

Abstract
SB-3CT is a thiirane-containing inhibitor of the gelatinase class of matrix metalloprotease enzymes. In support of the mechanistic study of this inhibition, the conformational analyses of SB-3CT (and of two methyl-substituted derivatives) were undertaken using x-ray crystallography and molecular dynamics simulation.
Keywords: Gelatinase Inhibitors, Matrix metalloproteases, x-ray crystallography, molecular dynamics, aryl sulfone conformation
Gelatinases are members of the family of matrix metalloproteinases (MMPs), enzymes that have been implicated in many pathological and physiological functions.1–5 Compound 1, also referred to as SB-3CT, is a potent and selective gelatinase inhibitor both in vitro and in vivo.6–9 Compound 1 is active in rodent models for cancer and stroke, and is a useful tool for the elucidation of the functional properties of these enzymes in in vivo models. A recent metabolism study of compound 1 revealed that it is metabolized primarily by oxidation, mainly at the α-methylene to the sulfonyl group and at the para position of the terminal phenyl ring.10 Despite active metabolic turnover of 1, it shows potent activity in vivo and holds considerable promise in investigating the roles of gelatinases in biological systems.
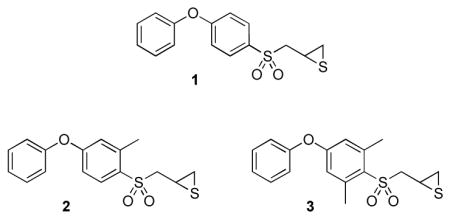
Attempts to determine the structure of compound 1 when bound to gelatinases by x-ray crystallography have failed. In order to understand the structural issues that govern the interactions between the inhibitor and these enzymes, we have resorted to x-ray absorption spectroscopy.11 While these studies have provided quantitative structural information concerning the inhibited enzyme (wherein the thiirane has undergone ring opening), an understanding of the structural aspects to the initial presentation of 1 to the catalytic zinc ion in the MMP active site is much less well understood. In this study, we expand our understanding of the structural chemistry of this inhibitor class. As both experimental and computational chemistry reveal a distinct conformational preference for the aryl sulfone, strongly favoring the conformation wherein the π orbital of the ipso carbon atom bisects the two sulfur-oxygen bonds,12 we wondered as to the importance of this preference to the inhibitory ability of compound 1. Furthermore, an understanding of the effect of structure alteration near the aryl sulfone on the conformational preferences was necessary to the interpretation of the structure-activity relationships within this inhibitor class. To address these issues, we synthesized compounds 2 and 3 for the purpose of structural comparison to 1 using crystallographic and molecular dynamics methods.
The synthetic route followed the methodology developed by our group (Scheme 1),13,14 which involves thiolate generation from methylated phenoxyphenyl bromide, followed successively by alkylation with epichlorohydrin, oxirane ring formation, oxidation to sulfone and conversion of the oxirane to the thiirane. The synthetic challenge with respect to 2 and 3 was the preparation of the methylated phenoxyphenyl bromides (5a and 5b) as key intermediates. Introduction of the single methyl group, and of the dimethyl groups, in the middle phenyl ring was accomplished using 3-methyl and 3,5-dimethyl-4-bromophenol (4a and 4b), respectively.
Scheme 1.
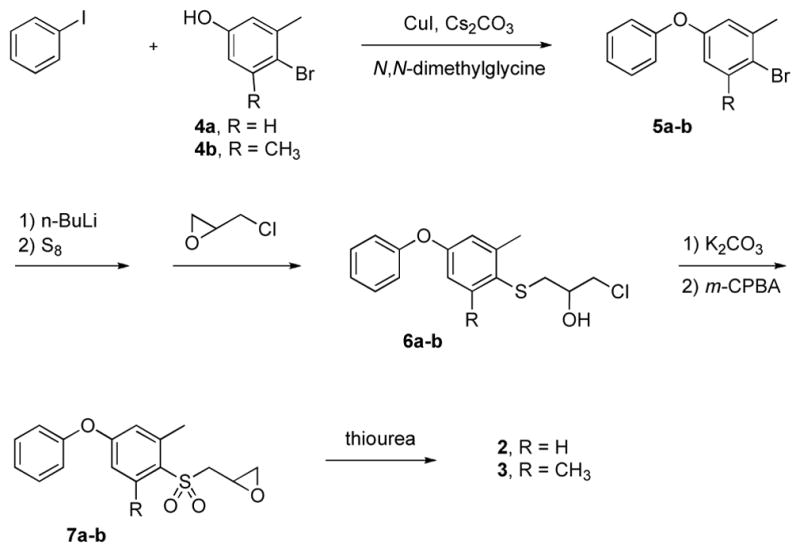
Syntheses of compounds (±)-2 and (±)-3.
These compounds were reacted separately with 4-iodobenzene under Ullmann conditions using copper(I) iodide, Cs2CO3 and N,N-dimethylglycine hydrochloride as a promoter.15 Under this Ullmann condition, self-condensation of the bromophenol moiety is considerably slower than the reaction with iodobenzene. By using limiting amounts of Cs2CO3 and of CuI, by strict control of the duration of the reaction, and by taking advantage of the favorable steric factors at the bromo position(s), the self-condensation reaction of the bromophenol was avoided completely. Elaboration at the bromo position in compounds 5a and 5b is problematic in general due to steric hindrance. According to literature precedents, lithiation of bromomesitylene requires treatment at room temperature16,17 or even reflux conditions.18 In our case, prolonged reaction time for lithiation at −78 °C and for the thiolate substitution gave access to compounds 6a and 6b in good yield. The transformations leading to (±)-2 (from 6a) and (±)-3 (from 6b) were done by the methodology developed by our group. 13,14,19
Compounds 1, 2, and 3 were crystallized as racemates. Compound 1 was crystallized from ethyl acetate and hexane, and compounds 2 and 3 were crystallized from methanol. The ORTEP diagrams of compounds 1, 2, and 3 are shown in Figure 1 and the full details on the crystal structures are given in the Supporting Information.19 Each compound crystallized with one molecule in the asymmetric unit. Compound 1 crystallized in the space group P21/c, while the other two structures both crystallized in the space group P1̄, with similar cell dimensions (Table 1).20 Disorder is seen in all three structures. Two orientations for the thiirane rings are seen for all three compounds. The thiirane groups of 1 and 3 are disordered about the sulfur atom. Compound 1 also shows a second disordered position for the C13 methylene. Compound 2 exhibits disorder in the positions of all three atoms of the thiirane. Last, there is orientational disorder in the two aromatic rings of 3. The angle between normals to the planes of the rings formed by C1 to C6 and the minor orientation of this ring is 163.8°. Similarly, the angle between ring C7 to C12 and its minor fraction is 12.9°.
Figure 1.
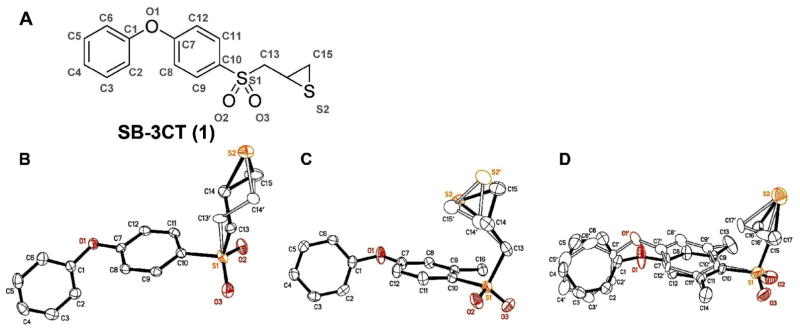
(A) The atom numbering is demonstrated on the structure of compound 1 (panel A). The ORTEP diagrams of compounds 1, 2, and 3 (panels B, C, and D, respectively) are shown at 50% probability level. Hydrogen atoms are omitted for clarity. The structures correspond to the R-stereoisomer. The alternate conformations of each are superimposed.
Table 1.
Crystallographic Details of compounds 1–3.
| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C15H14O3S2 | C16H16O3S2 | C17H18O3S2 |
| Mr | 306.38 | 320.41 | 334.43 |
| Space group | P21/c (No. 14) | P1̄ (No. 2) | P1̄ (No. 2) |
| a (Å) | 5.40350(10) Å | 5.7744(2) | 5.4528(3) |
| b (Å) | 28.1118(6) Å | 11.2258(4) | 11.4721(5) |
| c (Å) | 9.3269(2) Å | 12.0078(4) | 13.0581(6) |
| α (Å) | 90 | 85.695(2)° | 88.396(2) |
| β (Å) | 95.7320(10) | 78.737(2)° | 81.581(2) |
| γ (Å) | 90 | 85.483(2)° | 79.755(2) |
| V Å3 | 1409.69(5) | 759.59(5) | 795.16(7) |
| Z | 4 | 2 | 2 |
| T (°C) | 100(2) | 100(2) | 100 |
| λ (Å) | 1.54178 | 1.54178 | 1.54178 |
| Dobsvd (g cm−3) | 1.444 | 1.401 | 1.397 |
| μ (cm−1) | 3.464 | 3.239 | 3.117 |
| R1(F2, I>2σ(I)) | 0.0366 | 0.0423 | 0.0592 |
| wR2(F2) | 0.0969 | 0.1281 | 0.1566 |
| S | 1.049 | 1.247 | 1.057 |
; ;
n= number of reflections, p= number of parameters refined
The major conformers for compounds 1, 2, and 3 from the crystal structures are superimposed in Figure 2A as the R-enantiomers of each structure. The C13-S1-C10-C11 dihedral angle observed in the solid state for 1 is 94.0(4)°; for 2 is 102.6(2)°; and for 3 is 98.4°. These values correspond to stable conformations of the arylsulfone, as discussed by Hof et al.12 The structures of compounds 1 and 3 were additionally evaluated by molecular dynamics simulations in a solvated system.23 The results are shown in Figure 2B and 2C for compounds 1 and 3, respectively. The MD conformers encompass a C13-S1-C10-C11 dihedral angle of 90° ± 18° for 1, and of 90° ± 21° for 3. This relative motion is fully consistent with the previous study.12 Hence, the lowest energy conformations with respect to the aryl sulfone are seen in the crystal structures, and during the dynamics. As the structures reveal, the presence of the methyl groups in the middle ring moderates the degree of motion that the thiiranylmethyl segment experiences.
Figure 2.

(A) Superimposition of the stereo representation of the R-isomers from the crystal structures of 1 (green) 2 (blue) and 3 (orange) as capped stick representations, with superimposition centered around the central ring. (B, C) Superimposition of 16 molecular dynamics snapshots (each from the end of 0.1 ns of dynamics) for 1 (B) and for 3 (C). Hydrogen atoms are colored in grey, carbons in green (B) and blue (C), oxygens in red, and sulfurs in yellow.
The mechanism of gelatinase inhibition by 1 is characterized by a potent (low nanomolar), slow-binding kinetics progression to an enzyme-inhibitor complex, wherein its thiirane ring is opened. As this complex has not yet been amenable to characterization by crystallography, and as the Michaelis complex of 1 having the intact thiirane is transient, several efforts toward computational evaluation of both structures have been made. The recognition that the arylsulfone of 1 imparts significant conformational constraint,12 as confirmed here in the solid-state and by molecular dynamics simulations, provides strong guidance toward this structural understanding. The existence of this conformational constrain is implicit from the extensive crystallographic study of aryl sulfone-based hydroxamate inhibitors of MMP-9, as recently reported by Tochowicz et al., and also in their computational analysis of the 1-MMP-9 inhibited complex.29 All mechanistic postulates for the events following gelatinase complexation of 1 must originate from a Michaelis complex that accommodates a strong bias for the placement of the diarylether,29 a hydrogen bond between one of the oxygens of the sulfone and the MMP,29 the conformational constraint of the arylsulfone,12 and intimate contact between the thiirane and the active site zinc.11 Efforts toward a mechanistic postulate that embraces these criteria are in progress.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health by CA122417 (to MC and SM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Sternlicht MD, Coussens LM, Vu TH, Werb Z. Biology and regulation of the matrix metalloproteinases. Matrix Metalloproteinase Inhibitors in Cancer Therapy. 2001:1–37. [Google Scholar]
- 2.Stamenkovic I. J Pathol. 2003;200:448. doi: 10.1002/path.1400. [DOI] [PubMed] [Google Scholar]
- 3.Nagase H, Meng Q, Malinovskii V, Huang W, Chung L, Bode W, Maskos K, Brew K. Engineering of selective TIMPs. Inhibition of Matrix Metallo-proteinases: Therapeutic Applications. 1999;878:1–11. doi: 10.1111/j.1749-6632.1999.tb07670.x. [DOI] [PubMed] [Google Scholar]
- 4.Massova I, Kotra LP, Fridman R, Mobashery S. FASEB J. 1998;12:1075. [PubMed] [Google Scholar]
- 5.Lee M, Fridman R, Mobashery S. Chem Soc Rev. 2004;33:401. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- 6.Bonfil RD, Sabbota A, Nabha S, Bernardo MM, Dong Z, Meng H, Yamamoto H, Chinni SR, Lim IT, Chang M, Filetti LC, Mobashery S, Cher ML, Fridman R. Int J Cancer. 2006;118:2721. doi: 10.1002/ijc.21645. [DOI] [PubMed] [Google Scholar]
- 7.Kruger A, Arlt MJE, Gerg M, Kopitz C, Bernardo MM, Chang M, Mobashery S, Fridman R. Cancer Res. 2005;65:3523. doi: 10.1158/0008-5472.CAN-04-3570. [DOI] [PubMed] [Google Scholar]
- 8.Gu ZZ, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY, Lipton SA. J Neurosci. 2005;25:6401. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown S, Bernardo MM, Li ZH, Kotra LP, Tanaka Y, Fridman R, Mobashery S. J Am Chem Soc. 2000;122:6799. [Google Scholar]
- 10.Lee M, Villegas-Estrada A, Celenza G, Boggess B, Toth M, Kreitinger G, Forbes C, Fridman R, Mobashery S, Chang M. Chem Biol Drug Design. 2007;70:371. doi: 10.1111/j.1747-0285.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 11.Kleifeld O, Kotra LP, Gervasi DC, Brown S, Bernardo MM, Fridman R, Mobashery S, Sagi I. J Biol Chem. 2001;276:17125. doi: 10.1074/jbc.M011604200. [DOI] [PubMed] [Google Scholar]
- 12.Hof F, Schutz A, Fah C, Meyer S, Bur D, Liu J, Goldberg DE, Diederich F. Angew Chem Int Ed Engl. 2006;45:2138. doi: 10.1002/anie.200504119. [DOI] [PubMed] [Google Scholar]
- 13.Lee M, Bernardo MM, Meroueh SO, Brown S, Fridman R, Mobashery S. Org Lett. 2005;7:4463. doi: 10.1021/ol0517269. [DOI] [PubMed] [Google Scholar]
- 14.Lim IT, Brown S, Mobashery S. J Org Chem. 2004;69:3572. doi: 10.1021/jo049857v. [DOI] [PubMed] [Google Scholar]
- 15.Ma DW, Cai Q. Org Lett. 2003;5:3799. doi: 10.1021/ol0350947. [DOI] [PubMed] [Google Scholar]
- 16.Kajiyama K, Yoshimune M, Nakamoto M, Matsukawa S, Kojima S, Akiba K. Org Lett. 2001;3:1873. doi: 10.1021/ol015927y. [DOI] [PubMed] [Google Scholar]
- 17.Nudelman NS, Doctorovich F. Tetrahedron. 1994;50:4651. [Google Scholar]
- 18.Goldwhite H, Kaminski J, Millhauser G, Ortiz J, Vargas M, Vertal L, Lappert MF, Smith SJ. J Organomet Chem. 1986;310:21. [Google Scholar]
- 19.Synthetic procedures for compounds 2 and 3, Tables of atomic positions, thermal parameters, bond lengths and angles, and full crystallographic data for compounds 1, 2, and 3 are given in Supporting Information. Crystallographic data (excluding structure factors) for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 665003-665005. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44 (0)1223-336033 or by e-mail: deposit@ccdc.cam.ac.uk].
- 20.Crystals Growth and Analysis. Compound 1 (300 mg) in EtOAc (5 mL) was heated to produce a clear, colorless solution. Crystals of suitable size for single-crystal X-ray diffraction analysis were obtained by diffusion of n-hexane into the EtOAc solution at room temperature overnight. Compounds 2 and 3 were dissolved in hot MeOH and crystals of suitable size for single-crystal X-ray diffraction analysis were grown at room temperature overnight. Crystals were examined under a light hydrocarbon oil. The datum crystal was cut from a larger needle. It was then affixed to a 0.2 mm Mitegen loop mounted atop a tapered copper mounting-pin and transferred to the 100 K nitrogen stream of a Bruker SMART Apex diffractometer equipped with an Oxford Cryosystems 700 series low-temperature apparatus. Cell parameters were determined using reflections harvested from three orthogonal sets of 30 0.5° ϕ scans. The orientation matrix derived from this was passed to COSMO21 to determine the optimum data collection strategy. Minimum 4-fold redundancy was achieved using a combination of ω and ϕ scan series. Data were measured to 0.81 Å. Cell parameters were refined using reflections with I ≥ 10σ(I) harvested from the entire data collection. All data were corrected for Lorentz and polarization effects and runs were scaled using SADABS.22 The structures were solved using direct methods. All non-hydrogen atoms were assigned after the initial solution. Hydrogens were placed at calculated geometries and allowed to ride on the position of the parent atom. Hydrogen thermal parameters were set to 1.2× the equivalent isotropic U of the parent atom, 1.5× for methyl hydrogens. Non-hydrogen atoms were refined with parameters for anisotropic thermal motion.
- 21.COSMO. Bruker-AXS; Madison, WI: 2004. [Google Scholar]
- 22.Sheldrick GM. SADABS. University of Göttingen; Germany: 2004. [Google Scholar]
- 23.Computational Procedures. Molecular dynamics simulations24 were carried out with the crystal structures of 1 and 3. The structures were visualized using Sybyl (Tripos, Inc., 7.3).25 The 6-31G(d) Hartree-Fock method (Gaussian 03)26 was applied for generating electrostatic potential, which was combined with a two-stage potential fitting procedure27 for assigning atomic charges. Water molecules (TIP3P) were added to encompass each compound. The energy of the complex of water and the compound was minimized in 50,000 steps. Subsequently, 1.6 ns molecular dynamics simulations were undertaken. The conformations of 3,000 snapshots were analyzed. The stereo image was rendered using the program Pymol (0.99rev8).28
- 24.Case DA, et al. AMBER. Vol. 8. University of California; San Francisco: 2004. (see Supporting Information for full author list).
- 25.SYBYL 7.3. Tripos Inc; St. Louis, Misssouri: [Google Scholar]
- 26.Frisch MJ, et al. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford, CT: 2004. (see Supporting Information for full author list).
- 27.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269. [Google Scholar]
- 28.PyMOL 0.99rev8. DeLano Scientific LLC; Palo Alto, California: [Google Scholar]
- 29.Tochowicz A, Maskos K, Huber R, Oltenfreiter R, Dive V, Yiotakis A, Zanda M, Bode W, Goettig P. J Mol Biol. 2007;371:989. doi: 10.1016/j.jmb.2007.05.068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.