al. Nat Protoc. 2010;5:725. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Y Zhang. Proteins. 2009;9:100. [Google Scholar]
- 15.Y Zhang, et al. BMC Bioinformatics. 2008;9:40. [Google Scholar]
- 16.RA Laskowski, et al. J App Cryst. 1993;26:283. [Google Scholar]
- 17.J Dundas, et al. Nucleic Acids Res. 2006;34:W116. [Google Scholar]
- 18.RA Friesner, et al. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 19.TA Halgren, et al. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 20.RA Friesner, et al. J Med Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 21.Glide version 5.6, Schrödinger,Prime version 2.2, Schrödinger. New York, NY: LLC; 2010. Schrödinger Suite 2010 Induced Fit Docking protocol. [Google Scholar]
- 22.K Banerjee, et al. Bioinformation. 2011;7:5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.