

Abstract
An understanding of the distribution and spatial structure of the natural vectors of zoonothic pathogens is of interest for effective disease control and prevention. Here, we investigate the range-wide population genetic structure of common pochard (Aythya ferina), a long-distance migratory duck and potential vector of highly pathogenic avian influenza. We collected several hundred samples from breeding and wintering grounds across Eurasia including some H5N1-positive individuals and generated partial sequences of the mitochondrial control region and multilocus microsatellite genotypes. Genetic differentiation among breeding populations was significant for both marker types but higher for maternally inherited mtDNA than for biparentally inherited nuclear markers. There was only weak genetic divergence between ducks sampled in Europe and East Asia, and genetic differentiation between populations was not generally associated with geographical distance. No evidence of genetic substructure was detected for ducks sampled on the European wintering grounds. Our results suggest limited breeding-site fidelity, especially in females, but extensive population admixture on the wintering grounds. The specific role of pochards as natural vectors of zoonotic pathogens and in particular H5N1 remains to be clarified but our results point to wintering grounds as potential hotspots for disease transmission.
Keywords: Diving duck, H5N1, Microsatellites, Migration, Mitochondrial DNA, Population genetics
Introduction
Zoonoses are diseases that are transmitted from animals to humans (Jones et al. 2008). Most zoonoses (71.8%) originate in wildlife, and the incidence of zoonotic events has globally increased over recent time (Jones et al. 2008). Apart from being sources of zoonotic pathogens, wild animals can additionally play a significant role in the geographic spread of pathogens (Morens et al. 2004). Information about the distribution, abundance, and spatial structure of wild animal species may therefore be essential for effective disease control, especially in situations where the transmission of pathogens can be attributed to particular vector species (Kurtenbach et al. 2006). In most wild organisms, populations are spatially structured because of limited dispersal abilities or substantial natural or anthropogenic barriers to movements (e.g., rivers, mountains, habitat fragmentation), and the distribution and dynamics of associated pathogens may, in turn, be spatially heterogeneous (Biek and Real 2010). It is therefore not surprising that an increasing number of studies has taken into account the spatial organization of vector populations in order to understand patterns of infectious disease prevalence and transmission across different geographic scales (Biek and Real 2010, and examples therein).
Population genetics approaches provide a powerful toolbox for characterizing patterns of population structure in vector species and relating them to the spatial (and temporal) dynamics of associated pathogens (e.g., Kempf et al. 2009; Cullingham et al. 2010; Schmidt-Chanasit et al. 2010). In particular, it may be possible to identify certain geographic regions as potential transmission hotspots where different host populations come into contact. For example, the joint analysis of molecular and ringing data showed that northern pintail (Anas acuta) wintering in Japan and North America, respectively, share breeding areas in eastern Russia (Flint et al. 2009). This specific population structure thus points to a potential pathway for the exchange of pathogens between Asia and North America (Flint et al. 2009). At a more local scale, the identification of heterogeneous patterns of dispersal and gene flow among host populations may provide important information about environmental, ecological, and social factors affecting pathogen transmission. As an example, a relatively strong degree of female philopatry was shown by comparing genetic variation of maternally inherited mitochondrial DNA (mtDNA) and biparentally inherited microsatellite markers in white-tailed deer (Odocoileus virginianus), a vector of chronic wasting disease (Cullingham et al. 2010). Consequently, matrilineal social cohesion and social interactions among related females in the vector species may have significant effects on local transmission and dynamics of the disease (Cullingham et al. 2010).
Waterbirds have long been identified as natural reservoirs of various avian influenza (AI) virus subtypes (Webster et al. 1992), and they probably played a role in the spatial spread of highly pathogenic avian influenza (HPAI) H5N1 from south east Asia into Europe (Kilpatrick et al. 2006; Feare 2007; Starick et al. 2007). Common pochards (Aythya ferina, pochards hereafter) have been listed as a high-risk species in relation to AI by the European Union (2005) because they were repeatedly detected as carriers of H5N1 among a number of wild bird species. During the H5N1 outbreak in winter 2005/2006, pochards were either ranked first or second among wild bird species in terms of the number of birds found infected in France, Germany, and Switzerland (Starick et al. 2007; Hars et al. 2008; Baumer et al. 2010). The species is reported as sensitive to infection with HPAI viruses, and although the course of the infection varies between individuals (Keawcharoen et al. 2008), the discovery of an asymptomatic live H5N1-positive wild pochard in Switzerland in winter 2008 increased further the importance of the species for HPAI surveillance programs (Baumer et al. 2010). The very high abundance of pochards (Scott and Rose 1996) and their largely east–west oriented migration direction across Eurasia (Fig. 1) are of additional relevance in the context of AI surveillance in Europe. Against this background, it seems thus relevant to understand the global population structure of pochard and in particular the extent of mixing between East Asian and European ducks.
Figure 1.
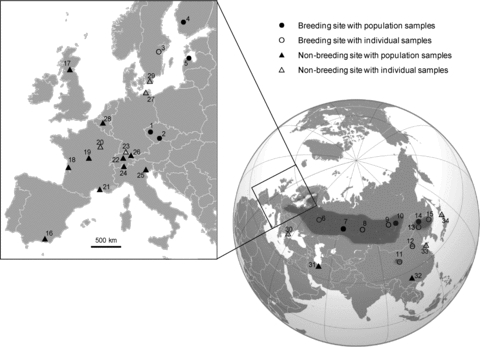
Sampling localities of the common pochard (Aythya ferina) across its distribution range in Eurasia. The species’ breeding range is highlighted in dark gray in the global view. Circles and triangles represent samples from the breeding and the nonbreeding colonies, respectively. Filled symbols indicate sites used for population genetic studies with sample size N≥ 9 at one of the marker sets and open ones represent locations with lower sample sizes used for phylogeographic analyses only. Coordinates and sample sizes for each site are displayed in Table A1.
Based on ring recovery data, Blums and Baumanis (1990) distinguished four geographical subpopulations of pochard, associated with different wintering grounds: (1) Japan, (2) Eastern Asia, (3) Caspian Sea and (4) Europe. Within Europe, a further subdivision has been proposed between a northwestern population wintering around the North Sea and a southeastern population wintering in central Europe and around the Mediterranean and Black Seas (Monval and Pirot 1989; Rose and Scott 1994). However, ring recovery data indicate that the two wintering populations overlap along the North Sea coast (Keller et al. 2009), and that individuals wintering in NW or SE Europe, respectively, may breed in the same areas extending into Western Siberia (Hofer et al. 2006). The situation on breeding grounds further east is relatively unclear, and it is possible that the birds wintering in different parts of Asia may breed at different average longitudes.
In this study, we use molecular data to describe the range-wide population structure of pochards. Our primary aim was to delineate the patterns of genetic differentiation between ducks on the breeding and wintering grounds across Eurasia, with a particular focus on assessing the previously postulated subdivisions. We further examine the congruence between nuclear and mitochondrial markers with different modes of inheritance to investigate sex-specific differences in the rates of gene flow (Prugnolle and de Meeus 2002). These analyses may further help to understand the interactions between population structure of an avian host and the potential spread of zoonotic pathogens across a wide geographical range.
Materials and Methods
Sample collection and DNA extraction
Samples were collected from across the entire Eurasian range of pochard during our field expeditions to the Baltic States, Russia, and China between 2008 and 2010, and through ringing schemes and hunting associations between 2006 and 2010. We collected mostly feathers in a noninvasive manner, but also some muscle, blood, or egg membrane samples. A few tissue samples were obtained from museum collections. In addition, samples of individual ducks identified as H5N1-positive were available from the AI reference laboratories in Switzerland (three individuals), and Germany (five individuals). In total, 345 samples were available from 34 localities in 16 countries (Fig. 1; Table A1). Localities with N≥ 9 for mitochondrial and/or nuclear markers were defined as population samples and included in all population genetic analyses while samples from other localities were included in the phylogeographic analyses only. Samples collected between June and August were considered to be from breeding populations, samples collected between November and February from wintering populations. For the breeding season, we obtained seven population samples plus smaller samples from eight additional locations. On the wintering grounds, population samples were obtained from 12 sites and additional samples from another four sites (Table A1). Three small samples were considered to be from migrating individuals (combined with winter samples as nonbreeding in Fig. 1). Genomic DNA was extracted using a standard phenol–chloroform extraction protocol (Sambrook et al. 1989).
Mitochondrial DNA sequencing
A fragment of the mitochondrial control region (ctr), including most of the hypervariable region (HVR) I, was amplified using the primers M1 (Sorenson and Fleischer 1996) and H774 (Sorenson et al. 1999). PCR amplification was performed in a GeneAmp® PCR System 9700 (Applied Biosystems) in a reaction volume of 25 μl, containing 12.5 μl dH2O, 4.8 μl dNTPs (2.5 mM), 1 μl MgCl2 (25 mM), 1 μl of each primer (10 pmol/μl), 2.5 μl 10× buffer (containing 1.5 mM MgCl2), and 0.2 μl Taq polymerase (5 unit/μl; QIAGEN) and 2 μl template DNA (20–100 ng/μl). The PCR amplification profile included an initial denaturation step of 93°C for 2 min, followed by 35 cycles of 93°C for 30 sec, 56°C for 30 sec, and 72°C for 1 min, and a final extension step of 72°C for 7 min. PCR products were purified with the GenElute™ PCR clean-up kit (Sigma) according to the manufacturer's protocol. Sequencing reactions were carried out in a volume of 10 μl with the Terminator Ready Reaction Mix “Big Dye” (v.3.1, Applied Biosystems) according to the guidelines of the manufacturer. The cycling protocol was as follows: 50 sec at 96°C, followed by 35 cycles of 10 sec at 96°C, 10 sec at 50°C, and 4 min and 30 sec at 60°C. The products were purified using sodium acetate precipitation, and were separated and detected on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems).
Nuclear pseudogenes of mitochondrial origin (numts) are a known issue in some diving ducks, including the genus Aythya (Sorenson and Fleischer 1996), and may cause problems in phylogenetic analysis (Zhang and Hewitt 1996). Here, we used primer M1 that is expected to amplify only the mitochondrial target in pochard (Sorenson and Fleischer 1996). Second, we excluded a few samples (five each from CZSb and SPSe) from further analyses for which multiple fragments were amplified (assessed on minigels) or for which the final sequence showed ambiguous nucleotides. This was mainly observed in blood samples, and may be related to the relatively high ratio of nuclear to mitochondrial DNA in the nucleated red blood cells of birds (Sorenson and Quinn 1998). Finally, we carefully examined all sequences by comparing the levels of variability to reference sequences of ctr from pochard reported in Sorenson and Fleischer (1996) and Sorenson (pers. comm.). Because of the above restrictions, we are confident that the sequences retained for further analyses are authentic mtDNA sequences.
Microsatellite genotyping
All samples were screened at 14 autosomal microsatellite loci initially developed for related species (Fields and Scribner 1997; Maak et al. 2003; Paulus and Tiedemann 2003; Stai and Hughes 2003; Denk et al. 2004; Hefti-Gautschi and Koller 2005; Huang et al. 2006). The 14 microsatellite loci were arranged into two multiplex reactions (Table A2) and the 5′-end of each reverse primer was modified with a pig-tail extension to facilitate genotyping (Brownstein et al. 1996). Amplification was carried out in a 10-μl reaction volume containing 5 μl of PCR mix (QIAGEN Multiplex Kit), 1 μl of a primer mix (Table A2), and 1 μl of template DNA. The PCR conditions were as follows: initial denaturation at 95°C for 15 min, followed by 30 cycles of 94°C for 30 sec, 57°C for 90 sec, and 72°C for 60 sec, and a final extension at 60°C for 15 min. Products were separated and detected on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems). Fragment length was determined in comparison to an internal size standard (GeneScanTM-500LIZ™, Applied Biosystems) using GeneMapper software v.3.7 (Applied Biosystems). Approximately 15% of the samples were re-amplified and genotyped independently to ensure genotyping repeatability (Schweizer et al. 2007).
Genetic diversity indices
The mtDNA sequences were aligned using the CLUSTALW algorithm (Thompson et al. 1994) implemented in the program BioEdit v.7.0 (Hall 1999), and were revised manually. The average number of pairwise nucleotide differences (k), the number of haplotypes (NH), haplotype diversity (H), and nucleotide diversity (π) were calculated for each population sample using DnaSP v.5.0 (Librado and Rozas 2009). For each microsatellite locus, we calculated allelic richness (AR), number of alleles (NA), and observed (HO) and expected heterozygosities (HE) in each population sample with FSTAT v.2.9.3.2 (Goudet 2002). The same program was used to test for deviations from Hardy–Weinberg equilibrium (HWE) and genotypic equilibrium, and to calculate FIS across loci for each population and assess its significance based on 10,000 permutations. Significance levels were adjusted for multiple testing using the Bonferroni procedure (Rice 1989).
Phylogeographic analyses based on all samples
Phylogenetic relationships among ctr haplotypes were reconstructed using Bayesian inference in MrBayes v.3.1.2 (BI; Ronquist and Huelsenbeck 2003). The Hasegawa–Kishino–Yano model assuming a gamma-shaped distribution across sites with an estimated proportion of invariant sites was selected as the best-fitting nucleotide substitution model based on the Akaike Information Criterion in jModelTest v.0.1.1 (Posada 2008). Four independent chains with default heating temperatures were run for 10 million steps and sampled every 1000th step. The first 25% of samples were discarded as burn-in. Convergence of the chains was assessed with the web-based program AWTY (Nylander et al. 2008). The trees were rooted using a homologous sequence from tufted duck (A. fuligula; Liu et al. unpubl. ms.). Further, haplotype networks were constructed based on a median-joining algorithm (Bandelt et al. 1999) in Network v.4.516 (http://www.fluxus-engineering.com). The phylogeographic structure at the nuclear microsatellite markers was investigated with principal coordinates analyses (PCoA) based on pairwise Euclidian distances between individual genotypes in GENALEX v.6.2 (Peakall and Smouse 2006), again separately for breeding and wintering birds.
Population genetic analyses
All population genetic analyses were carried out separately for the seven breeding populations and the 12 wintering populations. Analyses of molecular variance (AMOVA; Excoffier et al. 1992) implemented in Arlequin v.3.5 (Excoffier and Lischer 2010) were performed for both marker sets to assess the proportion of genetic variance explained by the hypothesized migratory divides among breeding or wintering grounds (breeding populations: Europe, Western Siberia, Eastern Siberia, China; wintering populations: SE Europe, NW Europe, Caspian Sea, Eastern Asia). For mtDNA, we calculated pairwise ΦST with the Tamura–Nei model (Tamura and Nei 1993) and for the microsatellite data, pairwise FST using the estimator of Weir and Cockerham (1984) in Arlequin. Significance was assessed based on 10,000 permutations, with significance levels adjusted for multiple testing using the Bonferroni procedure. Preliminary tests showed that our main results are unlikely to be biased by temporal effects. Specifically, the patterns of genetic differentiation observed between samples collected in the same year were consistent with the results of the full analyses (data not shown). For breeding populations only, we used Mantel tests in GENALEX to test for associations between genetic distance (ΦST or FST) and geographical distance in kilometers between pairs of populations. The significance of the association was assessed based on 1000 permutations.
We also used individual-based clustering approaches for the microsatellite data to investigate the genetic structure among breeding and wintering ducks. For Bayesian analyses with STRUCTURE v.2.3.1 (Pritchard et al. 2000), we used an admixture model with correlated allele frequencies (Falush et al. 2003) and a burn-in of 100,000 iterations followed by 500,000 Markov Chain Monte Carlo steps. The number of clusters (K) was varied between 1 and 7 for breeding and 1 and 12 for wintering ducks, respectively, and we carried out 10 independent runs per K. The most likely K was assessed as suggested by Evanno et al. (2005) using STRUCTURE Harvester v.0.56 (Earl 2009). We further used Discriminant Analysis of Principal Components (DAPC), a model-free multivariate method to identify genetic clusters when prior grouping information is lacking (Jombart et al. 2010). We performed DAPC and graphically displayed our results using the package “adegenet” (Jombart 2008) in R version 2.12.0 (R Development Core Team 2008). In all analyses, 33 principal components (PCs) were retained in the data transformation step, which accounted for more than 90% of the total genetic variability. The inference of the most likely number of clusters was based on the Bayesian Information Criterion (BIC; Schwarz 1978).
To assess the statistical power of our microsatellite markers to detect given levels of population differentiation, we performed power analyses in POWSIM v.4.0 (Ryman and Palm 2006). We generated 1000 datasets each for six predefined levels of population differentiation (FST = 0.001, 0.0025, 0.005, 0.01, 0.02, and 0.025) with samples sizes, numbers of markers, and allele frequencies corresponding to the empirical data. Statistical power was defined as the proportion of times the null hypothesis of equal allele frequencies across populations was rejected using a chi-square test or a Fisher's exact test.
Results
Genetic diversity
Sequences of 601 bp from the mitochondrial control region were obtained for 274 individuals, and 143 haplotypes were identified based on 80 polymorphic sites. Forty-seven haplotypes were shared by two to 36 individuals and the remaining 96 were singletons. The average number of pairwise nucleotide differences (k) was similar in breeding and wintering populations (4.4 and 4.38; Mann–Whitney U test, P = 0.96).
Microsatellite genotypes were obtained for 345 individuals. Loci Sfiu3 and MM07 were monomorphic across all populations and not considered for statistical analyses. At the remaining loci, the total number of alleles ranged from 4–17. The mean allelic richness per population was between 3.09 and 3.70 (Table 1), with no significant differences between breeding and wintering populations (Mann–Whitney U test, P = 0.14). There was no evidence of genotypic disequilibrium after Bonferroni correction. Significant heterozygote deficits were observed in 17 of 228 locus-specific tests with locus Caud13 being affected six times (Table A3). Exclusion of this locus from further statistical analyses had no significant quantitative or qualitative effect (results not shown). FIS ranged from –0.01 to 0.22, and values were not significantly different from zero except for one population from the Czech Republic and one from Russia (Table 1).
Table 1.
Estimates of genetic variability in 19 populations of common pochard analyzed for the mitochondrial control region and 12 microsatellite loci. The number of individuals analyzed for mtDNA (Nmt) and nuclear DNA (Nnuc) are given. Map ref. corresponds to the numbers provided in Figure 1, and bold font indicates breeding populations. For mtDNA, the average number of nucleotide differences (k), the number of haplotypes (NH), haplotype diversity (H ± SD), and nucleotide diversity (π± SD, in percent) were calculated. For microsatellites, mean allelic richness (AR), mean observed (HO), and mean expected (HE) heterozygosity were quantified. Multilocus inbreeding coefficients (FIS) were calculated for each population, and values in bold indicate significant deviations from Hardy–Weinberg equilibrium (HWE) after Bonferroni correction
| Mitochondrial DNA | Microsatellites | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Map ref. | Location | Nmt | K | NH | H ± SD | π± SD (%) | Nnuc | AR | HO | HE | FIS |
| 1 | CZSb | 24 | 4.71 | 19 | 0.98 ± 0.02 | 0.80 ± 0.01 | 29 | 3.64 | 0.48 | 0.51 | 0.06 |
| 2 | CZDi | 9 | 4.44 | 9 | 1.00 ± 0.05 | 0.75 ± 0.13 | 20 | 3.39 | 0.49 | 0.57 | 0.13 |
| 4 | FISo | 9 | 7.40 | 5 | 0.81 ± 0.12 | 1.25 ± 0.18 | 9 | 3.11 | 0.47 | 0.56 | 0.18 |
| 5 | LAKa | 12 | 2.53 | 7 | 0.83 ± 0.10 | 0.42 ± 0.11 | 16 | 3.56 | 0.47 | 0.50 | 0.07 |
| 7 | RUYe | 7 | 2.29 | 4 | 0.71 ± 0.18 | 0.39 ± 0.15 | 9 | 3.70 | 0.47 | 0.60 | 0.22 |
| 10 | RUBl | 21 | 4.26 | 15 | 0.92 ± 0.05 | 0.72 ± 0.10 | 21 | 3.43 | 0.43 | 0.49 | 0.12 |
| 14 | CNLf | 16 | 5.17 | 14 | 0.98 ± 0.04 | 0.87 ± 0.10 | 12 | 3.54 | 0.49 | 0.53 | 0.05 |
| 16 | SPSe | 6 | 5.93 | 6 | 1.00 ± 0.10 | 0.99 ± 0.24 | 11 | 3.26 | 0.60 | 0.63 | 0.06 |
| 17 | UKLe | 10 | 5.60 | 9 | 0.98 ± 0.05 | 0.94 ± 0.14 | 10 | 3.67 | 0.52 | 0.54 | 0.05 |
| 18 | FRSa | 12 | 4.64 | 12 | 1.00 ± 0.03 | 0.77 ± 0.13 | 13 | 3.30 | 0.49 | 0.52 | 0.05 |
| 19 | FRIn | 16 | 4.23 | 14 | 0.98 ± 0.03 | 0.71 ± 0.12 | 16 | 3.27 | 0.52 | 0.53 | 0.01 |
| 21 | FRCa | 7 | 2.48 | 6 | 0.95 ± 0.10 | 0.41 ± 0.10 | 10 | 3.2 | 0.5 | 0.54 | 0.08 |
| 22 | CHOb | 24 | 3.94 | 16 | 0.95 ± 0.03 | 0.66 ± 0.08 | 67 | 3.47 | 0.51 | 0.54 | 0.05 |
| 24 | ITVa | 10 | 3.44 | 9 | 0.98 ± 0.05 | 0.58 ± 0.10 | 9 | 3.48 | 0.56 | 0.6 | 0.06 |
| 25 | ITVe | 13 | 4.59 | 12 | 0.99 ± 0.04 | 0.76 ± 0.08 | 12 | 3.48 | 0.58 | 0.65 | 0.09 |
| 26 | DEBs | 6 | 4.60 | 6 | 1.00 ± 0.10 | 0.77 ± 0.09 | 10 | 3.09 | 0.48 | 0.56 | 0.12 |
| 28 | BEBr | 10 | 3.27 | 8 | 0.96 ± 0.10 | 0.55 ± 0.13 | 10 | 3.35 | 0.51 | 0.54 | 0.06 |
| 31 | IRCs | 9 | 4.56 | 7 | 0.94 ± 0.07 | 0.77 ± 0.10 | 10 | 3.18 | 0.48 | 0.48 | −0.01 |
| 32 | CNCh | 21 | 5.29 | 16 | 0.97 ± 0.09 | 0.90 ± 0.09 | 21 | 3.45 | 0.53 | 0.56 | 0.02 |
Phylogeographic inference
The haplotype networks show relatively little molecular differentiation in ducks sampled across Eurasia with most haplotypes separated by one to four mutational steps, and only a few unique haplotypes differing from the most frequent haplotypes by seven to nine mutational steps (Fig. 2). A few frequent haplotypes were shared by individuals from different breeding or wintering regions (Fig. 2), suggesting that no prominent genetic subdivision is associated with the major geographic regions sampled for breeding or wintering pochards. H5N1-positive individuals from Central European wintering grounds showed mostly frequent haplotypes that were shared with ducks sampled on different breeding grounds throughout Eurasia. Phylogenetic trees were star-like and did not show clusters containing individuals of a common geographical origin. Polytomies were frequent and only a few terminal nodes were well supported (posterior probability > 90%; trees not shown). Plots of the first two axes from the principal coordinates analysis based on individual microsatellite genotypes did not reveal distinct groups for breeding or wintering birds (Fig. 3). In both cases, large overlap was observed between individuals from the different geographic areas.
Figure 2.
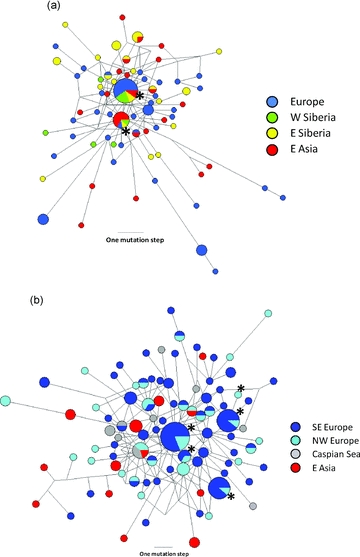
Unrooted median joining networks based on 601 bp sequences of the mitochondrial control region for (A) breeding (N = 119) and (B) wintering (N = 155) pochards. Samples from the breeding season were grouped as Europe, Western Siberia (west of Ural Mountains), Eastern Siberia, and Eastern Asia, following the population subdivision proposed by Blums and Baumanis (1990). Wintering ducks were partitioned into hypothetical subpopulations in southeastern and northwestern Europe (Scott and Rose 1996), around the Caspian Sea and in Eastern Asia. The size of the circles is proportional to the number of individuals with a particular haplotype. Asterisks mark those haplotypes that were found in H5N1-positive pochards sampled on European wintering grounds (see text).
Figure 3.
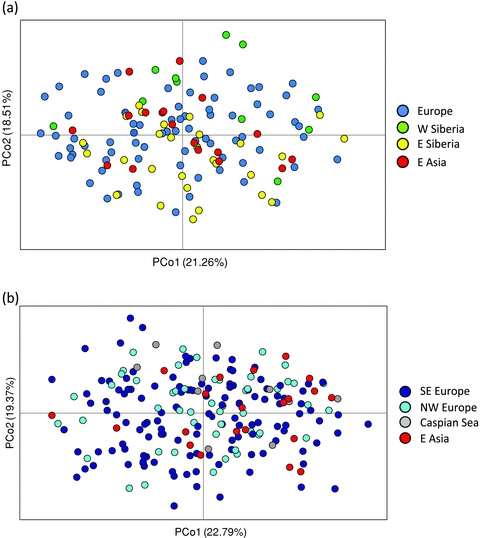
Plots of the first two coordinates from a principal coordinates analysis based on individuals genotyped at 12 microsatellite loci for (A) breeding individuals (N = 134) and (B) wintering individuals (N = 211) of pochard. Different colors represent postulated populations.
Genetic population structure across Eurasia
Breeding populations
The population-based analyses provided no evidence of major migratory divides across Eurasia. The four geographical groups (Europe, Western Siberia, Eastern Siberia, Eastern Asia) did not explain a significant proportion of the genetic variation for mtDNA or microsatellites (Table 2). The variation attributed to differences between populations within regions, however, was significant for both marker types but larger for mtDNA (mtDNA: 8.76%, P < 0.0001; microsatellites: 1.74%, P = 0.016). For mtDNA, five of 21 pairwise comparisons between populations were significant (Table A4), and these involved two far eastern populations (RUBl & CNLf) and two European populations (CZSb & FISo). There was no evidence that pairwise genetic distances (ΦST) increased with geographical distance between sampling locations (R2 = 0.034, P = 0.26). Based on the microsatellite markers, we obtained a global FST estimate of 0.018 (P < 0.0001), and five of 21 pairwise FST values were significant (Table A4). These comparisons mainly involved populations from Russia (RUYe & RUBl) and Latvia (LAKa), and two populations from the Czech Republic (Table A4). Again, there was no evidence that pairwise genetic differentiation increased with geographical distance (R2 = 0.005, P = 0.42).
Table 2.
Hierarchical analyses of molecular variance (AMOVA) based on mtDNA and microsatellite loci for common pochard. Breeding populations (top) were grouped by four regions (Europe, Western Siberia, Eastern Siberia, and Eastern Asia). Wintering ducks (bottom) were partitioned according to four major wintering grounds in northwestern and southeastern Europe, around the Caspian Sea, and in Eastern Asia. Numbers in brackets represent the map reference numbers (shown in Fig. 1 and Table 1) of the populations within each group
| Grouping | Markers | Variation among groups (%) | Variation among populations within groups (%) | Variation within populations (%) |
|---|---|---|---|---|
| [Europe (1, 2, 4, 5)] | mtDNA | −0.66 | 8.76** | 91.90 |
| [Western Siberia (7)] | Microsatellites | 0.09 | 1.74* | 98.17 |
| [Eastern Siberia (10)] | ||||
| [Eastern Asia (14)] | ||||
| [Northwestern Europe (17, 18, 19, 28)] | mtDNA | 1.83 | 0.70 | 97.48 |
| [Southeastern Europe (16, 21, 22, 24, 25, 26)] | Microsatellites | 0.33 | 0.61 | 99.06 |
| [Caspian Sea (31)] | ||||
| [Eastern Asia (32)] |
P < 0.05
P < 0.01.
Individual-based clustering approaches provided no evidence of genetic substructuring among ducks from breeding populations. STRUCTURE analyses showed highest support for one genetic cluster across Eurasia (Fig. S2). DAPC inferred the optimal number of genetic clusters as four, but each population contained components of each of these clusters (Fig. S3), and their relative frequencies in the populations did not show a geographical pattern.
Wintering populations
The proportion of genetic variation associated with the a priori defined wintering regions was not significantly different from zero for mtDNA or microsatellites (Table 2). There was also no evidence of significant substructure within wintering regions (both P > 0.15; Table 2). However, for mtDNA, we observed a global ΦST of 0.02 (P = 0.0156), and two of 66 pairwise comparisons were significantly different from zero (Table A4). If we excluded the Chinese and the Iranian population, the global ΦST value was reduced to 0.005, and no longer significantly different from zero (P = 0.15). For the microsatellite data, the overall population structure was very low but statistically significant (FST = 0.008; P = 0.0147), while none of the pairwise FST values were significant (Table A4). Exclusion of the Chinese and Iranian population resulted in an even lower FST of 0.004, which was not significantly different from zero (P = 0.17). No evidence of genetic substructure was detected by STRUCTURE (Fig. S2). DAPC indicated an optimal number of six clusters but, again, their relative frequencies within populations were not associated with geography (Fig. S3). The simulations performed in POWSIM showed that, with our specific microsatellite panel and sample sizes, we had high statistical power (>95%) to detect genetic substructure if the true FST≥ 0.01 (Fig. S1).
Discussion
Genetic structure and lack of isolation by distance among breeding grounds
Our results provide no direct evidence of the four subpopulations of pochard resulting from migratory divides suggested by Blums and Baumanis (1990) based on analyses of ringing data. It may be not surprising that our phylogeographic and population genetics analyses did not provide support for these specific subdivisions given the difficulty of making quantitative inferences from the typically very limited amount of spatially heterogeneous ringing data (Hofer et al. 2006; Korner-Nievergelt et al. 2010). Nevertheless, our analyses show that breeding populations of pochard are not genetically homogenous across their very large distribution range. This is consistent with other species of migratory ducks where genetic structure was detected at different geographical scales and was sometimes suggested to be associated with recent colonization events (Gay et al. 2004; Tiedemann et al. 2004; Pearce et al. 2009). In pochard, genetic subdivision is likely caused by relatively recent processes given that clear phylogeographic patterns were absent and only allele or haplotype frequency based analyses detected relatively subtle signals of subdivision. This low level of structure among breeding populations is consistent with the high genetic diversity detected here, the large census size of approximately 1,500,000 pairs of the species (Scott and Rose 1996), its largely continuous breeding distribution, and high mobility.
It would be interesting in regard to the potential role of the species in the transmission of pathogens (see below) to investigate its movement patterns and the specific factors causing restricted gene flow between some of the breeding populations in more detail. Given that individuals are capable of traveling thousands of kilometers during annual migration, we would not expect physical restrictions to limit dispersal between breeding grounds. Indeed, the apparent absence of an isolation by distance (IBD) pattern between populations across Eurasia may suggest that dispersal occurs at least partially over very large distances as IBD can only build up if dispersal is spatially restricted (Slatkin 1993). Unfortunately, dispersal events between different breeding grounds are very rarely documented through ring recoveries, but anecdotal evidence suggests that long-distance dispersal occurs at least occasionally (Blums et al. 2002; our own unpub. data). Satellite tracking of individual ducks would provide very detailed information on dispersal rates and distances, but the associated costs typically limit such studies to a very small number of individuals (e.g., Gaidet et al. 2008).
It is worth noting that IBD in pochard may not have built up because populations are not at mutation–drift equilibrium, for example, because of a recent range expansion of the species. At a local scale, such an expansion has occurred. During the last centuries, changes in the availability of suitable water bodies and food sources have led to a westward extension of the species’ breeding range into Central and Western Europe (Cramp and Simmons 1977; Scott and Rose 1996) However, for the remaining distribution range, there are no historical records supporting the possibility of massive population increases and associated range expansions. This possibility requires dedicated further investigations and could have significant consequences for the transmission dynamics of associated pathogens (see e.g., Biek et al. 2007).
In this context, attention should be paid to potential sex differences in colonization and dispersal patterns of pochard given the higher level of genetic differentiation among breeding grounds in maternally inherited mtDNA than in biparentally inherited microsatellite markers (Table 2). Of course, this difference may be related to elevated rates of genetic drift in mtDNA as the effective population size of the mitochondrial genome is only one-fourth of the nuclear genome (Ballard and Whitlock 2004). Additionally, the homogenizing effect of male-biased dispersal would contribute to lower differentiation among populations in nuclear markers but not mtDNA (e.g., Petit et al. 2001; Ruedi et al. 2008). Male-biased dispersal and female philopatry in pochard, if indeed confirmed in future analyses, would be consistent with anecdotal evidence from ringing data that showed that, among the birds observed in two different breeding seasons, 78% of the females had returned to the same location (N = 18), compared to 33% of the males (N = 24; our own unpub. data). Along similar lines, Blums et al. (2002) estimated very high breeding-site fidelity rates of 0.88 for 1-year-old females and 1 for older females using long-term mark-recapture data. Similar sex-specific differences in the level of philopatry are reported from other migratory ducks (reviewed by Greenwood 1980; see also Robertson and Cooke 1999; Gay et al. 2004; Tiedemann et al. 2004; Hefti-Gautschi et al. 2009; Liu et al. unpubl. ms.). Breeding-site fidelity of females may be particularly important in species with uniparental offspring care such as pochard (Cramp and Simmons 1977; Scott and Rose 1996) because of the advantages of local experience in the choice of feeding and breeding sites (Greenwood 1980; Newton 2008).
Population admixture at wintering grounds
Pochards sampled on the disjunct wintering grounds in Europe, the Caspian Sea, and eastern Asia show remarkably little genetic differentiation given the large geographical distances between them. Only population-based analyses including the wintering grounds in Asia revealed very weak signals of genetic subdivision. The latter result—taken alone—could be interpreted as reflecting a subdivision at least between the wintering grounds in Europe and East Asia at the western and eastern edges of the distribution range (Blums and Baumanis 1990). Indeed, this pattern is consistent with the general population structure detected among breeding grounds, where some significant pairwise FST values were observed between European and Asian populations (Table A4). More importantly, however, the much weaker genetic structure among wintering than among breeding grounds (Table 2) demonstrates extensive mixing of individuals from different origins during or after migration.
Given the very high levels of genetic diversity and relatively low genetic structure, it is not surprising that an aggregation of individuals from multiple breeding populations does not produce a classical population genetics signal for admixture among strongly subdivided populations such as elevated diversity or deviations from Hardy–Weinberg expectations (e.g., Wahlund effect; Wahlund 1928; reviewed by Manel et al. 2005). Large-scale admixture between wintering migratory ducks of the Northern Hemisphere is not unique to pochard as demonstrated by genetic analyses and satellite tracking for several species (Pearce et al. 2009; Liu et al. unpubl. ms.). Even ducks wintering several thousand kilometers apart, as for example in the case of northern pintail in Japan or North America, may be part of the same breeding population and even move between these wintering regions (Flint et al. 2009).
Extensive movements within and/or between winters are also the likely cause for the apparent genetic homogeneity among European wintering grounds of pochard. A subdivision was earlier proposed between a northwestern population wintering around the North Sea and a southeastern population wintering in central Europe and around the Mediterranean and Black Seas (Monval and Pirot 1989; Rose and Scott 1994). However, more recent ring recovery data indicate that individuals wintering in NW or SE Europe, respectively, may breed in the same areas extending into Western Siberia (Hofer et al. 2006), and that the two wintering populations overlap along the North Sea coast (Keller et al. 2009). Movements of pochard within a winter occur relatively frequently in response to environmental factors such as food availability and winter harshness (Keller et al. 2009), and may additionally promote contacts between individuals from different origins.
Implications for AI transmission
The absence of a clear association between genetic variation and geography and, in particular, the apparent mixing of ducks on wintering grounds imposes strong limitations on further analyses of the transmission and spread of pathogens of pochard. H5N1-positive individuals from European wintering grounds shared mtDNA haplotypes with ducks from several continental regions, and genetic differentiation was insufficient to use individual-based assignment methods to trace the geographic origin of pochard with highly variable microsatellite genotypes. However, complete panmixia is apparently prevented by low levels of breeding philopatry, which indicates that even higher genetic resolution might allow the identification of the large-scale region of origin of individuals gathering on wintering grounds. Such an analysis would require hundreds or thousands of markers distributed across the entire genome, as recently exemplified in humans, which also show globally low genetic population structure (see Novembre et al. 2008). Additionally, the extensive collection of reference samples from the regions of interest would considerably improve the precision of such assignments (Manel et al. 2005).
It must be noted, however, that direct links between the movement of individuals or populations of pochard and the transmission and spread of HPAI H5N1 will be difficult to establish without considerably improved knowledge on the natural host spectrum, infection pathways, and the consequences of infection. H5N1-positive individuals were repeatedly found among pochard wintering in Europe but infections have also been detected in a number of other waterfowl species (Kim et al. 2009; Kou et al. 2009). Six published sequences of viral isolates from pochard are available from Switzerland and Germany, which were all assigned to the same viral subclade, 2.2.1 (Starick et al. 2007; Hofmann et al. 2008). However, this variant has also been detected in a range of other bird species, which limits the potential for strong associations between the virus and pochard in particular. Moreover, human activities (e.g., poultry trade) certainly contribute to the global spread of the virus without involvement of wild birds (e.g., Gilbert et al. 2006; Kilpatrick et al. 2006; Feare 2007; Gauthier-Clerc et al. 2007). A central question in the context of natural AI transmission is whether infected birds rapidly succumb to the disease or if they are still able to move while already excreting the virus. Infection experiments have demonstrated large differences between and within different species in this respect (reviewed in Gaidet et al. 2010). In pochard, four experimentally infected individuals showed mild to severe clinical symptoms while another four were asymptomatic but excreted the virus (Keawcharoen et al. 2008). While these results may not be directly transferable to wild birds, they still suggest that pochard cannot be ruled out as potential vectors of HPAI. Even if the course of the disease is severe, individual ducks may be able to travel long distances within the period of latency (Gaidet et al. 2008, 2010).
Continent-wide transport of HPAI by individual birds may be rather unlikely but transfer in a stepping-stone fashion through successively infected birds appears nevertheless possible (Reluga et al. 2007; Gaidet et al. 2010). In pochard, the observed very weak population subdivision even between East Asia and Europe and the large number of migrating individuals suggest that such processes could operate at an intercontinental scale. The likelihood for disease transmission and local spread may be particularly high on the wintering grounds, because of the increased stability of RNA viruses at low temperatures (Brown et al. 2009), the aggregation of birds from different geographic regions, and frequent movements within wintering areas.
Acknowledgments
We acknowledge the following institutions and people for kindly providing samples: Swiss Ornithological Institute; Swedish Museum of Natural History; Max Planck Institute for Ornithology at Radolfzell, Germany; M. Boo, A. Boto, N. Bulatova, A. Caizergues, L. Chen, W.X. Dong, I. Fefelov, S. Fereidouni, J. Figuerola, M. Ilieva, M. Janaus, K. Kaisel, R. Hearn, J. Hofer, J. Kreisinger, R. Lezalova, S. Moskvitin, V. Ostapenko, K. Rabii, D. Rodrigues, A. Sauter, Y. Shi, T. Shimada, J. Squire, D. Vangeluwe, J. Viksne, V-M. Väänänen, X.M. Wang, J.H. Yang, S.P. Zhang, and Z.W. Zhang. We are grateful to the Swiss National Reference Laboratory for Poultry Science and the Friedrich-Löffler Institute in Germany for providing samples of H5N1 positive pochards. Y. Liu gratefully acknowledges V. Polyakov and S. Pyzhjanov, who assisted his fieldwork in Russia. We thank F. Korner-Nievergelt for sharing her unpublished manuscripts about analyses of ringing data of pochard. We thank E. Kindler, S. Tellenbach, T. Jenkins, and M. Fischer for technical assistance, discussion, and comments on an early version of the manuscript and the reviewers for helpful suggestions. This project was supported by a grant from the Swiss Veterinary Office (BVET) to G. Heckel and I. Keller.
Appendix
Table A1.
Sampling localities of the common pochard. Map ref. corresponds to numbers in Figure1. Coordinates, sample sizes for mtDNA (Nmt) and nucDNA (Nnuc) analyses, and migration status (breeding: June–August, wintering: November–February, and migrating: October or March) of each population are given
| Map ref. | Location label | Location | Migration status | Country | Latitude | Longitude | Nmt | Nnuc |
|---|---|---|---|---|---|---|---|---|
| 1 | CZSb | Southern Bohemia | Breeding | Czech Rep. | 49°08′30″N | 14°43′38″E | 24 | 29 |
| 2 | CZDi | Divcice, Southern Bohemia | Breeding | Czech Rep. | 49°06′33″N | 14°18′32″E | 9 | 20 |
| 3 | SWSö | Södermanland | Breeding | Sweden | 59°14′19″N | 15°57′30″E | 2 | 2 |
| 4 | FISo | Southern Finland | Breeding | Finland | 61°08′00″N | 24°15′00″E | 9 | 9 |
| 5 | LAKa | Kanieris Lake | Breeding | Latvia | 56°59′45″N | 23°27′45″E | 12 | 16 |
| 6 | RUMo | Moscow region | Breeding | Russia | 55°45′00″N | 37°34′00″E | 2 | 2 |
| 7 | RUYe | Yekaterinburg region | Breeding | Russia | 54°48′00″N | 64°09′00″E | 7 | 9 |
| 8 | RUTo | Tomsk region | Breeding | Russia | 56°27′00″N | 84°57′00″E | 1 | 1 |
| 9 | RUTv | Tunka Valley | Breeding | Russia | 51°50′00″N | 102°22′00″E | 6 | 6 |
| 10 | RUBl | Selenga Delta, Baikal Lake | Breeding | Russia | 52°22′05″N | 106°32′45″E | 21 | 21 |
| 11 | CNQh | Qinghai Lake, Qinghai | Breeding | China | 39°49′00″N | 99°47′00″E | 1 | 1 |
| 12 | CNHa | Hasu Lake, Inner Mongolia | Breeding | China | 40°36′00″N | 110°57′00″E | 4 | 1 |
| 13 | CNQa | Qian'an, Jilin | Breeding | China | 45°15′00″N | 124°11′00″E | 4 | 4 |
| 14 | CNLf | Longfeng Lake, Heilongjiang | Breeding | China | 46°30′19″N | 125°10′43″E | 16 | 12 |
| 15 | CNXk | Xinkai Lake, Heilongjiang | Breeding | China | 45°21′00″N | 132°21′00″E | 1 | 1 |
| 16 | SPSe | Sevillia | Wintering | Spain | 37°26′00″N | 05°22′00″E | 6 | 11 |
| 17 | UKLe | Loch Leven | Wintering | United Kingdom | 56°11′00″N | 03°22′00″W | 10 | 10 |
| 18 | FRSa | Saint-Philbert-de-Grand-Lieu | Wintering | France | 47°02′00″N | 01°38′00″W | 12 | 13 |
| 19 | FRIn | Indre | Wintering | France | 51°21′00″N | 00°11′00″E | 16 | 16 |
| 20 | FRVa | Vanne | Wintering | France | 47°37′00″N | 05°49′00″E | 1 | 1 |
| 21 | FRCa | Camargue | Wintering | France | 40°38′00″N | 08°44′00″E | 7 | 10 |
| 22 | CHOb | Oberkirch | Wintering | Switzerland | 47°09′00″N | 08°06′00″E | 24 | 67 |
| 23 | CHNe | Neuhausen | Wintering | Switzerland | 47°08′00″N | 08°11′00″E | 2 | 2 |
| 24 | ITVa | Varese | Wintering | Italy | 49°06′33″N | 14°18′32″E | 10 | 9 |
| 25 | ITVe | Valle Zignago, Venezia | Wintering | Italy | 45°40′00″N | 12°53′00″E | 13 | 12 |
| 26 | DEBs | Lake Constance | Wintering | Germany | 47°39′00″N | 09°25′00″E | 6 | 10 |
| 27 | DEOs | Baltic Sea coast | Migrating | Germany | 55°19′00″N | 12°05′00″E | 2 | 2 |
| 28 | BEBr | Brussels region | Wintering | Belgium | 50°43′00″N | 04°29′00″E | 10 | 10 |
| 29 | SWSk | Skåne | Migrating | Sweden | 55°59′59″N | 15°11′40″E | 1 | 1 |
| 30 | BUBs | Black Sea coast | Wintering | Bulgaria | 43°21′00″N | 28°02′00″E | 3 | 4 |
| 31 | IRCs | Caspian Sea coast | Wintering | Iran | 36°44′00″N | 53°00′00″E | 9 | 10 |
| 32 | CNCh | Caihai, Guizhou | Wintering | China | 26°51′22″N | 104°13′09″E | 21 | 21 |
| 33 | CNQd | Qingdao, Shandong | Migrating | China | 36°08′00″N | 120°38′00″E | 1 | 1 |
| 34 | JPIz | Izunuma Lake | Wintering | Japan | 38°42′00″N | 141°08′00″E | 1 | 1 |
| Total | 274 | 345 |
Table A2.
Microsatellite loci and design of multiplex reactions: repeat motif, sequences of forward, F (fluorescently labeled) and reverse, R (pig-tailing: GTTTCTT added at 5′-end) primer, fluorescent dye, primer final concentration, and the original reference describing the primers are given for each locus
| Mix | Locus | Repeat motif | Primer sequences (5'-3') | Dye | Concentration (μM) | Reference |
|---|---|---|---|---|---|---|
| Caud13 | (AC)n | F: ACAATAGATTCCAGATGCTGAA | PET | 0.5 | Huang et al. 2006 | |
| R: ATGTCTGAGTCCTCGGAGC | ||||||
| MM07 | (CA)n | F: GCAAAAGGGGCTATGAACAC | NED | 1.25 | Hefti-Gautschi & Koller 2005 | |
| R: GGAGGCAGAGCTGGTTAGTG | ||||||
| Sfiμ3 | (GA)nN2(GA)n | F: TATTCCAATATTCTGCAGGGAGG | 6-FAM | 1.25 | Fields and Scribner 1997 | |
| R: TCCAAGTTAATCAATTATCTGAT | ||||||
| SET1 | Smo11 | (TG)nGA | F: AAATCAACCAAAGAGGCATAGCC | 6-FAM | 2.0 | Paulus and Tiedemann 2003 |
| R: GCAGTTGTTTTGGAGGACAGACA | ||||||
| Sfiμ4 | (GA)n | F: CTGAGGGGGAAGAGAATAAGAGA | PET | 3.0 | Fields and Scribner 1997 | |
| R: CAGGGCAGTATTTTCAGGACATT | ||||||
| MM05 | (AC)n | F: CCAAATCTGACCACCAGGAG | VIC | 3.0 | Hefti-Gautschi and Koller 2005 | |
| R: GCCGTCAGGCAAATAGGAAC | ||||||
| CmAAT28 | (AAT)n | F: TGAAAAGGGTCTTTACCTTATAT | 6-FAM | 2.0 | Stai and Hughes 2003 | |
| R: TTCCACATAAAAATTCATTCAGT | ||||||
| Apl12 | (GA)n | F: AGTTGACCCTAATGTCAGCATC | 6-FAM | 1.25 | Denk et al. 2004 | |
| R: AAGAGACACTGAGAAGTGCTATTG | ||||||
| CmAAT38 | (AAT)n | F: TCCCGAGCTACCACATTGAC | NED | 3.0 | Stai and Hughes 2003 | |
| R: AGCAACTGGAAGGCATTTATCT | ||||||
| Smo4 | (AG)nA | F: ACTTTCCACAGCCTCTTTCACAA | VIC | 2.5 | Paulus and Tiedemann 2003 | |
| R: GACAGTGTTTGTCAATGGATTTT | ||||||
| SET2 | Aph13 | (GA)n | F: CAACGAGTGACAATGATAAAA | 6-FAM | 1.25 | Maak et al. 2003 |
| R: CAATGATCTCACTCCCAATAG | ||||||
| MM03 | (CA)n | F: AAGTACATGTAAAAGCTGAAGTTGC | PET | 3.0 | Hefti-Gautschi and Koller 2005 | |
| R: TTGCCTGATAAAAGGAATGC | ||||||
| Apl36 | (CA)n | F: ATGCTTTGCTGTTGGAGAGC | NED | 3.0 | Denk et al. 2004 | |
| R: TCCACTGGGTGCAAACAAG | ||||||
| Sfiμ2 | (A)n(CA)n | F: CATAAACGGCTAATATGAAGTCT | 6-FAM | 2.0 | Fields and Scribner 1997 | |
| R: AGGCTAGATATTGCTCTTATCCT |
Table A3.
Estimates of genetic variability at 12 polymorphic microsatellite loci in common pochard. For each population and each microsatellite locus, the sample size (N), the number of alleles (NA), the observed (HO) and the expected (HE) heterozygosity is indicated. The HO values shown in bold indicate statistically significant deviations from Hardy–Weinberg equilibrium (HWE) based on 10,000 permutations
| Pop | N | Parameter | Caud13 | SMo11 | Sfiμ04 | MM05 | CmAAT28 | Apl12 | CmAAT38 | Smo4 | Aph13 | MM03 | Apl36 | Sfiμ02 | Mean |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CZSb | 29 | NA | 7 | 4 | 2 | 12 | 10 | 7 | 7 | 3 | 8 | 4 | 2 | 3 | 5.750 |
| HO | 0.448 | 0.517 | 0.138 | 0.862 | 1.000 | 0.655 | 0.552 | 0.069 | 0.724 | 0.276 | 0.345 | 0.138 | 0.477 | ||
| HE | 0.682 | 0.516 | 0.128 | 0.868 | 0.846 | 0.715 | 0.700 | 0.067 | 0.707 | 0.303 | 0.328 | 0.131 | 0.508 | ||
| CZDi | 20 | NA | 5 | 4 | 3 | 10 | 9 | 6 | 5 | 1 | 8 | 4 | 4 | 1 | 5.000 |
| HO | 0.250 | 0.600 | 0.200 | 0.900 | 0.800 | 0.350 | 0.350 | 0.000 | 0.550 | 0.333 | 0.600 | 0.000 | 0.493 | ||
| HE | 0.539 | 0.536 | 0.261 | 0.831 | 0.845 | 0.684 | 0.411 | 0.000 | 0.555 | 0.444 | 0.446 | 0.000 | 0.469 | ||
| FISo | 9 | NA | 5 | 3 | 2 | 8 | 5 | 5 | 4 | 1 | 4 | 3 | 2 | 1 | 3.583 |
| HO | 0.667 | 0.333 | 0.111 | 0.778 | 0.111 | 0.556 | 0.444 | 0.000 | 1.000 | 0.444 | 0.222 | 0.000 | 0.467 | ||
| HE | 0.580 | 0.549 | 0.105 | 0.821 | 0.673 | 0.716 | 0.519 | 0.000 | 0.599 | 0.426 | 0.346 | 0.000 | 0.565 | ||
| LAKa | 16 | NA | 5 | 3 | 3 | 8 | 10 | 7 | 4 | 3 | 9 | 3 | 3 | 2 | 5.000 |
| HO | 0.500 | 0.313 | 0.214 | 0.813 | 0.875 | 0.813 | 0.375 | 0.188 | 0.750 | 0.313 | 0.375 | 0.067 | 0.466 | ||
| HE | 0.635 | 0.506 | 0.309 | 0.865 | 0.836 | 0.773 | 0.363 | 0.174 | 0.725 | 0.271 | 0.354 | 0.064 | 0.503 | ||
| RUYe | 9 | NA | 5 | 4 | 4 | 6 | 8 | 5 | 3 | 1 | 6 | 3 | 2 | 2 | 4.083 |
| HO | 0.667 | 0.444 | 0.556 | 0.667 | 0.625 | 0.556 | 0.286 | 0.000 | 0.667 | 0.167 | 0.143 | 0.111 | 0.474 | ||
| HE | 0.747 | 0.691 | 0.562 | 0.741 | 0.828 | 0.765 | 0.439 | 0.000 | 0.772 | 0.486 | 0.133 | 0.105 | 0.606 | ||
| RUBl | 21 | NA | 7 | 3 | 2 | 11 | 9 | 7 | 5 | 1 | 10 | 4 | 3 | 2 | 5.333 |
| HO | 0.238 | 0.524 | 0.200 | 0.900 | 0.810 | 0.714 | 0.143 | 0.000 | 0.571 | 0.381 | 0.190 | 0.048 | 0.429 | ||
| HE | 0.407 | 0.500 | 0.180 | 0.865 | 0.825 | 0.760 | 0.466 | 0.000 | 0.647 | 0.396 | 0.176 | 0.046 | 0.490 | ||
| CNLf | 12 | NA | 7 | 2 | 2 | 9 | 8 | 7 | 3 | 2 | 8 | 3 | 4 | 1 | 4.667 |
| HO | 0.500 | 0.583 | 0.091 | 0.818 | 0.833 | 0.667 | 0.250 | 0.083 | 0.750 | 0.364 | 0.500 | 0.000 | 0.494 | ||
| HE | 0.580 | 0.469 | 0.087 | 0.835 | 0.840 | 0.740 | 0.344 | 0.080 | 0.691 | 0.376 | 0.521 | 0.000 | 0.528 | ||
| SPSe | 11 | NA | 7 | 3 | 1 | 6 | 6 | 7 | 5 | 1 | 4 | 4 | 3 | 1 | 4.000 |
| HO | 0.727 | 0.727 | 0.000 | 0.545 | 1.000 | 0.545 | 0.364 | 0.000 | 0.636 | 0.545 | 0.273 | 0.000 | 0.596 | ||
| HE | 0.785 | 0.541 | 0.000 | 0.748 | 0.793 | 0.756 | 0.504 | 0.000 | 0.517 | 0.492 | 0.310 | 0.000 | 0.633 | ||
| UKLe | 10 | NA | 6 | 2 | 2 | 9 | 6 | 8 | 3 | 2 | 8 | 4 | 2 | 1 | 4.417 |
| HO | 0.500 | 0.400 | 0.100 | 0.900 | 1.000 | 0.900 | 0.500 | 0.100 | 0.700 | 0.500 | 0.100 | 0.000 | 0.519 | ||
| HE | 0.625 | 0.420 | 0.095 | 0.855 | 0.800 | 0.800 | 0.505 | 0.095 | 0.775 | 0.625 | 0.095 | 0.000 | 0.544 | ||
| FRSa | 13 | NA | 5 | 2 | 1 | 10 | 8 | 6 | 3 | 2 | 7 | 4 | 3 | 1 | 6.727 |
| HO | 0.385 | 0.538 | 0.000 | 0.769 | 0.769 | 0.615 | 0.417 | 0.077 | 0.615 | 0.500 | 0.231 | 0.000 | 0.492 | ||
| HE | 0.444 | 0.488 | 0.000 | 0.898 | 0.837 | 0.722 | 0.344 | 0.074 | 0.547 | 0.462 | 0.210 | 0.000 | 0.519 | ||
| FRIn | 16 | NA | 4 | 3 | 2 | 8 | 11 | 6 | 3 | 2 | 7 | 3 | 2 | 1 | 4.333 |
| HO | 0.250 | 0.563 | 0.200 | 1.000 | 0.813 | 0.750 | 0.188 | 0.063 | 0.813 | 0.563 | 0.500 | 0.000 | 0.518 | ||
| HE | 0.556 | 0.490 | 0.180 | 0.822 | 0.871 | 0.750 | 0.361 | 0.061 | 0.672 | 0.432 | 0.430 | 0.000 | 0.526 | ||
| FRCa | 10 | NA | 5 | 2 | 1 | 10 | 8 | 5 | 4 | 1 | 4 | 4 | 2 | 1 | 5.182 |
| HO | 0.200 | 0.600 | 0.000 | 0.900 | 0.700 | 0.600 | 0.300 | 0.000 | 0.500 | 0.500 | 0.200 | 0.000 | 0.500 | ||
| HE | 0.513 | 0.480 | 0.000 | 0.860 | 0.780 | 0.675 | 0.270 | 0.000 | 0.415 | 0.465 | 0.180 | 0.000 | 0.539 | ||
| CHOb | 67 | NA | 8 | 6 | 4 | 13 | 10 | 9 | 8 | 3 | 8 | 4 | 5 | 1 | 6.583 |
| HO | 0.493 | 0.493 | 0.090 | 0.821 | 0.836 | 0.791 | 0.365 | 0.030 | 0.582 | 0.523 | 0.545 | 0.000 | 0.506 | ||
| HE | 0.608 | 0.520 | 0.087 | 0.867 | 0.840 | 0.786 | 0.553 | 0.030 | 0.612 | 0.532 | 0.430 | 0.000 | 0.537 | ||
| ITVa | 9 | NA | 4 | 4 | 1 | 8 | 7 | 6 | 4 | 1 | 6 | 3 | 3 | 2 | 4.083 |
| HO | 0.444 | 0.667 | 0.000 | 0.889 | 0.778 | 0.889 | 0.500 | 0.000 | 0.667 | 0.556 | 0.222 | 0.000 | 0.561 | ||
| HE | 0.574 | 0.599 | 0.000 | 0.784 | 0.796 | 0.778 | 0.414 | 0.000 | 0.642 | 0.475 | 0.364 | 0.198 | 0.595 | ||
| ITVe | 12 | NA | 7 | 2 | 1 | 8 | 7 | 6 | 6 | 1 | 8 | 3 | 2 | 1 | 4.333 |
| HO | 0.750 | 0.333 | 0.000 | 0.818 | 1.000 | 0.500 | 0.455 | 0.000 | 0.583 | 0.333 | 0.417 | 0.000 | 0.576 | ||
| HE | 0.771 | 0.486 | 0.000 | 0.818 | 0.809 | 0.646 | 0.711 | 0.000 | 0.667 | 0.426 | 0.330 | 0.000 | 0.658 | ||
| DEBs | 10 | NA | 5 | 2 | 2 | 8 | 7 | 5 | 1 | 1 | 4 | 3 | 3 | 1 | 3.500 |
| HO | 0.500 | 0.300 | 0.143 | 0.800 | 0.667 | 0.875 | 0.000 | 0.000 | 0.400 | 0.200 | 0.400 | 0.000 | 0.476 | ||
| HE | 0.712 | 0.495 | 0.133 | 0.825 | 0.772 | 0.641 | 0.000 | 0.000 | 0.480 | 0.340 | 0.340 | 0.000 | 0.555 | ||
| BEBr | 10 | NA | 3 | 3 | 2 | 6 | 9 | 6 | 5 | 1 | 8 | 3 | 2 | 1 | 4.083 |
| HO | 0.400 | 0.500 | 0.100 | 0.700 | 0.900 | 0.700 | 0.600 | 0.000 | 0.700 | 0.200 | 0.300 | 0.000 | 0.510 | ||
| HE | 0.340 | 0.485 | 0.095 | 0.720 | 0.850 | 0.710 | 0.590 | 0.000 | 0.615 | 0.460 | 0.255 | 0.000 | 0.539 | ||
| IRCS | 10 | NA | 5 | 2 | 3 | 6 | 7 | 5 | 3 | 2 | 5 | 5 | 2 | 1 | 3.833 |
| HO | 0.500 | 0.600 | 0.286 | 0.700 | 0.700 | 0.700 | 0.300 | 0.100 | 0.800 | 0.444 | 0.100 | 0.000 | 0.475 | ||
| HE | 0.420 | 0.480 | 0.255 | 0.705 | 0.740 | 0.725 | 0.395 | 0.095 | 0.585 | 0.525 | 0.095 | 0.000 | 0.481 | ||
| CNCh | 21 | NA | 5 | 4 | 2 | 12 | 11 | 6 | 4 | 2 | 5 | 3 | 4 | 1 | 4.917 |
| HO | 0.381 | 0.476 | 0.111 | 0.850 | 0.905 | 0.619 | 0.474 | 0.048 | 0.762 | 0.550 | 0.667 | 0.000 | 0.531 | ||
| HE | 0.600 | 0.541 | 0.105 | 0.843 | 0.858 | 0.774 | 0.676 | 0.046 | 0.675 | 0.411 | 0.484 | 0.000 | 0.556 |
Table A4.
Estimates of pairwise genetic differentiation among 19 populations of common pochard. Location abbreviations are given in Table A1, and are shown in bold for breeding populations. Above the diagonal are pairwise ΦST values for mtDNA and below the diagonal are pairwise FST values for microsatellites. Values shown in bold indicate significant genetic differentiation after Bonferroni correction
| CZSb | CZDi | FISo | LAKa | RUYe | RUBl | CNLf | SPSe | UKLe | FRSa | FRIn | FRCa | CHOb | ITVa | ITVe | DEBs | BEBr | IRCs | CNCh | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CZSb | − | 0.0697 | 0.1328 | 0.0773 | 0.0982 | 0.0695 | 0.0743 | 0.0349 | 0.0116 | 0.0503 | 0.0386 | 0.0502 | 0.1281 | 0.0466 | 0.0680 | −0.0100 | 0.0684 | 0.0777 | 0.0202 |
| CZDi | 0.0229 | − | 0.1521 | −0.0154 | −0.0162 | 0.0196 | 0.0085 | −0.0119 | 0.0144 | 0.0138 | 0.0389 | 0.0052 | 0.0262 | 0.0035 | −0.0006 | −0.0378 | −0.0096 | −0.0044 | 0.0306 |
| FISo | 0.0137 | 0.0239 | − | 0.1984 | 0.1942 | 0.1718 | 0.1423 | 0.1118 | 0.0726 | 0.1198 | 0.1437 | 0.1255 | 0.2342 | 0.1728 | 0.1497 | 0.0654 | 0.1638 | 0.1418 | 0.1259 |
| LAKa | 0.0082 | 0.0183 | 0.0088 | − | −0.0076 | 0.0242 | −0.0011 | 0.0417 | 0.0053 | 0.0356 | −0.0050 | 0.0654 | 0.0396 | 0.0131 | −0.0189 | −0.0304 | −0.0338 | −0.0152 | 0.0257 |
| RUYe | 0.0363 | 0.0357 | 0.0299 | 0.0194 | − | 0.0604 | 0.0263 | 0.0256 | 0.0151 | 0.0578 | 0.0072 | 0.0605 | 0.0365 | 0.0026 | 0.0268 | −0.0044 | 0.0196 | ––0.0013 | 0.0426 |
| RUBl | 0.0205 | 0.0148 | 0.0167 | 0.0219 | 0.0542 | − | −0.0016 | 0.0167 | 0.0214 | 0.0308 | 0.0457 | 0.0322 | 0.0455 | 0.0787 | −0.0219 | −0.0507 | −0.0144 | 0.0155 | 0.0342 |
| CNLf | 0.0036 | −0.0038 | 0.0097 | 0.0047 | 0.0301 | 0.0089 | − | −0.0219 | 0.0068 | 0.0113 | 0.0206 | 0.0288 | 0.0133 | 0.0309 | −0.0176 | −0.0571 | −0.0214 | −0.0270 | 0.0376 |
| SPSe | 0.0182 | 0.0142 | 0.0080 | 0.0112 | 0.0184 | 0.0323 | 0.0148 | − | 0.0137 | 0.0455 | 0.0126 | 0.0387 | 0.0494 | 0.0151 | 0.0240 | −0.0574 | −0.0028 | −0.0146 | 0.0478 |
| UKLe | 0.0006 | 0.0200 | 0.0104 | 0.0051 | 0.0249 | 0.0005 | −0.0082 | 0.0098 | − | −0.0233 | −0.0258 | −0.0347 | 0.0537 | −0.0140 | −0.0005 | −0.0700 | 0.0105 | −0.0070 | −0.0085 |
| FRSa | 0.0144 | −0.0048 | 0.0190 | 0.0152 | 0.0536 | −0.0044 | −0.0078 | 0.0088 | −0.0139 | − | 0.0220 | −0.0312 | 0.0380 | 0.0199 | 0.0037 | −0.0471 | 0.0441 | 0.0075 | 0.0273 |
| FRIn | 0.0270 | 0.0020 | 0.0085 | 0.0144 | 0.0211 | 0.0231 | 0.0018 | 0.0133 | 0.0229 | 0.0103 | − | 0.0262 | 0.0767 | 0.0141 | 0.0075 | −0.0533 | 0.0052 | 0.0101 | −0.0020 |
| FRCa | 0.0305 | 0.0078 | 0.0514 | 0.0344 | 0.0710 | 0.0103 | −0.0020 | 0.0365 | 0.0099 | −0.0213 | 0.0325 | − | 0.0319 | 0.0284 | 0.0176 | −0.0350 | 0.0425 | 0.0164 | 0.0460 |
| CHOb | 0.0066 | 0.0072 | 0.0117 | 0.0150 | 0.0366 | 0.0151 | −0.0065 | 0.0081 | −0.0036 | −0.0011 | 0.0129 | 0.0143 | − | 0.0092 | 0.0257 | 0.0160 | 0.0371 | 0.0032 | 0.1067 |
| ITVa | −0.0020 | −0.0024 | −0.0009 | −0.0139 | 0.0113 | 0.0116 | −0.0129 | −0.0134 | −0.0116 | −0.0102 | −0.0104 | 0.0048 | −0.0116 | − | 0.0412 | −0.0121 | 0.0515 | −0.0011 | 0.0496 |
| ITVe | −0.0045 | 0.0131 | 0.0309 | 0.0233 | 0.0311 | 0.0293 | 0.0086 | 0.0059 | 0.0012 | 0.0164 | 0.0263 | 0.0176 | −0.0001 | −0.0068 | − | −0.0628 | −0.0410 | −0.0063 | 0.0215 |
| DEBs | 0.0241 | 0.0090 | 0.0375 | 0.0071 | 0.0453 | 0.0086 | −0.0044 | 0.0080 | 0.0169 | 0.0087 | 0.0148 | 0.0050 | 0.0169 | 0.0001 | 0.0302 | − | −0.0526 | −0.0422 | −0.0372 |
| BEBr | −0.0030 | 0.0005 | 0.0177 | 0.0186 | 0.0426 | 0.0010 | −0.0122 | 0.0231 | −0.0061 | −0.0142 | 0.0166 | −0.0027 | −0.0079 | −0.0145 | −0.0027 | 0.0213 | − | −0.0115 | 0.0232 |
| IRCs | 0.0317 | 0.0080 | 0.0132 | 0.0153 | 0.0117 | 0.0103 | 0.0202 | 0.0198 | 0.0124 | 0.0036 | 0.0065 | 0.0309 | 0.0215 | −0.0057 | 0.0519 | 0.0404 | 0.0046 | − | 0.0411 |
| CNCh | 0.0041 | 0.0050 | 0.0169 | 0.0223 | 0.0174 | 0.0247 | −0.0043 | 0.0268 | 0.0120 | 0.0172 | 0.0211 | 0.0227 | 0.0049 | 0.0010 | −0.0017 | 0.0400 | −0.0008 | 0.0250 | − |
Data archiving
Data were deposited in the Dryad Repository: http://dx.doi.org/10.5061/dryad.vt642
Supporting information
Additional Supporting Information may be found in the online version of this article.
Figure S1. Analysis of statistical power to detect significant differentiation between pochard populations based on the 12 microsatellite markers used in this study.
Figure S2. Analyses with the program STRUCTURE v.2.3.1 provide no evidence of genetic substructure within (A) breeding or (B) wintering pochard.
Figure S3. Inference of genetic clusters using discriminant analysis of principal components (DAPC) for breeding (A) and wintering (B) populations of common pochard, respectively.
Please note: Wiley-Blackwell Publishing is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol. Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294x.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Baumer A, Feldmann J, Renzullo S, Muller M, Thur B, Hofmann MA. Epidemiology of avian influenza virus in wild birds in Switzerland between 2006 and 2009. Avian Dis. 2010;54:875–884. doi: 10.1637/9119-110209-Reg.1. [DOI] [PubMed] [Google Scholar]
- Biek R, Real LA. The landscape genetics of infectious disease emergence and spread. Mol. Ecol. 2010;19:3515–3531. doi: 10.1111/j.1365-294X.2010.04679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biek R, Henderson JC, Waller LA, Rupprecht CE, Real LA. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proc. Natl. Acad. Sci. USA. 2007;104:7993–7998. doi: 10.1073/pnas.0700741104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blums P, Baumanis J. Migration and geographical distribution of pochard and tufted duck populations in the USSR. Baltic Birds. 1990;5:49–57. [Google Scholar]
- Blums P, Nichols J, Hines J, Mednis A. Sources of variation in survival and breeding site fidelity in three species of European ducks. J. Anim. Ecol. 2002;71:438–450. [Google Scholar]
- Brown JD, Goekjian G, Poulson R, Valeika S, Stallknecht DE. Avian influenza virus in water: infectivity is dependent on pH, salinity and temperature. Vet. Microbiol. 2009;136:20–26. doi: 10.1016/j.vetmic.2008.10.027. [DOI] [PubMed] [Google Scholar]
- Brownstein M, Carpten D, Smith J. Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques. 1996;20:1004–1010. doi: 10.2144/96206st01. [DOI] [PubMed] [Google Scholar]
- Commission of the European Communities. Commission decision 2005/726/EC of 17 October 2005 amending Decision 2005/464/EC on the implementation of survey programmes for avian influenza in poultry and wild birds to be carried out in the Member States. Journal of the European Union. 2005;273:21–24. [Google Scholar]
- Cramp S, Simmons K. Handbook of the birds of Europe, the Middle East and North Africa: the birds of the Western Palearctic. Vol. 1. Oxford: Oxford Univ. Press; 1977. [Google Scholar]
- Cullingham CI, Merrill EH, Pybus MJ, Bollinger TK, Wilson GA, Coltman DW. Broad and fine-scale genetic analysis of white-tailed deer populations: estimating the relative risk of chronic wasting disease spread. Evol. Appl. 2010;4:116–131. doi: 10.1111/j.1752-4571.2010.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk AG, Gautschi B, Carter K, Kempenaers B. Seven polymorphic microsatellite loci for paternity assessment in the mallard (Anas platyrhynchos. Mol. Ecol. Notes. 2004;4:506–508. [Google Scholar]
- Earl DA. 2009. Structure Harvester v0.3. Available at http://users.soe.ucsc.edu/~dearl/software/struct_harvest/
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010;3:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feare CJ. The role of wild birds in the spread of HPAI H5N1. Avian Dis. 2007;51:440–447. doi: 10.1637/7575-040106R1.1. [DOI] [PubMed] [Google Scholar]
- Fields RL, Scribner KT. Isolation and characterization of novel waterfowl microsatellite loci: cross-species comparisons and research applications. Mol. Ecol. 1997;6:199–202. doi: 10.1046/j.1365-294x.1997.d01-123.x. [DOI] [PubMed] [Google Scholar]
- Flint P, Ozaki K, Pearce J, Guzzetti B, Higuchi H, Fleskes J, Shimada T, Derksen D. Breeding season sympatry facilitates genetic exchange among allopatric wintering population of northern pintails in Japan and California. The Condor. 2009;111:591–598. [Google Scholar]
- Gaidet N, Cattoli G, Hammoumi S, Newman SH, Hagemeijer W, Takekawa JY, Cappelle J, Dodman T, Joannis T, Gil P. Evidence of infection by H5N2 highly pathogenic avian influenza viruses in healthy wild waterfowl. PLoS Pathogens. 2008;4:e1000127. doi: 10.1371/journal.ppat.1000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidet N, Cappelle J, Takekawa JY, Prosser DJ, Iverson SA, Douglas DC, Perry WM, Mundkur T, Newman SH. Potential spread of highly pathogenic avian influenza H5N1 by waterfowl: dispersal ranges and rates determined from large-scale satellite telementry. J. Appl. Ecol. 2010;47:1147–1157. [Google Scholar]
- Gauthier-Clerc M, Lebarbenchon C, Thomas F. Recent expansion of highly pathogenic avian influenza H5N1: a critical review. Ibis. 2007;149:202–214. [Google Scholar]
- Gay L, Defos Du Rau P, Mondain-Monval J, Crochet P. Phylogeography of a game species: the red-crested pochard (Netta rufina) and consequences for its management. Mol. Ecol. 2004;13:1035–1045. doi: 10.1111/j.1365-294X.2004.02117.x. [DOI] [PubMed] [Google Scholar]
- Gilbert M, Xiao X, Domenech J, Lubroth J, Martin V, Slingenbergh J. Anatidae migration in the western Palearctic and spread of highly pathogenic avian influenza H5N1 virus. Emerg. Infect. Dis. 2006;12:1650–1656. doi: 10.3201/eid1211.060223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3.2) Switzerland: Lausanne; 2002. Available at: http://www2.unil.ch/popgen/softwares/fstat.htm. Updated from Goudet (1995) [Google Scholar]
- Greenwood PJ. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 1980;28:1140–1162. [Google Scholar]
- Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Hars J, Ruette S, Benmergui M, Fouque C, Fournier J-Y, Legouge A, Cherbonnel M, Daniel B, Dupuy C, Jestin V. The epidemiology of the highly pathgenic H5N1 avian influenza in mute swan (Cygnus olor) and other Anatidae in the Dombes region (France), 2006. J. Wildl. Dis. 2008;44:811–823. doi: 10.7589/0090-3558-44.4.811. [DOI] [PubMed] [Google Scholar]
- Hefti-Gautschi B, Koller B. Polymorphic microsatellite markers for the goosander (Mergus merganser. Mol. Ecol. Notes. 2005;5:133–134. [Google Scholar]
- Hefti-Gautschi B, Pfunder M, Jenni L, Keller V, Ellegren H. Identification of conservation units in the European Mergus merganser based on nuclear and mitochondrial DNA markers. Conserv. Genet. 2009;10:87–99. [Google Scholar]
- Hofer J, Korner-Nievergelt F, Korner-Nievergelt P, Kestenholz M, Jenni L. Herkunft und Zugverhalten von in der Schweiz überwinternden oder durchziehenden Tafelenten Aythya ferina. Ornithologischer Beobachter. 2006;103:65–86. [Google Scholar]
- Hofmann MA, Renzullo S, Baumer A. Phylogenetic characterization of H5N1 highly pathogenic avian influenza viruses isolated in Switzerland in 2006. Virus Genes. 2008;37:407–413. doi: 10.1007/s11262-008-0285-2. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhao Y, Haley CS, Hu S, Hao J, Wu C, Li N. A genetic and cytogenetic map for the duck (Anas platyrhynchos. Genetics. 2006;173:287–296. doi: 10.1534/genetics.105.053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K, Patel N, Levy M, Storeygard A, Balk D, Gittleman J, Daszak P. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keawcharoen J, van Riel D, van Amerongen G, Bestebroer T, Beyer WE, van Lavieren R, Osterhaus A, Fouchier RAM, Kuiken T. Wild ducks as long-distance vectors of highly pathogenic avian influenza virus (H5N1) Emerg. Infect. Dis. 2008;14:600–607. doi: 10.3201/eid1404.071016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempf F, Boulinier T, de Meeûs T, Aanathau C, McCoy K. Recent evolution of host-associated divergence in the seabird tick Ixodes uriae. Mol. Ecol. 2009;18:4450–4462. doi: 10.1111/j.1365-294X.2009.04356.x. [DOI] [PubMed] [Google Scholar]
- Keller I, Korner-Nievergelt F, Jenni L. Within-winter movements: a common phenomenon in the Common Pochard Aythya ferina. J. Ornithol. 2009;150:483–494. [Google Scholar]
- Kilpatrick AM, Chmura AA, Gibbons DW, Fleischer RC, Marra PP, Daszak P. Predicting the global spread of H5N1 avian influenza. Proc. Natl. Acad. Sci. USA. 2006;103:19368–19373. doi: 10.1073/pnas.0609227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Negovetich NJ, Forrest HL, Webster RG. Ducks: the “Trojan horses” of H5N1 influenza. Influenza and Other Respiratory Viruses. 2009;3:121–128. doi: 10.1111/j.1750-2659.2009.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korner-Nievergelt F, Sauter A, Atkinson P, Guélat J, Kania W, Kéry M, Köppen U, Robinson R, Schaub M, Thorup K. Improving the analysis of movement data from marked individuals through explicit estimation of observer heterogeneity. J. Avian Biol. 2010;41:8–17. [Google Scholar]
- Kou Z, Li Y, Yin Z, Guo S, Wang M, Gao X, Li P, Tang L, Jiang P, Luo Z. The survey of H5N1 flu virus in wild birds in 14 provinces of China from 2004 to 2007. PLoS ONE. 2009;4:e6926. doi: 10.1371/journal.pone.0006926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach K, Hanincová K, Tsao J, Margos G, Fish D, Ogden N. Fundamental processes in the evolutionary ecology of Lyme borreliosis. Nat. Rev. Microbiol. 2006;4:660–669. doi: 10.1038/nrmicro1475. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Maak S, Wimmers K, Weigend S, Neumann K. Isolation and characterization of 18 microsatellites in the Peking duck (Anas platyrhynchos) and their application in other waterfowl species. Mol. Ecol. Notes. 2003;3:224–227. [Google Scholar]
- Manel S, Gaggiotti OE, Waples RS. Assignment methods: matching biological questions with appropriate techniques. Trends Ecol. Evol. 2005;20:136–142. doi: 10.1016/j.tree.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Monval JY, Pirot JY. Results of the IWRB International Waterfowl Census 1967–1986: population estimates, trends and distribution in selected species of ducks, swans, and coot Fulica atra wintering in the Western Palearctic and West Africa. Slimbridge: The International Waterfowl and Wetlands Research Bureau; 1989. [Google Scholar]
- Morens D, Folkers G, Fauci A. The challenge of emerging and re-emerging infectious diseases. Nature. 2004;430:242–249. doi: 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton I. The migration ecology of birds. London: Academic Press; 2008. [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander JAA, Wilgenbusch JC, Warren DL, Swofford DL. AWTY (are we there yet?): a system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics. 2008;24:581–583. doi: 10.1093/bioinformatics/btm388. [DOI] [PubMed] [Google Scholar]
- Paulus K, Tiedemann R. Ten polymorphic autosomal microsatellite loci for the eider duck Somateria mollissima and their cross-species applicability among waterfowl species (Anatidae) Mol. Ecol. Notes. 2003;3:250–252. [Google Scholar]
- Peakall ROD, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce J, McCracken K, Christensen T, Zhuravlev Y. Migratory patterns and population structure among breeding and wintering red-breasted mergansers (Mergus serrator) and common mergansers (M. merganser. The Auk. 2009;126:784–798. [Google Scholar]
- Petit E, Balloux F, Goudet J. Sex-biased dispersal in a migratory bat: a characterization using sex-specific demographic parameters. Evolution. 2001;55:635–640. doi: 10.1554/0014-3820(2001)055[0635:sbdiam]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1258. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prugnolle F, de Meeus T. Inferring sex-biased dispersal from population genetic tools: a review. Heredity. 2002;88:161–165. doi: 10.1038/sj.hdy.6800060. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. ISBN 3–900051-07–0. Available at: http://www.R-project.org. [Google Scholar]
- Reluga T, Meza R, Walton DB, Galvani AP. Reservoir interactions and disease emergence. Theor. Popul. Biol. 2007;72:400–408. doi: 10.1016/j.tpb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Robertson G, Cooke F. Winter philopatry in migratory waterfowl. The Auk. 1999;116:20–34. [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Rose P, Scott D. Waterfowl population estimates. Slimbridge: The International Waterfowl and Wetlands Research Bureau; 1994. [Google Scholar]
- Ruedi M, Walter S, Fischer MC, Scaravelli D, Excoffier L, Heckel G. Italy as a major ice age refuge area for the bat Myotis myotis (Chiroptera: Vespertilionidae) in Europe. Mol. Ecol. 2008;17:1801–1814. doi: 10.1111/j.1365-294X.2008.03702.x. [DOI] [PubMed] [Google Scholar]
- Ryman N, Palm S. POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol. Ecol. Notes. 2006;6:600–602. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schmidt-Chanasit J, Essbauer SS, Petraityte R, Yoshimatsu K, Tackmann K, Conraths FJ, Sasnauskas K, Arikawa J, Thomas A, Pfeffer M, et al. Extensive host sharing of Central European Tula virus. J. Virol. 2010;84:459–474. doi: 10.1128/JVI.01226-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz G. Estimating the dimension of a model. Annal Stat. 1978;6:461–464. [Google Scholar]
- Schweizer M, Excoffier L, Heckel G. Fine-scale genetic structure and dispersal in the common vole (Microtus arvalis. Mol. Ecol. 2007;16:2463–2473. doi: 10.1111/j.1365-294X.2007.03284.x. [DOI] [PubMed] [Google Scholar]
- Scott D, Rose P. Atlas of Anatidae populations in Africa and Western Eurasia. Wageningen: Wetlands International; 1996. [Google Scholar]
- Slatkin M. Isolation by distance in equilibrium and non-equilibrium populations. Evolution. 1993;47:264–279. doi: 10.1111/j.1558-5646.1993.tb01215.x. [DOI] [PubMed] [Google Scholar]
- Sorenson MD, Fleischer RC. Multiple independent transpositions of mitochondrial DNA control region sequences to the nucleus. Proc. Natl. Acad. Sci. USA. 1996;93:15239–15243. doi: 10.1073/pnas.93.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson MD, Quinn TW. Numts: a challenge for avian systematics and population biology. The Auk. 1998;115:214–221. [Google Scholar]
- Sorenson MD, Ast JC, Dimcheff DE, Yuri T, Mindell DP. Primers for a PCR-based approach to mitochondrial genome sequencing in birds and other vertebrates. Mol. Phylogenet. Evol. 1999;12:105–114. doi: 10.1006/mpev.1998.0602. [DOI] [PubMed] [Google Scholar]
- Stai SM, Hughes CR. Characterization of microsatellite loci in wild and domestic muscovy ducks (Cairina moschata. Anim. Genet. 2003;34:387–389. doi: 10.1046/j.1365-2052.2003.01038.x. [DOI] [PubMed] [Google Scholar]
- Starick E, Beer M, Hoffmann B, Staubach C, Werner O, Globig A, Strebelow G, Grund C, Durban M, Conraths FJ. Phylogenetic analyses of highly pathogenic avian influenza virus isolates from Germany in 2006 and 2007 suggest at least three separate introductions of H5N1 virus. Vet. Microbiol. 2007;128:243–252. doi: 10.1016/j.vetmic.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- Thompson J, Higgins D, Gibson T. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann R, Paulus KB, Scheer M, Von Kistowski KG, Skirnisson K, Bloch D, Dam M. Mitochondrial DNA and microsatellite variation in the eider duck (Somateria mollissima) indicate stepwise postglacial colonization of Europe and limited current long-distance dispersal. Mol. Ecol. 2004;13:1481–1494. doi: 10.1111/j.1365-294X.2004.02168.x. [DOI] [PubMed] [Google Scholar]
- Wahlund S. Zusammensetzung von Populationen und Korrelationserscheinungen vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas. 1928;11:65–106. [Google Scholar]
- Webster R, Bean W, Gorman O, Chambers T, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol. Mol. Biol. Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Zhang D-X, Hewitt GM. Nuclear integrations: challenges for mitochondrial DNA markers. Trends Ecol. Evol. 1996;11:247–251. doi: 10.1016/0169-5347(96)10031-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.