

Background: The molecular basis by which BubR1 is post-translationally modified during the cell cycle remains poorly understood.
Results: BubR1 is modified by sumoylation and lysine 250 is crucial for its SUMO-modification.
Conclusion: A new type of post-translational modification is identified that is essential for BubR1 function.
Significance: An important molecular mechanism is identified that inactivates the spindle checkpoint.
Keywords: Cell Cycle, Checkpoint Control, Chromosomes, Mitosis, Sumoylation, Kinetochore, Post-translational Modification
Abstract
BubR1 functions as a crucial component that monitors proper chromosome congression and mitotic timing during cell division. We investigated molecular regulation of BubR1 and found that BubR1 was modified by an unknown post-translation mechanism during the cell cycle, resulting in a significant mobility shift on denaturing gels. We termed it BubR1-M as the nature of modification was not characterized. Extended (>24 h) treatment of HeLa cells with a microtubule disrupting agent including nocodazole and taxol or release of mitotic shake-off cells into fresh medium induced BubR1-M. BubR1-M was derived from neither phosphorylation nor acetylation. Ectopic expression coupled with pulling down analyses showed that BubR1-M was derived from SUMO modification. Mutation analysis revealed that lysine 250 was a crucial site for sumoylation. Significantly, compared with the wild-type control, ectopic expression of a sumoylation-deficient mutant of BubR1 induced chromosomal missegregation and mitotic delay. Combined, our study identifies a new type of post-translational modification that is essential for BubR1 function during mitosis.
Introduction
BubR1 is highly conserved protein that functions as a key component of the spindle checkpoint. Because both the Bub1 and MAD3 ortholog in mammals share significant sequence homology, MAD3 was therefore named BubR1 (Bub1-related)3 (1). Fluorescence microscopy reveals that BubR1 colocalizes with CREST, kinetochore antigens, during late G2 and early mitosis, but not after metaphase (2, 3). Despite its marked sequence homology to yeast MAD3, human BubR1 possesses a unique C-terminal extension that contains a serine-threonine kinase domain. BubR1 interacts with components of anaphase-promoting complex/cyclosome (APC/C) and this interaction is enhanced in response to spindle checkpoint activation (3–5).
BubR1 (Bub1b)-null mice are embryonically lethal (6). BubR1+/− mouse embryonic fibroblasts are defective in spindle checkpoint activation, contain a significantly reduced amount of securin and Cdc20, and exhibit a greater level of micronuclei than wild-type cells do (7). BubR1 insufficiency also causes infertility as well as phenotypes characteristic of early aging (8, 9). Given its importance in the regulation of mitotic progression, BubR1 expression and activity are tightly regulated during the cell cycle. At the protein level, BubR1 is modified by several types of post-translational modification (4, 10, 11). BubR1 is extensively phosphorylated on many sites (11–13). Plk1 appears to play an important role in phosphorylation of BubR1 although additional kinases including Cdk1 and Mps1 are also involved in phosphorylating BubR1 (11–13). Hyper-phosphorylated BubR1, as well as other components of the checkpoint machinery including Bub1, Bub3, Mad1, Mad2, and CENP-E, is associated with unattached kinetochores and regulates the stability of kinetochore microtubule interactions (14–16). Although BubR1 and Mad2 appear to function in the same signaling pathway after spindle checkpoint activation, BubR1 is a much more potent inhibitor of APC/C than Mad2 (31). In addition to phosphorylation, BubR1 is also subjected to posttranslational modifications including acetylation (10). The acetylated BubR1 is thought important for checkpoint function by inhibition of the ubiquitin-dependent degradation of this protein (10).
We have recently demonstrated that BubR1 was modified by sumoylation during the cell cycle, resulting in a distinct mobility shift on denaturing gels. Lysine 250 is a crucial site for sumoylation. Ectopic expression of a sumoylation-deficient BubR1 mutant but not the corresponding wilt-type control induced mitotic arrest coupled with a significant chromosomal missegregation. Our study reveals a new type of molecular mechanism that regulates the activity of BubR1 during mitosis.
EXPERIMENTAL PROCEDURES
Cell Culture
HeLa and U2OS cell lines were obtained from the American Type Culture Collection. Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Invitrogen) and antibiotics (100 μg/ml of penicillin and 50 μg/ml of streptomycin sulfate, Invitrogen) at 37 °C under 5% CO2. Mitotic shake-off cells were obtained from gentle tapping of either normally growing mitotic (rounded up) cells or cells treated with nocodazole (40 ng/ml) (Sigma-Aldrich) for 14 h. Both types of shake-off cells were used for mitotic release in the presence or absence of nocodazole (or taxol), caffeine (Sigma-Aldrich), and/or MG132 (Sigma-Aldrich) as specified in each experiment.
Antibodies
Antibodies for HA, p-H3S10, and β-actin were purchased from Cell Signaling Technology Inc. Rabbit polyclonal antibodies (#32, #33, and #35) for BubR1 were developed in the laboratory. An independent antibody against BubR1 was purchased from Santa Cruz. GFP and SUMO-1 antibodies were purchased from Santa Cruz Biotechnology. Rabbit anti-ubiquitin antibodies were from Abcam (Boston). Mouse anti-FLAG antibody was purchased from Sigma-Aldrich. Mouse anti-SUMO2/3 antibodies were kindly provided by Dr. Michael J. Matunis (Johns Hopkins University). Human IgGs (CREST) against centromere proteins were purchased from Antibodies Incorporated (Davis, CA).
Plasmids, Mutagenesis, and Transfection
The original plasmid for cloning the full-length BubR1 expression plasmid or making BubR1 deletion constructs was described previously (4). An N-terminal fragment (610 amino acids) of BubR1 which corresponded to the caspase 3-cleaved fragment (18) was cloned into a GFP-expression plasmid. BubR1 mutation at lysine K250 was carried out using the QuickChange Lightning Multi Site-directed Mutagenesis kit (Stratagene) using the N-terminal fragment as a template. Individual mutations were confirmed by DNA sequencing. BubR1 and its truncated fragment were expressed as HA- or GFP-tagged fusion proteins. HA-UBC9 and His6-SUMO-1 plasmids were purchased from Addgene. SENP-1 and its mutant expression plasmids were kindly provided by J. Cheng (19). Transfection of plasmids or siRNAs was carried out using Lipofectamine 2000 according to the instruction provided by the supplier (Invitrogen).
Western Blot
SDS-PAGE was carried out using the mini gel system from Bio-Rad. Proteins were transferred to PVDF membranes. After blocking with TBST containing 5% nonfat dry milk for 1 h, the membranes were incubated overnight with primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. After thorough washing with TBST buffer, signals on the membranes were developed with an enhanced chemiluminescent system (Pierce).
Immunoprecipitation and Pull-down Assays
For immunoprecipitation, cells were lysed in a lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% Triton, 2 mm sodium pyrophosphate and 1 mm EDTA, 1 mm NaF, 1 mm sodium orthovanadate, 500 μm PMSF, 2 μm pepstatin A, 10 units/ml aprotinin, 20 mm NEM), and cleared by centrifugation. 1 μg of antibody and 40 μl of protein G-agarose resin (50/50, Millipore) were then added to 1–3 mg cell lysates and incubated at 4 °C overnight followed by extensive washing with the lysis buffer. Proteins bound to resin were eluted with SDS sample buffer and then subjected to analysis by SDS-PAGE followed by Western blot with appropriate antibodies.
For pull-down assays, HeLa cells transfected with plasmids expressing BubR1, HA-Ubc9 and/or His6-SOMU-1 were lysed in a lysis buffer as described (20). Ni2+-NTA-agarose resin (Qiagen) was then added to the cell lysates and incubated with gentle agitation at 4 °C overnight. The resin was successively washed at room temperature with four different buffers (Washing buffer 1: 6 m guanidine-HCl, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 8.0 plus 10 mm β-mercaptoethanol; Washing buffer 2: 8 m urea, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 8.0, 10 mm β-mercaptoethanol; Washing buffer 3: 8 m urea, 0.1 m Na2HPO4/NaH2PO4, 0.01 m Tris/HCl, pH 6.3, 10 mm β-mercaptoethanol plus 0.2% Triton X-100; Washing buffer 4: buffer A plus 0.1% Triton X-100). After last wash, His6-tagged products were eluted in the following buffer (200 mm imidazole, 0.15 m Tris/HCl pH 6.7, 30% glycerol, 0.72 m β-mercaptoethanol, 5% SDS), and samples were Western-blotted for BubR1 or for the HA tag.
Fluorescence Microscopy
Fluorescence microscopy was performed as described in our early studies (21, 22). Briefly, HeLa cells seeded on chamber slides were transfected with various expression constructs for 48 h. At the end of transfection, cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. After permeabilization by incubation with 0.5% Triton X-100 in PBS for 20 min, cells were incubated with 2% bovine serum albumin (BSA) in PBS for 1 h, followed by incubating overnight with the antibody to CREST. Cells were stained with Alex Fluor 555-conjugated goat anti-human IgG (Invitrogen) for 1 h. Cellular DNA was finally stained with 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probe, Eugene, OR). Fluorescence signals were detected on a Leica TCS SP5 confocal microscope or on a Leica AF6000 fluorescence microscope.
Flow Cytometry
Cells were initially fixed in 75% ethanol, then suspended in a solution of PBS containing 100 μg/ml of RNase A (Sigma) and 10 μg/ml of propidium iodide (Molecular Probes) and kept at room temperature for 1 h. Cellular fluorescence was then measured using Beckman Coulter® Epics XL-MCL™ Flow Cytometer (Fullerton, CA). DNA frequency distribution dot-blots were deconvoluted using Muticycle software (Phoenix Flow System, San Diego, CA) to estimate percent of cells in different phases of the cell cycle. Other details of cell staining and flow cytometric analysis are given elsewhere (23).
Statistical Analysis
The Student's t test was used to evaluate the difference between two groups. A value of p < 0.05 was considered to be statistically significant.
RESULTS
During the course of our study of BubR1, we observed that mitotic lysates often contained a slow mobility band that was immunoreactive to the BubR1 antibody on denaturing blots (Fig. 1A). Given that several BubR1 antibodies from independent sources all detected this band (supplemental Fig. S1), we named it as a modified form of BubR1 (BubR1-M). The molecular mass of the band was about 170 kDa, ∼40 kDa larger than the unphosphorylated, interphase form of BubR1. BubR1-M increased during mitotic release and inclusion of caffeine accelerated and enhanced BubR1 (Fig. 1A). The increase of BubR1-M was inversely correlated with levels of BubR1 and p-BubR1, suggesting a direct conversion of BubR1 and p-BubR1 into BubR1-M. The presence of BubR1-M was not HeLa cell-specific as U2OS cells, as well as A549 and HCT116 cells (data not shown), treated with nocodazole for over 28 h also contained this signal (Fig. 1B). Treatment with taxol, another mitotic inducer, for 24 h or longer also induced BubR1-M (Fig. 1C), suggesting a possible consequence of prolonged mitotic arrest. The presence of BubR1-M did not depend on BubR1 phosphorylation as MG132 treatment stabilized BubR1 phosphorylation but failed to enrich BubR1-M (Fig. 1D).
FIGURE 1.

BubR1 is modified, resulting in a significant mobility shift on denaturing gels. A, HeLa cells were treated with nocodazole for (40 ng/ml) 14 h after which mitotic cells were collected by shake-off. These cells were then re-cultured in the presence or absence of nocodazole and/or caffeine for various times, as indicated. Equal amounts of cell lysates were blotted for BubR1 and β-actin. B, U2OS cells were cultured in the presence of nocodazole for various times as indicated. Equal amounts of cell lysates were blotted for BubR1 and β-actin. Lysates of nocodazole- and caffeine-treated cells (N+C) were used as a positive control. C, HeLa cells were cultured in the presence or absence of taxol (40 nm) for 14 or 24 h. Equal amounts of cell lysates were blotted for BubR1 and β-actin. D, shake-off mitotic cells from HeLa treated with nocodazole for 18 h were released into fresh medium supplemented with or without MG132 for various times. Equal amounts of cell lysates were blotted for BubR1.
When normal mitotic shake-off cells without any drug treatment were re-cultured into fresh medium, BubR1-M was detected within 24 min after the release (Fig. 2A). BubR1-M peaked around 92 min after the release when the signal of phospho-histone H3 (p-H3S10), a mitotic marker, started to decline (Fig. 2A). These results suggest that BubR1-M is a physiological form that occurs during normal cell cycle progression. Immunoprecipitation revealed that BubR1-M was specifically brought down by the BubR1 antibody, but not by control IgG (Fig. 2B). RNA interference experiments further confirmed that BubR1-M was BubR1-specific as BubR1 siRNA, but not a control siRNA, almost completely depleted it and greatly reduced the interphase and the mitotic forms of BubR1 as well (Fig. 2C).
FIGURE 2.

BubR1-M is specific. A, mitotic shake-off cells collected from exponentially growing HeLa cells were re-cultured in fresh medium. Cell lysates were prepared at various times of culture. Equal amounts of cell lysates were blotted for BubR1, phosphorylated histone H3 (p-H3S10) and β-actin. B, asynchronized and mitotic (synchronized with nocodazole) cell lysates were immunoprecipitated with the BubR1 antibody or with a control IgG. Immunoprecipitates, along with cell lysate inputs, were blotted for BubR1. Blots of both short exposure and long exposure are shown. C, HeLa cells were transfected with BubR1 or luciferease (Luc) siRNA for 24 h followed by treatment with nocodazole and/or caffeine overnight. At the end of treatment, equal amounts of cell lysates were blotted for BubR1.
BubR1 is subjected to extensive phosphorylation during mitosis (4, 11). However, protein phosphatase treatment did not reduce the intensity of the BubR1-M band although it completely collapsed the phosphorylated form of BubR1 (Fig. 3A). As BubR1 is also reported to be acetylated at prometaphase (10), we examined cells treated with trichostatin A (TSA), a universal histone deacetylase inhibitor, in the presence or absence of a mitotic inducer for 40 h. Whereas TSA significantly stabilized the acetylated form of histones in cells treated with nocodazole or taxol, it did not enhance the signal of BubR1-M induced by an extended treatment with mitotic inducers (Fig. 3B).
FIGURE 3.

BubR1 is modified by sumoylation. A, HeLa cells treated with or without taxol and/or caffeine for 40 h were collected for lysate preparation. Equal amounts of lysates were incubated with or without λ phosphatase for 1 h at room temperature before analysis by SDS-PAGE followed by immunoblotting with antibodies to BubR1 and β-actin. B, HeLa cells were treated with nocodazole or taxol for 18 h followed by treatment with TSA (100 nm) for 24 h. Equal amounts of cell lysates from various treatments were blotted for BubR1, acetylated histones, and β-actin. C, HeLa cells treated with taxol for 40 h were collected for lysate preparation. Equal amounts of cell lysates were incubated with or without NEM (20 mm) in vitro for 1 h on ice, after which the lysates, along with asynchronized and mitotic cell lysate inputs, were blotted for BubR1 and β-actin. D, HeLa cells transfected with plasmids expressing FLAG-tagged SENP-1 or with an enzymatically defective SENP1 (SENP-1-Mut) for 18 h were treated with taxol for an additional 24 h. Equal amounts of lysates were blotted for BubR1 and β-actin.
To understand the biochemical nature of BubR1-M, we first incubated taxol-treated cell lysates with or without N-ethylmaleimide (NEM), a chemical capable of inhibiting isopeptidases for de-sumoylation and de-ubiquitination. Immunoblotting revealed that NEM significantly enhanced BubR1-M (Fig. 3C), suggesting the involvement of either sumoylation or ubiquitination.
As the first step to determine whether sumoylation resulted in BubR1-M, HeLa cells were transfected with a plasmid construct expressing either FLAG-tagged wild-type sentrin-specific peptidase 1 (SENP1) or FLAG-tagged enzymatically defective SENP1 (SENP1-Mut). The transfected cells were then treated with taxol. Compared with parental cells, expression of FLAG-SENP1 almost completely eliminated BubR1-M (Fig. 3D). Supporting this, the mutant SENP1 was not effective in suppression of BubR1-M than the wild-type SENP1 (Fig. 3D). Expression of both SENP1 and its mutant was confirmed by blotting with the anti-FLAG antibody (Fig. 3D). These results strongly suggest that BubR1-M is derived from modification by sumoylation
We next tested if BubR1 was modified by SUMO-1 using the co-immunoprecipitation (Co-IP) approach. The SUMO-1 antibody detected a band that migrated at the same position as BubR1-M in the precipitates brought down by BubR1 antibody but not by the control IgG (Fig. 4A). On the other hand, when BubR1 immunoprecipitates were blotted for ubiquitin, no specific signals that migrated at the position of BubR1-M were detected (Fig. 4B). To further confirm SUMO-1 modification of BubR1, HeLa cells were co-transfected with plasmids expressing His6-SUMO-1 and FLAG-UBC9, the latter being the essential SUMO E2-conjugating enzyme. The transfected cells were then treated with nocodazole and/or caffeine. Affinity pull-down of SUMO-1-tagged proteins using nickel (Ni-NTA) resin followed by immunoblotting with the BubR1 antibody revealed one band that migrated at the same position as BubR1-M, which was further enriched after nocodazole treatment (Fig. 4C). An intermediate BubR1 band (marked by *) was also pulled down and its signal was significantly higher in interphase cell lysates than that in mitotic ones.
FIGURE 4.

BubR1 can be modified by SUMO-1. A, mitotic cell lysates prepared from taxol-treated cells were immunoprecipitated with the anti-BubR1 antibody or with the control IgG. Immunoprecipitates, along with asynchronized and mitotic cell lysates, were blotted for BubR1 and SUMO-1. B, mitotic cell lysates prepared from taxol-treated (or vehicle-treated) cells were immunoprecipitated with the anti-BubR1 antibody or with the control IgG. Immunoprecipitates, along with interphase and mitotic cell lysates, were blotted with the antibody to ubiquitin. C, HeLa cells were transfected with FLAG-UBC9 and His6-SUMO-1 for 24 h followed by treatment with nocodazole and/or caffeine for an additional 18 h. Equal amounts of cell lysates of various treatments were incubated with Ni-NTA resin. After extensive washing, proteins specifically bound to the resin, along with lysate inputs, were blotted for BubR1. D, HeLa cells were co-transfected with HA-BubR1 (or vector), FLAG-UBC9 and His6-SUMO-1 for 48 h. Equal amounts of cell lysates were incubated with Ni-NTA resin. Proteins specifically bound to the resin, along with lysate inputs, were blotted for BubR1 and the HA tag.
We next transfected HeLa cells with a plasmid construct expressing HA-tagged BubR1, along with plasmids expressing His6-SUMO-1 and FLAG-UBC9. Consistent with our prediction, sumoylated bands were detected by the BubR1 antibody in transfected cells. These bands were significantly enriched by affinity pull-down with Ni-NTA resin (Fig. 4D). Blotting with the anti-HA antibody confirmed that HA-BubR1 was specifically pulled down by nickel resin (Fig. 4D).
As we have obtained HeLa cell lines that constitutively expressed transfected His6-SUMO-1 and His6-SUMO-2 (Fig. 5A), we asked if BubR1 could be modified by SUMO-2, as well as SUMO-1. After treatment with taxol for 28 h, cell lysates prepared from parental HeLa cells, as well as from His6-SUMO-1- and His6-SUMO-2-expressing HeLa cells, were collected and equal amounts of lysates were blotted for BubR1. Immunoblotting revealed that BubR1-M was accumulated to higher levels in SUMO-1-expressing cells than in parental cells (Fig. 5A). BubR1-M was also induced to a level comparable to that observed in SUMO-1 cells. These data suggest that BubR1 can be modified by SUMO-2.
FIGURE 5.

BubR1 K250 is crucial for sumoylation. A, HeLa cells constitutively expressing His6-SUMO-1 or His6-SUMO-2, as well as parental HeLa cells, were collected and equal amounts of cell lysates were blotted with the antibody to the His6 tag. These cells were also treated with taxol for 18 h after which cell lysates were prepared. Equal amounts of cell lysates were blotted for BubR1. B, schematic presentation of BubR1 (Wt) and its N-terminal fragment (610 amino acids) with (N-K250R) or without (N-Wt) K250 replaced with R250. GFP was fused in-frame with BubR1 N-terminal fragment. C, HeLa cells transfected with a plasmid construct expressing GFP-tagged N-terminal fragment of BubR1 (GFP-N-Wt) or its mutant counterpart (GFP-N-K250R) for 24 h followed by taxol treatment for 18 h. Equal amounts of cell lysates were blotted for BubR1, GFP, and β-actin. Arrow N-Wt-M denotes the sumoylated, GFP-tagged N-terminal fragment of BubR1. Arrow GFP-N denotes both the wild-type N-terminal fragment of BubR1 and its mutant counterpart. Vehicle and T+C denote lysate inputs that were derived from cells treated with vehicle or taxol (T) plus caffeine (C) for 18 h, respectively. D, HeLa cells constitutively expressing His6-SUMO-1 were transfected with His6-HA-N-Wt or His6-HA-N-K250R expression plasmids for 48 h. Ectopically expressed proteins were enriched by incubation with Ni-NTA resin and analyzed, along with lysate inputs, by Western blotting using the antibody to HA tag. His6-HA-N-M denotes the sumoylated BubR1 N-terminal fragment.
To identify the potential lysine residues of BubR1 that were sumoylated, we analyzed BubR1 amino acid sequences for optimal sumoylation sites using the criteria available at Abgent Inc. Three lysines sites (K5, K250, and K769) with the highest scores were subjected to mutagenic analysis. The relative position of these sites to other domains is shown in supplemental Fig. S2. We first made plasmid constructs expressing a GFP-tagged N-terminal fragment (610 amino acids) of BubR1 (termed BubR1 N-Wt) and its corresponding lysine mutant (termed BubR1 N-K250R) (Fig. 5B), as early studies show that the N terminus retains the checkpoint function of BubR1 (24, 25). Immunoblotting with antibodies to BubR1 and GFP confirmed that both N-Wt and N-K250R were efficiently expressed (Fig. 5C, arrows GFP-N and GFP). A new band (arrow N-Wt-M) was detected in cells transfected with GFP-N-Wt expression construct, and the intensity of the band increased after treatment with taxol, suggesting a sumoylated form of the ectopically expressed GFP-N-Wt. Consistent with this prediction, the molecular mass of the band was about 117 kDa, approximately one SUMO-1 size larger than GFP-N-Wt, predicted to be 97 kDa. Further supporting this, cells transfected with GFP-N-K250R mutant expression construct did not contain this form of BubR1 even after taxol treatment (Fig. 5C). Thus, these data suggest the importance of K250 for sumoylation.
We were unable to detect a shifted band when lysates from GFP-N-Wt transfected cells were probed with the anti-GFP antibody. We reasoned that the bulky GFP moiety might interfere with the detection. To further study whether K250 is a crucial site for sumoylation, we made a plasmid construct expressing N-terminal fragment of BubR1 or its corresponding K250R mutant that was fused in-frame with both His6 and HA tags. After transfection into HeLa cells constitutively expressing His6-SUMO-1, both BubR1 fragments were efficiently expressed (Fig. 5D, Input). After enrichment with Ni-NTA resin, a modified BubR1 with a predicted mobility was easily detected only in cells transfected with the plasmid expressing the wild-type protein (Fig. 5D, Ni-NTA), strongly suggesting a sumoylated form of ectopically expressed wild-type BubR1 (arrow His6-HA-N-M).
Early studies show that the N terminus of BubR1 retains the checkpoint function of BubR1 (24, 25). Thus, we asked whether BubR1 sumoylation played a role in regulating mitotic progression. HeLa cells ectopically expressing GFP-tagged BubR1 N-Wt or its mutant counterpart N-K250R were examined via time-lapse confocal microscopy. Both GFP-BubR1 N-Wt and GFP-N-K250R localized normally to kinetochores during early mitosis (Fig. 6A). BubR1 N-K250R appeared to be associated with the kinetochores longer than BubR1 N-Wt (Fig. 6A). Significantly, expression of BubR1 N-K250R frequently caused the formation of lagging chromosomes during anaphase (Fig. 6A, arrow in 1:04 time frame). Further analysis of fixed and stained mitotic cells revealed that a much higher percentage of anaphase and telophase cells expressing BubR1 N-K250R contained lagging chromosomes than their counterparts expressing BubR1 N-Wt (Fig. 6, B and C), strongly suggesting that sumoylation plays an important role in regulating normal nuclear division. Intriguingly, a significant fraction of cells expressing BubR1 N-K250R exhibited a delayed progression through mitosis, leading to enrichment of mitotic cells (supplemental Fig. S3 and Table S1). This observation apparently differs from an early study that expression of DsRed-tagged BubR1-K250R mutant induces mitotic slippage (10). One explanation is that expression of DsRed-BubR1 K250R was coupled with depletion of endogenous BubR1 in that study (10). The accelerated mitotic exit could result from lack of endogenous BubR1 as it is known that deficiency in BubR1 function (e.g. haplo-insufficiency) can induce mitotic slippage (7).
FIGURE 6.

Sumoylation-resistant mutant of BubR1 induces mitotic delay and chromosomal missegregation. A, HeLa cells transfected with GFP-tagged BubR1 N-Wt and BubR1 N-K250R were subjected to time-lapse videography. Time of each frame is indicated (upper left corner). Paired representative cells are shown. Arrows in 0:30 GFP time-frame denote BubR1 N-K250R signals that remain on kinetochores. Arrow in 1:04 DIC time-frame denotes mis-segregated chromosomes. B, HeLa cells transfected with a plasmid construct expressing GFP-tagged N-terminal fragment of BubR1 (GFP-N-Wt) or its mutant counterpart (GFP-N-K250R) for 48 h were fixed and stained with antibody to CREST (red). DNA was stained with DAPI (blue). Representative cell images are shown. Lagging chromosomes are indicated by arrows. C, HeLa cells were transfected with a plasmid expressing either GFP-tagged wild-type of N-terminal BubR1 fragment (N-Wt) or its mutant counterpart with K250 replaced with R (N-K250R). After 48 h transfection, HeLa cells were fixed and stained with the antibody to CREST. DNA was stained with DAPI. The percentage of anaphase/telophase cells with mis-segregated chromosomes was determined for each group. Data are summarized from three independent experiments.
BubR1 is inactivated during late mitosis via both proteasome- and caspase-mediated degradation (10, 18). It has been suggested capspase-mediated cleavage of BubR1 functions as an important internal cue for mitotic progression (18). We then examined the relationship between BubR1 sumoylation and the appearance of degradation products. BubR1-M was closely associated with the levels of two major degradation products (Fig. 7A, arrows BubR1-D1 and BubR1-D2). The patterns and molecular weights suggested that they were derived from a caspase-mediated degradation process (18). Consistent with this notion, we detected enhanced cleavage of PARP-1. We then further examined the effect of MG132 (a proteasome inhibitor) and DEVD-CHO (an inhibitor for caspase 3 or caspase 3-like activities) on BubR1 degradation during mitosis. Both MG132 and DEVD-CHO enriched BubR1-D1 compared with the control, suggesting its ability to inhibit its further degradation; moreover, and DEVD-CHO completely blocked the appearance of BubR1-D2 (Fig. 7B). In the meantime, sumoylated BubR1 was not significantly affected by treatment with either inhibitor (Fig. 7B). These results suggest that BubR1 sumoylation is associated with its degradation, which depends at least partially on the proteasome and caspase 3-mediated pathways.
FIGURE 7.
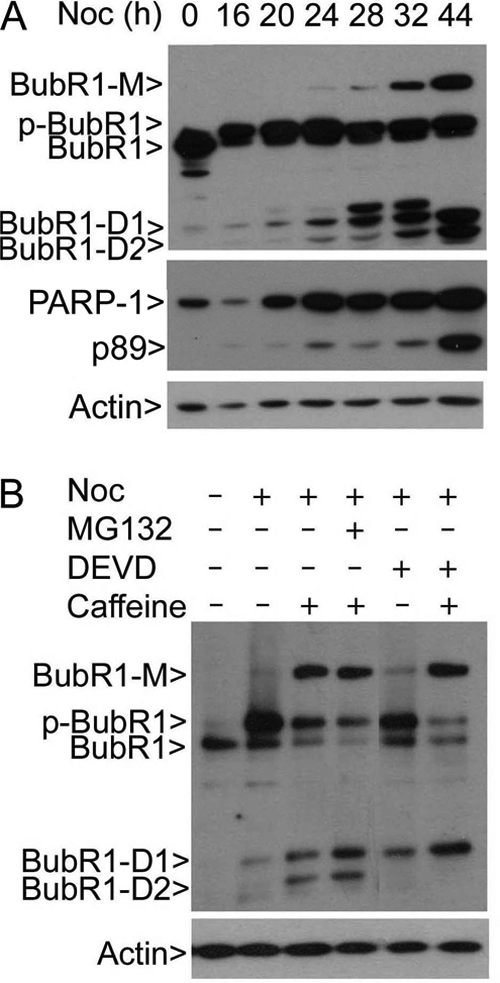
BubR1 sumoylation is associated with its degradation. A, HeLa cells treated with nocodazole for various times as indicated after which equal amounts of cell lysates were blotted for BubR1, PARP-1, and β-actin. p89 represents a cleaved fragment of PARP-1. BubR1-D1 and BubR1-D2 denote two major BubR1 degradation products. B, HeLa cells treated with various agents as indicated for 24 h after which equal amounts of cell lysates were blotted for BubR1 and β-actin. MG132 and DEVD-CHO (20 μm) are proteasome and caspase 3 inhibitor, respectively. BubR1-D1 and BubR1-D2 denote two major degradation products of BubR1, respectively.
DISCUSSION
This studt demonstrates that BubR1 is modified via sumoylation and that BubR1 sumoylation is important for regulating normal chromosome segregation. Sumoylated BubR1 is strongly induced after an extended mitotic arrest as the result of treatment with nocodazole or taxol. BubR1 sumoylation appears to be independent of the activation/phosphorylation status of BubR1 because the level of p-BubR1 is not correlated with sumoylated BubR1 during mitotic release. It has been reported that CENP-E, a motor protein capable of interacting with BubR1 (26), is also modified by sumoylation (27). It is conceivable that there is a coordinated mechanism that regulates BubR1 and CENP-E, both being spindle checkpoint components, during cell division.
Our molecular analyses identify that K250 is crucial for BubR1 sumoylation. A fraction of interphase BubR1 appears to be modified by mono-sumoylation as a distinct form of BubR1 with about one SUMO size larger than the unmodified form is detected in interphase cell lysates (Fig. 4C). Sumoylation is known to be essential for the regulation of many mitotic processes. An early mouse genetic study shows that sumoylation is required for development as mouse embryos deficient in the SUMO E2-conjugating enzyme UBC9 die at the early post-implantation stage (28). UBC9-null blastocysts fail to expand in culture with major defects in chromosome condensation and segregation (28). The importance of sumoylation for mitotic processes has been recapitulated in a separate study showing that RanBP2, a nucleoporin with SUMO E3 ligase activity, plays a major role in chromosome segregation and suppressing chromosomal instability (29).
It seems that BubR1 is subjected to the modification by both SUMO-1 and SUMO-2. At present, we do not know whether SUMO-1 modification is a predominant form. However, the observations that a comparable level of BubR1-M is detected in both SUMO-1- and SUMO-2-expressing cell lines and that transfected SUMO-1 is expressed at a much lower level than SUMO-2 suggest that BubR1 is primarily SUMO-1 modified. It is known that common enzymes are involved in conjugation of SUMO-1 and SUMO-2/3 (30). A mouse genetic study shows that disruption of sumo-1 alleles does not cause noticeable developmental defects (31), further supporting a compensatory role of SUMO-2/3 in these mice. At present, we are not certain whether SUMO-1 forms polymeric chains on BubR1 although SUMO-1 chain has been described before (32, 33).
BubR1 is a crucial component of the spindle checkpoint. Our current study indicates that BubR1 sumoylation is independent of its checkpoint activation. Cells treated with nocodazole overnight contain a robust spindle checkpoint but little sumoylated BubR1. It seems that BubR1 sumoylation is a later event of mitosis, correlating with a decreased level of p-H3S10 (Fig. 2A). A prolonged nocodazole treatment (which is likely to result in “mitotic escape”) or the mitotic release of shake-off cells collected after overnight nocodazole treatment causes significant BubR1 sumoylation. Therefore, a likely function of BubR1 sumoylation may be associated with the inactivation of the spindle checkpoint. Nevertheless, it would be interesting to examine whether sumoylation-deficiency affects the interaction between BubR1 and Mad2 or Cdc20. It is somewhat puzzling that caffeine enhances BubR1 sumoylation induced by nocodazole or taxol. Our explanation is that caffeine-mediated stimulation of BubR1 sumoylation is due to its known effect on promoting S phase cells into mitosis, resulting in a net increase of the mitotic population (17).
It has been shown that BubR1 is subjected to modification by acetylation (10). As Myc-tagged K250R mutant of BubR1 was non-detectable when ectopically expressed (10), it was proposed that acetylation might be involved in stabilizing BubR1 and positively regulating the spindle checkpoint (10). On the other hand, we have shown that BubR1 K250R mutant protein is rather stable as compared with the wild-type counterpart (Fig. 5, C and D; supplemental Movie S1). One possible explanation for this discrepancy is that the anti-Myc antibody may have failed to detect the tagged mutant protein of BubR1 in that study. Given that acetylation appears to also occur on K250, we propose that acetylation and sumoylation regulate BubR1 stability/activity in an opposing manner. Supporting this, we have shown that BubR1 sumoylation induced by extended treatment with nocodazole is correlated with the appearance of its degradation products (Fig. 7A).
Prolonged mitotic arrest after treatment with nocodazole or taxol can induce apoptosis. Therefore, an alternative explanation is that BubR1 sumoylation may provide an internal cue for initiation of apoptosis. However, treatment with caspase-3 inhibitor DEVD-CHO does not diminish the level of sumoylated BubR1 although it completely suppresses the generation of BubR1-D2 (Fig. 7B). Consistent with this observation, expression of sumoylation-deficient mutant of BubR1 fails to significantly suppress the apoptotic population as compared with the wild-type counterpart (supplemental Fig. S3). These results thus suggest that apoptosis may not be the direct result of BubR1 sumoylation. Obviously, additional studies are necessary to clarify the roles of BubR1 sumoylation and acetylation in regulating mitosis and/or apoptosis.
Supplementary Material
Acknowledgments
We thank co-workers in the laboratory for valuable discussions and assistance during the course of the study. We are grateful to Dr. Zbigniew Darzynkiewicz at New York Medical College for assisting in flow cytometry analysis and Dr. Jingke Cheng at Shanghai Jiaotong University School of Medicine for providing us with FLAG-Ubc9, His6-SUMO1, SENP-1, and SENP-1 mutant expression constructs. We also thank Dr. Michael J. Matunis at the Johns Hopkins University for providing us with antibodies to SUMO-2/3 and Dr. Ronald Hay at University of Dundee for HeLa cell lines constitutively expressing His6-SUMO-1 and SUMO-2.
This work was supported in part by United States Public Service Awards (to W. D.) (CA090658 and CA113349) and (to M. P.) (R01-GM057587 and R21-AG032560).

This article contains supplemental Figs. S1–S4 and Table S1.
- BubR1
- Bub1-related
- APC
- anaphase-promoting complex
- TSA
- trichostatin A
- NEM
- N-ethylmaleimide.
REFERENCES
- 1. Cahill D. P., Lengauer C., Yu J., Riggins G. J., Willson J. K., Markowitz S. D., Kinzler K. W., Vogelstein B. (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392, 300–303 [DOI] [PubMed] [Google Scholar]
- 2. Hoffman D. B., Pearson C. G., Yen T. J., Howell B. J., Salmon E. D. (2001) Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell 12, 1995–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan G. K., Schaar B. T., Yen T. J. (1998) Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J. Cell Biol. 143, 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W., Lan Z., Wu H., Wu S., Meadows J., Chen J., Zhu V., Dai W. (1999) BUBR1 phosphorylation is regulated during mitotic checkpoint activation. Cell Growth Differ. 10, 769–775 [PubMed] [Google Scholar]
- 5. Chan G. K., Jablonski S. A., Sudakin V., Hittle J. C., Yen T. J. (1999) Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J. Cell Biol. 146, 941–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Q., Liu T., Fang Y., Xie S., Huang X., Mahmood R., Ramaswamy G., Sakamoto K. M., Darzynkiewicz Z., Xu M., Dai W. (2004) BUBR1 deficiency results in abnormal megakaryopoiesis. Blood 103, 1278–1285 [DOI] [PubMed] [Google Scholar]
- 7. Dai W., Wang Q., Liu T., Swamy M., Fang Y., Xie S., Mahmood R., Yang Y. M., Xu M., Rao C. V. (2004) Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res. 64, 440–445 [DOI] [PubMed] [Google Scholar]
- 8. Baker D. J., Jeganathan K. B., Cameron J. D., Thompson M., Juneja S., Kopecka A., Kumar R., Jenkins R. B., de Groen P. C., Roche P., van Deursen J. M. (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 36, 744–749 [DOI] [PubMed] [Google Scholar]
- 9. Baker D. J., Perez-Terzic C., Jin F., Pitel K., Niederlander N. J., Jeganathan K., Yamada S., Reyes S., Rowe L., Hiddinga H. J., Eberhardt N. L., Terzic A., van Deursen J. M. (2008) Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 10, 825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi E., Choe H., Min J., Choi J. Y., Kim J., Lee H. (2009) BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J. 28, 2077–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang H., Yen T. J. (2009) BubR1 is an effector of multiple mitotic kinases that specifies kinetochore: microtubule attachments and checkpoint. Cell Cycle 8, 1164–1167 [DOI] [PubMed] [Google Scholar]
- 12. Elowe S., Hummer S., Uldschmid A., Li X., Nigg E. A. (2007) Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 21, 2205–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang H., Hittle J., Zappacosta F., Annan R. S., Hershko A., Yen T. J. (2008) Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J. Cell Biol. 183, 667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu H. (2002) Regulation of APC-Cdc20 by the spindle checkpoint. Curr. Opin. Cell Biol. 14, 706–714 [DOI] [PubMed] [Google Scholar]
- 15. Yu H. (2007) Cdc20: a WD40 activator for a cell cycle degradation machine. Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- 16. Wu H., Lan Z., Li W., Wu S., Weinstein J., Sakamoto K. M., Dai W. (2000) p55CDC/hCDC20 is associated with BUBR1 and may be a downstream target of the spindle checkpoint kinase. Oncogene 19, 4557–4562 [DOI] [PubMed] [Google Scholar]
- 17. Bode A. M., Dong Z. (2007) The enigmatic effects of caffeine in cell cycle and cancer. Cancer Lett. 247, 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim M., Murphy K., Liu F., Parker S. E., Dowling M. L., Baff W., Kao G. D. (2005) Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol. Cell. Biol. 25, 9232–9248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng J., Kang X., Zhang S., Yeh E. T. (2007) SUMO-specific protease 1 is essential for stabilization of HIF1α during hypoxia. Cell 131, 584–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu J., Zhang S. S., Saito K., Williams S., Arimura Y., Ma Y., Ke Y., Baron V., Mercola D., Feng G. S., Adamson E., Mustelin T. (2009) PTEN regulation by Akt-EGR1-ARF-PTEN axis. EMBO J. 28, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu X. S., Zhao X. D., Wang X., Yao Y. X., Zhang L. L., Shu R. Z., Ren W. H., Huang Y., Huang L., Gu M. M., Kuang Y., Wang L., Lu S. Y., Chi J., Fen J. S., Wang Y. F., Fei J., Dai W., Wang Z. G. (2010) Germinal cell aplasia in Kif18a mutant male mice due to impaired chromosome congression and dysregulated BubR1 and CENP-E. Genes Cancer 1, 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang X., Yang Y., Duan Q., Jiang N., Huang Y., Darzynkiewicz Z., Dai W. (2008) sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev. Cell 14, 331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao H., Traganos F., Dobrucki J., Wlodkowic D., Darzynkiewicz Z. (2009) Induction of DNA damage response by the supravital probes of nucleic acids. Cytometry A 75, 510–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malureanu L. A., Jeganathan K. B., Hamada M., Wasilewski L., Davenport J., van Deursen J. M. (2009) BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev. Cell 16, 118–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris L., Davenport J., Neale G., Goorha R. (2005) The mitotic checkpoint gene BubR1 has two distinct functions in mitosis. Exp. Cell Res. 308, 85–100 [DOI] [PubMed] [Google Scholar]
- 26. Mao Y., Abrieu A., Cleveland D. W. (2003) Activating and silencing the mitotic checkpoint through CENP-E-dependent activation/inactivation of BubR1. Cell 114, 87–98 [DOI] [PubMed] [Google Scholar]
- 27. Zhang X. D., Goeres J., Zhang H., Yen T. J., Porter A. C., Matunis M. J. (2008) SUMO-2/3 modification and binding regulate the association of CENP-E with kinetochores and progression through mitosis. Mol. Cell 29, 729–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nacerddine K., Lehembre F., Bhaumik M., Artus J., Cohen-Tannoudji M., Babinet C., Pandolfi P. P., Dejean A. (2005) The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev. Cell 9, 769–779 [DOI] [PubMed] [Google Scholar]
- 29. Dawlaty M. M., Malureanu L., Jeganathan K. B., Kao E., Sustmann C., Tahk S., Shuai K., Grosschedl R., van Deursen J. M. (2008) Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIα. Cell 133, 103–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tatham M. H., Kim S., Yu B., Jaffray E., Song J., Zheng J., Rodriguez M. S., Hay R. T., Chen Y. (2003) Role of an N-terminal site of Ubc9 in SUMO-1, -2, and -3 binding and conjugation. Biochemistry 42, 9959–9969 [DOI] [PubMed] [Google Scholar]
- 31. Evdokimov E., Sharma P., Lockett S. J., Lualdi M., Kuehn M. R. (2008) Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell Sci. 121, 4106–4113 [DOI] [PubMed] [Google Scholar]
- 32. Pichler A., Gast A., Seeler J. S., Dejean A., Melchior F. (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120 [DOI] [PubMed] [Google Scholar]
- 33. Hammer E., Heilbronn R., Weger S. (2007) The E3 ligase Topors induces the accumulation of polysumoylated forms of DNA topoisomerase I in vitro and in vivo. FEBS Lett. 581, 5418–5424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.