

Abstract
The development of an efficient diastereoselective method that permits rapid construction of the tetracyclic core 17 of the Strychnos-Aspidosperma alkaloids is described. Enaminone 16, synthesized in high yield, has been cyclized under the influence of a Brønsted acid to provide the core tetracyclic framework 17 of the Strychnos alkaloids in optically active form or alternatively to the β-ketoester tetrahydro-β-carboline (THBC) unit 18, by varying the equivalents of acid and the molar concentration. Attempts to utilize 18 to form the C(7)-C(16) bond of the akuammiline related alkaloids represented by strictamine (22), using metal-carbenoid chemistry are also described.
The pentacyclic 3,5-ethano-3H-pyrrolo[2,3-d]carbazole framework is common to a number of Strychnos alkaloids, of which (−)-strychnine (1) and (−)-akuammicine (2) are the most distinct members.1 This pentacyclic unit is also found in a number of bisindoles including (−)-geissospermine (3),2 (+)- geissolosimine (4),3 and (+)-divaricine (5),4 as shown in Figure 1. (+)-Geissoschizoline (6a)5 which has been previously reported as a constituent of Geissospermum vellosii (G. vellosii), is the pentacyclic strychnine-related monomer of these dimeric indole alkaloids 3–5.
Figure 1.

Strychnos-related indole alkaloids.
The stem bark extracts of G. vellosii commonly known as Pao periera are used to treat malaria, poor digestion, constipation, dizziness, and as a febrifuge and tonic or stimulant.6 In a study carried out by Lima et al.7 this plant extract, rich in alkaloids, exhibited potent anticholinesterase activity in vitro and memory enhancing effects in vivo, in a model of cholinergic deficit that has been validated for the development of drugs for the symptomatic treatment of Alzheimer’s disease. The principal alkaloid with anticholinesterase activity in the extract was identified as geissospermine (3), which has also been known to exhibit actions at the autonomic nervous system.8 Because of their complex structural framework and important pharmacological activity coupled with the fact that the southern hemisphere of bisindoles 3–5, had been synthesized in an enantiospecific sense by us,9 efforts have been directed toward the total synthesis of 6a,b as part of a doubly convergent strategy.10 To this end a concise and rapid route via the asymmetric Pictet-Spengler reaction11 to generate the core tetracyclic framework 712 which contains rings ABCE, crucial to the synthesis of the northern hemisphere of these bisindoles 3–5, as well as other complex members of the Strychnos-Aspidosperma class of indole alkaloids was explored. Herein, a brief and rapid entry into this tetracyclic core 7 by a Brønsted acid mediated cyclization of an enaminone 16 is described.
The acid catalyzed coupling of 6a with geissoschizine (8) or vellosimine (9), respectively, to form 32d and 4,3 has been documented and suggests a potential coupling of geissoschizoline Nb-oxide (6b) with vellosimine (9) to form 5. Since the southern hemispheres 8 and 9 of the bisindoles 3–5 have been synthesized from D-tryptophan,9 a doubly convergent strategy could be employed for the synthesis of both the hemispheres of the bisindoles 3–5. As illustrated in Scheme 1, retrosynthetically, the northern hemisphere 6a/6b might be obtained on further functionalization of the tetracyclic Strychnos unit 17′ which in turn might arise from an α-diazo β-ketoester tetrahydro-β-carboline unit 19 by making use of transition metal mediated carbenoid chemistry.13 The nucleophilic attack of the indole double bond onto the electrophilic center of the carbene generated from 19 might lead to the tetracyclic core 17′. Formation of such a tetracyclic framework from an α-ketocarbocation intermediate was originally studied by Takano et al.,14 albeit in poor yields, in an approach toward Aspidosperma alkaloids.15 In order to explore the diazonium approach via 19, the stereoselective synthesis of the key β-ketoester 18 (see Scheme 5) was required. The strategy was to generate this intermediate 18 by carrying out Cacylation on an acid moiety of a suitably substituted tetrahydro-β-carboline (THBC) such as 11 (see Scheme 2), which in turn could be synthesized from 1016 via the asymmetric Pictet-Spengler reaction.
Scheme 1.

Retrosynthetic Analysis
Scheme 5.

Acid Mediated Cyclization of Enaminone 16
Scheme 2.

Synthesis of C(1)-Substituted THBCa
aDST = Dean-Stark Trap
The synthesis began with the D-(+)-Nb-benzyltryptophan methyl ester (10) which served as the starting material and the chiral transfer agent. Based on the conditions developed by Soerens et al.,17 the asymmetric Pictet-Spengler reaction of 10 with 3,3-dimethoxypropanoic acid 1318 under acidic conditions or with commercially available oxaloacetic acid under thermal conditions, was carried out to form the 1-hydroxycarbonyl THBC 11. In both the cases, however, no formation of the target β-carboline 11 was observed. Instead, the trans isomer 12a was isolated under acidic conditions, and a mixture of cis and trans diastereomers 12a,b were formed under thermal nonacidic conditions (Scheme 2).19 Not unexpectedly, after initial formation of the iminium ion I, the β-carboxylic acid, presumably, underwent decarboxylation to give intermediate II, which tautomerized to the iminium ion III, which cyclized to provide the 1,3-disubstituted β-carboline 12a. Alternatively, oxaloacetic acid could have undergone decarboxylation under thermal conditions to generate CO2 and pyruvic acid, which on condensation with 10, would provide 12a,b via intermediate IV. The spectroscopic data of 1- methylTHBC’s 12a,b was identical to those of racemic cis/trans-1-methylTHBC’s.20
Based on the above results, an alternate approach was employed. The β-ketoester fragment 14 was synthesized first followed by the Pictet-Spengler reaction of β-ketoester 14 with 10 in order to form the target β-carboline 18 (Scheme 3). The acid- and base-sensitive β-ketoester 14, was thus synthesized via C-acylation under the conditions developed by Brooks et al.21 As illustrated in Scheme 3, the hydroxycarbonyl group in 13 was activated as an imidazolide with N,N′-carboxyldiimidazole (CDI), and condensed with the magnesium salt of methyl malonate under essentially neutral conditions. Spontaneous decarboxylation occurred during the acidic workup to afford the methyl 5,5-dimethoxy-3- oxopentanoate (14) in 82% yield.22
Scheme 3.

Synthesis of the Michael Adduct 15
Attempts to generate β-carboline 18, by condensation of 10 with 14 under the acid-mediated Pictet- Spengler reaction conditions (TFA/CH2Cl2) failed. Addition of excess reagent, continued stirring for extended periods at room temperature, change of solvent to MeCN and varying the temperature from room temperature to 50 °C either resulted in no reaction or decomposition of the starting material 10. The inability of acetal 14 to react with 10 in the presence of TFA, prompted the use of a milder acid and the more reactive Michael acceptor 15, synthesized in a single step from 14. The ability of trimethylsilyl trifluoromethanesulfonate (TMSOTf), presumably, to bind to a single heterofunctional group and to form a supercationic species23a was utilized to synthesize β-ketoester 15 from the dimethoxy-β-ketoester 14 in 87% yield.23b This represents an important route to synthesize methyl (E)-5-methoxy-3-oxopent-4- enoate (15) in an overall yield of 88% from methyl 3,3-dimethoxypropionate (Scheme 3).
When the β-ketoester 15, was stirred with 10 in the presence of 20 eq. HOAc in refluxing MeCN (Scheme 4), cyclization to the β-carboline 18 was not observed, however, the enaminone 16,24 present as the carbonyl tautomer, was formed in 91% yield. Enaminones25 such as 16 have been previously synthesized albeit in lower yield (34%) from L-tryptophan methyl ester by Pandit et al.26 by using an aziridine-based folate model, and by Singh et al.24 in a moderate yield (60%) by using oxazolidine-based folate models. The route toward the synthesis of the synthetically useful enaminone26–28 16 utilized the readily accessible Michael acceptor 15 and is an important improvement over the previous methods,24,26 especially in yield and scale-up. The reaction between 10 and the β-ketoester 14 also furnished the enaminone 16, but required longer reaction times with a considerable amount of starting material 10 remaining.
Scheme 4.

Synthesis of Enaminone 16a
abrsm: based on recovered starting material 10.
With the enaminone 16 in hand, the acid-mediated cyclization of 16 to the THBC framework in 18 was attempted (Scheme 5). Stirring the enaminone 16 in TFA/CH2Cl2, resulted in no reaction, whereas treatment in HCl/MeOH28 gave a mixture of compounds. Analysis of this crude mixture by NMR spectroscopy revealed the presence of a tetracyclic core structure of the Strychnos alkaloids (17 or 17′), accompanied by the THBC 18. Since both the cyclized products, 17/17′ and 18, are potentially important intermediates in the doubly convergent strategy to the total synthesis of indole alkaloids,29,30 it was decided to pursue selectivity in this process in order to obtain each exclusively. Methanesulfonic acid (MeSO3H, pKa: 1.6 in DMSO)31 which was a stronger acid than TFA, but equivalent in strength to HCl (pKa: 1.8 in DMSO)31 in an aprotic medium presented an attractive option to HCl/MeOH, the acidity of which was difficult to control. The use of MeSO3H would permit one to achieve the exact concentration of the acid in the reaction mixture unlike HCl (g) whose solutions in aprotic media are less accurate and cumbersome to generate.
Treatment of enaminone 16 with 8 or 10 eq. of MeSO3H in CH2Cl2 (0.1 M) at 0 °C, followed by removal of the ice bath immediately after the addition of the acid, gave a single spot at a higher Rf in 0.5 h. This indolenine was isolated and identified as the dihydroindole derivative 17. Encouraged by this result, the amount of acid was altered in order to obtain the β-ketoester THBC 18 as the sole product. It was found that addition of 4 eq. of MeSO3H and performing the reaction under more dilute conditions (0.05 M) at 0 °C (0.5 h) gave the THBC 18, exclusively in 86% yield, as shown in Scheme 5. Separate recrystallization of analytical samples of tetracyclic 17 and the THBC 18 from a mixture of dichloromethane: hexanes afforded crystalline material for single crystal X-ray analysis, as shown in Figure 2. The X-ray analysis confirmed the structural assignment and chirality of the ABCE Strychnos tetracyclic structure as 17, which is opposite to the tetracyclic Strychnos template 17′ (see Scheme 1).
Figure 2.

ORTEP view of the crystal structures of 17 and 18.
The stereochemistry of 17 and 18 obtained during the acid mediated cyclization of 16 suggested11a–c the formation of two different spiroindolenine intermediates resulting from attack of the indole double bond at C-3 from the Si-face to give the syn-spiroindolenine B1 (syn to the ester function) and from the Re-face to give the anti-spiroindolenine B2 (anti to the ester function) of the iminium ion double bond (Scheme 6). It is well known that in acidic medium wherein the enaminone is protonated, the nucleophilic addition occurred preferentially at the iminic sp2 carbon atom, thereby decreasing the possibility of a direct Michael addition.32 Enaminones form stable salts with strong acids and these salts have been found to be protonated at the oxygen atom, with only a few examples suggesting C-protonation. 33 Nevertheless, the five atom π-electron system of enaminone 16, in the presence of excess acid, presumably results in a push-pull system wherein the higher concentration of acid, temperature, and running the reaction more concentrated results in O-protonation (see A1) to generate the enol B1. In the presence of less acid (4 eq.), higher dilution, and lower temperature, the inherent reactivity of the enamine was sufficient to give an intermediate with C-protonation (see A2). The O-protonated intermediate A1, presumably, facilitated the intramolecular attack from the indole nucleus to the Si-face of the C=N double bond to give the syn spiroindolenine intermediate B1 wherein the stereochemistry of the final product was irreversibly established. The C(16)-C(2) bond in 17 was formed by attack of the enolate carbon atom on the iminium ion bond, which resulted in the formation of the tetracyclic Strychnos template 17 with rings C and E fused in a cis-fashion.30 It is possible that hydrogen bonding between the enolate double bond and the indole Na-H group might have resulted in some stabilization of A1 in contrast to that observed in intermediate A2. On the other hand, the C-protonated intermediate A2 gave the classical Pictet-Spengler product 18 with the targeted trans-stereoselectivity. Based on this hypothesis, the use of Nb-benzyl-L-tryptophan methyl ester in the same process should provide framework (17′) with all the stereogenic centers in the correct disposition. To confirm, whether the formation of 17 was not via an acid-catalyzed rearrangement of 18, the β-carboline 18 was stirred at room temperature with 8 eq. of MeSO3H for 24 h. Analysis of the crude reaction mixture by 1H NMR spectroscopy after work-up indicated the presence of only starting 18. Any attempt to heat the reaction mixture resulted in decomposition of the material.
Scheme 6.

Possible Mechanism of the Acid Mediated Cyclization of the Enaminone 16
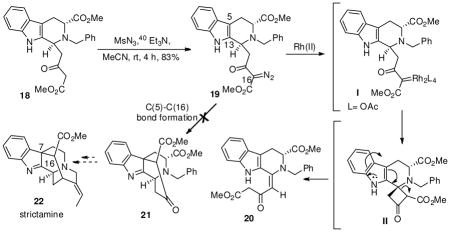
With the important tetracyclic Strychnos template 17 and the THBC unit 18 synthesized in just two steps from 10, the diazonium approach via 18, to form the C(7)-C(16) bond present in the akuammiline series, represented by strictamine 22,34 was attempted. The required diazo compound 19 was readily obtained using standard diazo transfer conditions from 18. With the α-diazo β-keto ester 19 in hand, the efficiency of transition metal salts as catalysts in diazo decompositions, to effect the formation of the C(5)-C(16) bond in 19 was explored (Table 1). Diazo decomposition12 of 19 in benzene in the presence of 5–10 mol % of Rh2(OAc)4 at room temperature, 40 or 75 °C (Table 1, entries 1–3), gave compound 20 (lower Rf) with complete consumption of the starting material 19. Based on 2D NMR experiments on 20, it appeared that the electrophilic Rh(II) carbene35 I, formed by extrusion of nitrogen,36 had inserted into the electron-rich tertiary C-H bond at the benzylic position35 via a single three-centered transition state37 to form the cyclobutanone38 II. This was, presumably, followed by opening of the cyclobutanone ring and intramolecular proton transfer to provide the olefinic THBC 20 as an E-isomer. This was confirmed by analysis of the 1H NMR spectrum which showed the indole Na-H proton at δ 13.8 because of hydrogen bonding with the C-15 carbonyl group. The effect of a CuOTf-mediated diazo decomposition of 19 to provide the C(5)-C(16) bond was next attempted. Initial trials (not shown) using catalytic amounts (5–10 mol %) of the catalyst at room temperature or under refluxing conditions gave only starting material 19. A new product was obtained only after increasing the amount of CuOTf39 to 40 mol % (at reflux) and 80 mol % at room temperature to furnish the β-keto ester THBC 18 (Table 1, entries 4 & 5). With the failure of the transition metal mediated process to give the product with C(5)- C(16) bond formation in 21, attention turned to the use of Brønsted acid catalysis (Table 1, entries 7 & 8). Diazocarbonyl compounds with suitably positioned internal nucleophilic sites permit cyclizations to occur with new bond formation. Treatment of the α-diazo β-keto ester 19 with 4 or 8 eq. of tetrafluoroboric acid at room temperature, provided only starting material 19 (TLC, silica gel) after 4 h. Continued stirring for prolonged periods of time resulted in decomposition of 19 with no new components observed by TLC. All attempts to heat the solution also resulted in decomposition of 19. Photolysis experiments on the diazo compound 19 also failed (Table 1, entry 9). Based on these results it is apparent that the inability to achieve the formation of the C(5)-C(16) bond in indole 21 presumably via a highly strained cyclopropane ring intermediate followed by ring opening, is less favorable, as a result of which the electrophilic carbene inserts into the readily accessible benzylic C-H bond thereby forming 20. In addition, formation of a Strychnos tetracycle like 17′ presumably via the nucleophilic attack of C-13 of the indole nucleus on the electrophilic carbene followed by rearrangement was also not observed. This suggests that the formed electrophilic carbenoid orients itself relatively far from the indole double bond in the β-carboline system 19.
Table 1.
Diazo Decomposition of 19
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp (°C) | Time (h) | Product | %Yield |
| 1 | Rh2(OAc)4 (10 mol %) | benzene | rt | 4 | 20 | 67% |
| 2 | Rh2(OAc)4 (10 mol %) | benzene | 40 | 1.5 | 20 | 67% |
| 3 | Rh2(OAc)4 (10 mol %) | benzene | 75 | 2 | 20 | 75% |
| 4 | CuOTf (80 mol %) | CH2Cl2 | rt | 1 | 18 | 51% |
| 5 | CuOTf (40 mol %) | CH2Cl2 | reflux | 1 | 18 | 47% |
| 6 | CuOTf (60–80 mol %) | benzene | rt | 2 | 18 | 51% |
| 7 | HBF4 (4 - 8 eq.) | CH2Cl2 | rt | > 24 | decomposition | - |
| 8 | HBF4 (4 - 8 eq.) | CH2Cl2 | reflux | 4 | decomposition | - |
| 9 | h (λ ≥ 254 nm) | CCl4 | rt | 12 | 19 + baseline | - |
In summary, an efficient, rapid synthesis of the highly functionalized tetracyclic core 17 of the Strychnos alkaloids was developed. This permits stereospecific formation of rings C and E in a single step, which also generates the all important C-7 quaternary center in a stereospecific fashion. The tetracyclic core 17 of the Strychnos alkaloids and the β-ketoester THBC unit 18 were synthesized exclusively by simply varying the equivalents of MeSO3H, molar concentration, and the reaction temperature, from a common enaminone intermediate 16, which in turn was obtained in high yield by making use of a novel Michael acceptor 15. We believe that by employing L-tryptophan as the starting material, stereospecific formation of the tetracycle 17′ could be readily achieved. Further functionalization of this core should provide ready access to the northern hemispheres of bisindoles 3–5, as well as other complex members of this series of alkaloids.
EXPERIMENTAL SECTION
General Experimental Procedures
Melting points were taken on a Stuart melting point apparatus SMP3 manufactured by Barloworld Scientific US Ltd. Optical rotations were measured on a JASCO Model DIP-370 digital polarimeter. IR spectra were recorded on a Thermo Nicolet Nexus 870 FT-IR or a Perkin Elmer 1600 series FT-IR spectrometer. Proton (1H NMR) and carbon high resolution nuclear magnetic resonance spectra (13C NMR) were obtained on a Bruker 300-MHz or a GE 500-MHz NMR spectrometer. The low resolution mass spectra (LRMS) were obtained as electron impact (EI, 70eV) mass spectrometer, which were recorded on a Hewlett-Packard 5985B gas chromatography-mass spectrometer, while high resolution mass spectra (HRMS) were recorded on a VG Autospec (Manchester England) mass spectrometer. HRMS recorded by electrospray ionization (ESI) methods were performed on a API QStar Pulsar model, manufactured by MDS Sciex. Flash and gravity chromatography was performed using silica gel P60A, 40–63 μm purchased from Silicycle. Basic alumina (Act I, 50–200 μm) for chromatography was purchased from Dynamic Adsorbents. Neutral alumina (Brockman I, ~150 mesh) for chromatography was purchased from Sigma-Aldrich. All reactions were carried out under an argon atmosphere with dry solvents using anhydrous conditions unless otherwise stated.
(E)-Ethyl-5-methoxy-3-oxo-4-pentenoate (15)
To a solution of methyl-5,5-dimethoxy-3-oxo-4- pentenoate 14 (3.6 g, 19.1 mmol) in dry CH2Cl2 (80 mL) was added 2,6-lutidine (6.1 g, 57.11 mmol). The reaction flask was cooled to 0 °C with ice, followed by dropwise addition of TMSOTf (8.5 g, 38.1 mmol). After 10 min, the ice bath was removed and the reaction mixture was allowed to stir at rt for 3 h or until analysis by TLC indicated disappearance of the starting material 14 (silica gel, EtOAc/hexanes, 13:7, run the TLC on a longer plate). After the acetal 14 was completely consumed (TLC), H2O was added to the reaction mixture at rt, after which it was stirred for 5 min and the two layers were separated. The aqueous layer was extracted with CH2Cl2 and the combined organic layers were washed with brine and dried (Na2SO4). The solvent was removed under reduced pressure to afford the crude product 15, which was dried in vacuo to remove the excess 2,6-lutidine before the liquid was purified by flash chromatography on silica gel (EtOAc/hexanes = 1: 9) to provide the enol ether 15 (2.6 g, 87%) as a pale yellow oil. IR (NaCl) νmax 2954, 1743, 1684, 1438 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.66 (1H, d, J = 12.6 Hz), 5.68 (1H, d, J = 12.6 Hz), 3.75 (6H, s), 3.52 (2H, s) (Proton NMR contains 4.8% of enol); 13C NMR (75 MHz, CDCl3) δ 190.7 (C), 167.9 (C), 164.2 (CH), 104.7 (CH), 57.7 (CH3), 52.3 (CH3), 47.8 (CH2); EIMS m/z 158 [M+] (100), 127 (17); HRESIMS m/z 159.0654 [M + H]+ (calcd for C7H11O4, 159.0657).
Alkylation of 10 to provide (R,E)-Methyl-5-((3-(1H-indol-3-yl)-1-methoxy-1-oxo-propan-2- yl)(benzyl)amino)-3-oxopent-4-enoate (16)
The Nb-benzyl-D-trypthophan methyl ester 10 (1.8 g, 5.8 mmol) and the enol ether 15 (1.6 g, 9.9 mmol) were dissolved in CH3CN (65 mL). The mixture was cooled to 0 °C with ice, followed by the dropwise addition of glacial acetic acid (7.0 g, 117 mmol). After stirring the reaction mixture for 15 min at 0 °C the ice bath was removed after which the reaction mixture was stirred for another 15 min and then the flask was transferred to a preheated oil bath (70 – 75 °C). The reaction mixture was stirred at this temperature under argon for 15 h, after which analysis by TLC (silica gel) indicated the disappearance of the starting material 10 and a new component was observed at a lower Rf. The reaction mixture was concentrated under reduced pressure, diluted with CH2Cl2 and neutralized with ice cold 10% aqueous NH4OH. The two layers were separated and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried (Na2SO4) and the solvent concentrated under reduced pressure. The brown colored residue was purified by flash column chromatography (silica gel, hexanes/EtOAc, 1:1), which provided enaminone 16 (2.3 g, 91%) as a light brown solid. Rf 0.49 silica gel, (EtOAc/hexanes, 4: 1); IR (KBr) νmax 3403, 3303, 2951, 1740, 1653, 1558, 743 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.27 (1H, s), 7.88 (1H, d, J = 10.4 Hz), 7.39 (2H, d, J = 8.3 Hz), 7.22 (4H, m), 7.09 (1H, t, J = 7.5 Hz), 7.04-6.94 (3H, m), 5.30 (1H, d, J = 12.8 Hz), 4.36-4.21 (3H, m), 3.71 (3H, s), 3.65 (3H, s), 3.51 (1H, dd, J = 14.8, 6.5 Hz), 3.39 (2H, dd, J = 17.0, 14.4 Hz), 3.26 (1H, dd, J = 14.8, 8.8 Hz); 13C NMR (75 MHz, CDCl3) δ 189.0, 170.6, 169.0, 151.2, 136.1, 134.7, 128.5, 127.6, 127.2, 126.7, 123.4, 122.2, 119.6, 118.0, 111.3, 109.6, 97.6, 65.7, 53.4, 52.4, 52.0, 48.0, 27.1; EIMS m/z 434 [M]+ (14), 361 (10), 334 (12), 319 (16), 305 (31), 275 (14), 243 (11), 234 (41), 201 (30), 170 (10), 130 (100), 91 (87); anal. C 68.74, H 6.10, N 6.29%, calcd for C25H26N2O5, C 69.11, H 6.03, N, 6.45%.
Brønsted acid mediated cyclization of 16 to provide (2R,3aR,6S,6aR,11bR)-Dimethyl 3-benzyl-5- oxo-2,3,3a,4,5,6,6a,7-octahydro-1H-pyrrolo[2,3-d]carbazole-2,6-dicarboxylate (17)
To a solution of enaminone 16 (725 mg, 1.7 mmol) in dry CH2Cl2 (15 mL), methanesulfonic acid (0.87 mL, 13.4 mmol) was added dropwise under an atmosphere of argon at 0 °C (ice bath). After addition of the MeSO3H, the ice bath was removed immediately and the reaction mixture was allowed to stir for 30 min, at which point analysis by TLC indicated the disappearance of the starting material 16 and a new spot at higher Rf. The reaction mixture was brought to pH 8 with a saturated cold aqueous solution of NaHCO3. The two layers were separated. The organic layer was washed with brine, dried (Na2SO4) and the solvent concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc: hexanes) to give 17 (518 mg, 71% yield) as a light yellow solid part of which was recrystallized from a mixture of CH2Cl2: hexanes to give 17 as white needle shaped crystals for X-ray analysis. Rf 0.54 (silica gel, EtOAc/hexanes, 3: 5); mp 142–143 °C; [α]26 D +81.64 (c 0.73, CHCl3); IR (KBr) νmax 3383, 2951, 1738, 1723, 1608, 1203, 751 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.35-7.26 (3H, m), 7.23-7.20 (2H, m), 7.14-7.06 (2H, m), 6.79 (1H, t, J = 7.4 Hz), 6.65 (1H, d, J = 7.5 Hz), 5.14 (1H, d, J = 2.0 Hz), 4.92 (1H, d, J = 3.2 Hz), 4.24 (1H, t, J = 2.0 Hz), 3.91-3.86 (4H, m), 3.62-3.54 (2H, m), 3.48 (3H, s), 2.90-2.82 (2H, m), 2.55 (2H, ddd, J = 43.1, 18.6, 2.8 Hz), 2.09 (1H, dd, J = 14.4, 6.6 Hz) (Proton NMR contains 10.5% of enol); 13C NMR (75 MHz, CDCl3) δ 202.9 (C), 174.0 (C), 170.9 (C), 149.9 (C), 135.8 (C), 131.1 (C), 129.4 (2 × CH), 128.8 (CH), 128.2 (2 × CH), 127.5 (CH), 122.8 (CH), 118.9 (CH), 109.5 (CH), 70.1 (CH), 67.2 (CH), 65.1 (CH), 55.9 (CH2), 54.6 (CH), 52.7 (C), 52.3 (CH3), 51.8 (CH3), 42.2 (CH2), 37.5 (CH2); EIMS m/z 434 [M+] (85), 403 (17), 375 (100), 343 (62), 319 (19), 284 (12), 259 (41), 242 (19), 204 (24), 154 (24), 143 (18), 130 (44), 91 (91); anal. C 68.76, H 6.19, N 6.28%, calcd for C25H26N2O5, C 69.11, H 6.03, N 6.45%.
Brønsted acid mediated cyclization of 16 to provide (1S,3R)-Methyl-2-benzyl-1-(4-(methoxy-2,4- dioxobutyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (18)
To a solution of enaminone 16 (840 mg, 1.94 mmol) in dry CH2Cl2 (38 mL) was added dropwise methanesulfonic acid (0.50 mL, 7.74 mmol) under an atmosphere of argon at 0 °C (ice bath). After stirring the reaction mixture for 30 min at 0 °C, analysis by TLC indicated the disappearance of the starting material 16 and a new spot at higher Rf. The reaction mixture was neutralized with an ice cold saturated solution of NaHCO3. The two layers were separated. The organic layer was washed with brine, dried (Na2SO4) and the solvent concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc: hexanes) to give 18 (730 mg, 86% yield) as a light yellow solid part of which was recrystallized from a mixture of CH2Cl2: hexanes to give 18 as yellow needle shaped crystals for X-ray analysis. For subsequent batches, the crude residue obtained after work up was directly subjected to the diazo transfer reaction without any further purification. Rf 0.55 (silica gel, EtOAc/hexanes, 2: 3); mp 126–127 °C; [α]26 D −10.71 (c 1.12, CHCl3); IR (KBr) νmax 3335, 3034, 2949, 2880, 1757, 1732, 1359, 1048 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.42 (1H, s), 7.56 (1H, d, J = 7.6 Hz), 7.39-7.25 (6H, m), 7.23-7.11 (2H, m), 4.38 (1H, dd, J = 9.4, 3.4 Hz), 4.01 (1H, dd, J = 9.5, 4.8 Hz), 3.94-3.77 (4H, m), 3.70 (3H, s), 3.62 (1H, d, J = 14.0 Hz), 3.45 (2H, s), 3.30-3.02 (4H, m); 13C NMR (75 MHz, CDCl3) δ 203.2 (C), 172.9 (C), 167.0 (C), 138.9 (C), 135.9 (C), 133.5 (C), 128.5 (2 × CH), 128.3 (2 × CH), 127.2 (CH), 126.5 (C), 121.9 (CH), 119.4 (CH), 118.0 (CH), 111.0 (CH), 106.8 (C), 57.7 (CH), 53.4 (CH2), 52.4 (CH3), 52.0 (CH3), 51.4 (CH), 49.8 (CH2), 49.2 (CH2), 20.8 (CH2); EIMS m/z 434 [M+] (14), 375 (10), 343 (23), 319 (100), 257 (24), 183 (8), 169 (15), 156 (19), 91(84); anal. C 68.92, H 6.14, N 6.24%, calcd for C25H26N2O5, C 69.11, H 6.03, N 6.45%.
Diazo transfer reaction of 18 to provide (1S,3R)-Methyl-2-benzyl-1-(3-diazo-4-methoxy-2,4- dioxobutyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (19)
To the above keto ester 18 (390 mg, 0.90 mmol) in CH3CN (12 mL), Et3N (0.20 mL, 1.44 mmol) was added at rt. The solution which resulted was allowed to stir for 15 min at which time, mesyl azide (192 mg, 1.8 mmol) was added, and the reaction mixture was allowed to stir at rt for 4 h. The reaction mixture was quenched with H2O and the mixture was partitioned between ether and H2O. The aqueous layer was extracted with ether and the combined organic layers were washed with brine, dried (MgSO4), and the solvent concentrated under reduced pressure. The residue was subjected to flash silica gel column chromatography to afford the diazo compound 19 as a yellow solid (343 mg, 83%). Rf 0.61 (silica gel, EtOAc/hexanes, 2: 3); IR (neat) νmax 3369, 2952, 2136, 1718, 1641, 1436, 741 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.48 (1H, s), 7.58 (1H, d, J = 7.4 Hz), 7.39-7.28 (5H, m), 7.25-7.12 (3H, m), 4.34 (1H, t, J = 6.7 Hz), 4.15 (1H, dd, J = 10.1, 5.2 Hz), 3.90-3.79 (7H, m), 3.52 (1H, d, J = 13.6 Hz), 3.33 (2H, ddd, J = 22.1, 14.3, 7.5 Hz), 3.17 (1H, dd, J = 16.0, 10.2 Hz), 3.08 (1H, dd, J = 16.0, 5.3 Hz); 13C NMR (75 MHz, CDCl3) δ 191.3 (C), 173.0 (C), 161.5 (C), 139.3 (C), 135.8 (C), 133.5 (C), 128.9 (2 × CH), 127.9 (2 × CH), 127.0 (CH), 126.7 (C), 121.8 (CH), 119.3 (CH), 118.1 (CH), 110.9 (CH), 107.2 (CH), 90.5 (C), 57.4 (CH), 53.1 (CH2), 52.2 (CH), 52.0 (2 × CH3), 46.8 (CH2), 20.4 (CH2); HRESIMS m/z 461.1824 [M + H]+ (calcd for C25H25N4O5, 461.1825); anal. C 64.93, H 5.42, N 11.90%, calcd. for C25H24N4O5, C 65.21, H 5.25, N 12.17%.
(R,E)-Methyl-2-benzyl-1-(4-methoxy-2,4-dioxobutylidene)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b] indole-3-carboxylate (20)
Procedure described for Entry 1 (Table 1): To a solution of 19 (207 mg, 0.45 mmol) in dry benzene (6 mL) under nitrogen, rhodium (II) acetate (20 mg, 0.045 mmol) was added. The mixture was allowed to stir at rt for 4.0 h. The solution was then filtered through a pad of celite and washed with ether. The filtrate was concentrated under reduced pressure and the residue was subjected to flash silica gel column chromatography (EtOAc: hexanes) to furnished 20 (130 mg, 67% yield) as a light yellow oil. Rf 0.41 (silica gel, EtOAc/hexanes, 2: 3); IR (neat) νmax 2952, 1735, 1508, 1463, 733 cm−1; 1H NMR (300 MHz, CDCl3) δ 13.8 (1H, s), 7.58 (1H, d, J = 8.0 Hz), 7.50 (1H, d, J = 8.3 Hz), 7.44-7.31 (6H, m), 7.14 (1H, t, J = 7.7 Hz), 5.55 (1H, s), 5.13 (1H, d, J = 16.2 Hz), 4.42-4.40 (2H, m), 3.68-3.61 (7H, m), 3.50 (2H, t, J = 14.6 Hz), 3.39 (1H, dd, J = 16.5, 6.6 Hz); 13C NMR (75 MHz, CDCl3) δ 187.0 (C), 171.4 (C), 169.0 (C), 152.3 (C), 136.5 (C), 135.8 (C), 129.0 (2 × CH), 127.9 (CH), 127.7 (C), 126.9 (2 × CH), 125.0 (CH), 124.6 (C), 119.9 (CH), 119.2 (CH), 113.0 (CH), 112.1 (C), 94.7 (CH), 62.4 (CH), 56.7 (CH2), 52.8 (CH3), 52.1 (CH3), 50.6 (CH2), 23.8 (CH2); EIMS m/z 432 [M+] (17), 373 (10), 359 (58), 341 (17), 331 (100), 299 (24), 271 (84), 257 (20), 226 (22), 209 (53), 195 (19), 181 (31), 154 (12); HREIMS m/z 432.1672 (calcd for C25H24N2O5, 432.1685).
Supplementary Material
Acknowledgments
We wish to acknowledge the NIMH (in part) and the Lynde and Harry Bradley Foundation for support of this work. X-ray crystallographic studies were supported by NIDANRL Interagency Agreement Number Y1-DA1101.
Footnotes
Supporting Information Experimental procedures for compounds 12a,b, 13, 14 and spectral data for all the compounds including X-ray data for 17 and 18 (CIF files). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.For a review, see: Bosch J, Bonjoch J, Amat M. The Strychnos Alkaloids. In: Cordell GA, editor. The Alkaloids. Vol. 48. Academic Press; New York: 1996. pp. 75–189.
- 2.(a) Wiesner K, Rideout W, Manson JA. Experientia. 1953;9:369. [Google Scholar]; (b) Rapoport H, Windgassen RJ, Jr, Huges NA, Onak TP. J Am Chem Soc. 1960;82:4404–4414. [Google Scholar]; (c) Rapoport H, Onak TP, Hughes NA, Reinecke MG. J Am Chem Soc. 1958;80:1601–1604. [Google Scholar]; (d) Janot MM. Tetrahedron. 1961;14:113–125. [Google Scholar]
- 3.Rapoport H, Moore RE. J Org Chem. 1962;27:2981–2985. [Google Scholar]
- 4.Mukherjee R, Da Silva BA, Bagnolia A, Das BC, Keifer PA, Shoolery JN. Heterocycles. 1991;32:985–990. [Google Scholar]
- 5.(a) Steele JCP, Veitch NC, Kite GC, Simmonds MSJ, Warhurst DC. J Nat Prod. 2002;65:85–88. doi: 10.1021/np0101705. [DOI] [PubMed] [Google Scholar]; (b) Dadson BA, Harley-Mason J. J Chem Soc [Section D], Chem Commun. 1969:665b. [Google Scholar]
- 6.Dos Santos NP, Pinto AC, Alencastro RB. Quim Nova. 1998;21:666–670. [Google Scholar]
- 7.(a) Araújo JQ, Lima JA, Pinto AC, de Alencastro RB, Albuquerque MG. J Mol Model. 2011;17:1401–1412. doi: 10.1007/s00894-010-0841-2. [DOI] [PubMed] [Google Scholar]; (b) Lima JA, Costa RS, Epifânio RA, Castro NG, Rocha MS, Pinto AC. Pharmacol Biochem Behav. 2009;92:508–513. doi: 10.1016/j.pbb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 8.(a) Aurousseau M. Ann Pharm Fr. 1961;19:175–189. [PubMed] [Google Scholar]; (b) Aurousseau M. Ann Pharm Fr. 1961;19:104–116. [PubMed] [Google Scholar]
- 9.For the synthesis of (+)-geissoschizine (6a) see: Yu S, Berner OM, Cook JM. J Am Chem Soc. 2000;122:7827–7828.For the synthesis of (+)-vellosimine (9) see: Wang T, Cook JM. Org Lett. 2000;2:2057–2059. doi: 10.1021/ol000095+.
- 10.(a) Edwankar CR, Edwankar RV, Rallapalli SK, Cook JM. Nat Prod Commun. 2008;3:1839–1870. [Google Scholar]; (b) Edwankar CR, Edwankar RV, Namjoshi OA, Rallapalli SK, Yang J, Cook JM. Curr Opin Drug Discovery Dev. 2009;12:752–771. [PubMed] [Google Scholar]
- 11.(a) Cox E, Cook JM. Chem Rev. 1995;95:1797–1842. [Google Scholar]; (b) Czerwinski KM, Cook JM. Sterochemical Control of the Pictet-Spengler Reaction in the Synthesis of Natural Products. In: Pearson W, editor. Advances in Heterocyclic Natural Product Synthesis. Vol. 3. Academic Press; San Diego: 1996. pp. 217–277. [Google Scholar]; (c) Lorenz M, Van Linn ML, Cook JM. Curr Org Synth. 2010;7:189–223. [Google Scholar]; (d) Lewis SE. Tetrahedron. 2006;62:8655–8681. [Google Scholar]
- 12.For most recent approaches to the ABCE tetracyclic core of the Strychnos alkaloids, see: Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Nature. 2011;475:183–188. doi: 10.1038/nature10232.Delgado R, Blakey SB. Eur J Org Chem. 2009;10:1506–1510.Sirasani G, Andrade RB. Org Lett. 2009;11:2085–2088. doi: 10.1021/ol9004799.Martin DBC, Vanderwal CD. J Am Chem Soc. 2009;131:3472–3473. doi: 10.1021/ja900640v.Pereira J, Barlier M, Guillou C. Org Lett. 2007;9:3101–3103. doi: 10.1021/ol0712576.Beniazza R, Dunet J, Robert F, Schenk K, Landais Y. Org Lett. 2007;9:3913–3916. doi: 10.1021/ol701493k.Lynch SM, Bur SK, Padwa A. Org Lett. 2002;4:4643–4645. doi: 10.1021/ol027024q.Hilton ST, Jones K, Ho TCT, Pljevaljcic G, Schulte M. Chem Commun. 2001:209–210.Mori M, Nakanishi M, Kajishima D, Sato Y. Org Lett. 2001;3:1913–1916. doi: 10.1021/ol0159571.
- 13.Doyle MP, Anthony Mckervey M, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley-Interscience; New York: 1998. [Google Scholar]
- 14.(a) Takano S, Shishido K, Sato M, Yuta K, Ogasawara K. J Chem Soc, Chem Commun. 1978:943–944. [Google Scholar]; (b) Takano S, Shishido K, Sato M, Ogasawara K. Heterocycles. 1977;6:1699–1704. [Google Scholar]; (c) Takano S, Shishido K, Matsuzaka J, Sato M, Ogasawara K. Heterocycles. 1979;13:307–320. [Google Scholar]
- 15.For a similar approach to the synthesis of Aspidosperma alkaloids via a Lewis acid catalyzed rearrangement through a carbocation intermediate, see: Harley-Mason J, Kaplan M. Chem Commun. 1967:915–916.
- 16.Yu P, Wang T, Li J, Cook JM. J Org Chem. 2000;65:3173–3191. doi: 10.1021/jo000126e. [DOI] [PubMed] [Google Scholar]
- 17.For synthesis of similar 1-hydroxycarbonyl-substituted THBC’s see: Soerens D, Sandrin J, Ungemach F, Mokry P, Wu GS, Yamanaka E, Hutchins L, DiPierro M, Cook JM. J Org Chem. 1979;44:535–545.Sandrin J, Soerens D, Hutchins L, Richfield E, Ungemach F, Cook JM. Heterocycles. 1976;4:1101–1105.
- 18.Valade A-G, Dugat D, Jeminet G, Royer J, Husson H-P. Eur J Org Chem. 2001:2041–2053.Acetal 13 was synthesized following a procedure by: Zaragoza F, Stephensen H, Peschke B, Rimvall K. J Med Chem. 2005;48:306–311. doi: 10.1021/jm040873u.
- 19.Kumpaty HL, Van Linn ML, Kabir MS, Försterling FH, Deschamps JR, Cook JM. J Org Chem. 2009;74:2771–2779. doi: 10.1021/jo8028168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandrin J, Hollinshead SP, Cook JM. J Org Chem. 1989;54:5636–5640. [Google Scholar]
- 21.Brooks DW, Lu LD-L, Masamune S. Angew Chem Int Ed. 1979;18:72–74. [Google Scholar]
- 22.Poupardin O, Greck C, Genêt J-P. Synlett. 1998:1279–1281. [Google Scholar]
- 23.Goundry WRF. Synlett. 2003;12:1940–1941.The trans configuration of enol ether 15 is based on the coupling constant reported for commercially available trans-4-methoxybut-3-en-2-one: Konkol M, Schmidt H, Steinborn D. J Mol Catal A-Chem. 2007;269:119–124.
- 24.The trans-configuration of enaminone 16 is based on the coupling constant reported for the ethyl β-ketoester of ent-16 as shown in Singh K, Deb PK, Venugopalan P. Tetrahedron. 2001;57:7939–7949.
- 25.(a) Elassar A-ZA, El-Khair AA. Tetrahedron. 2003;59:8463–8480. [Google Scholar]; (b) Lu P, Greenhill JV. Enaminones in Heterocyclic Synthesis. In: Katritzky AR, editor. Advances in Heterocyclic Chemistry. Vol. 67. Academic Press; San Diego: 1997. pp. 207–343. [Google Scholar]
- 26.Huizenga RH, Pandit UK. Tetrahedron. 1991;47:4155–4164. [Google Scholar]
- 27.For the first use of enaminones in total synthesis of alkaloids of the Aspidosperma series, see: Büchi G, Matsumoto KE, Nishimura H. J Am Chem Soc. 1971;93:3299–3301. doi: 10.1021/ja00742a042.Ando M, Büchi G, Ohnuma T. J Am Chem Soc. 1975;97:6880–6881.
- 28.Hiemstra HC, Bieräugel H, Pandit UK. Tetrahedron Lett. 1982;23:3301–3304. [Google Scholar]
- 29.For the conversion of THBC (18)-related compounds to potential intermediates for the synthesis of yohimbine alkaloids, see: Akhrem AA, Chernov YG. Synthesis. 1985:411–412.and references cited therein.
- 30.For a detailed study of Lewis acid mediated cyclizations of enaminones and enamidinones to the tetracyclic framework: see references 26 and 28 and also Huizenga RH, van Wiltenburg J, Bieräugel H, Pandit UK. Tetrahedron. 1991;47:4165–4174.Huizenga RH, Van Wiltenburg J, Pandit UK. Tetrahedron Lett. 1989;30:7105–7106.Pandit UK, Bieräugel H, Stoit AR. Tetrahedron Lett. 1984;25:1513–1516.
- 31. [accessed Dec 22, 2010];pKa Table1. http://evans.harvard.edu/pdf/evans_pKa_table.pdf.
- 32.Palmieri G, Cimarelli C. Arkivoc. 2006:104–126. [Google Scholar]
- 33.Greenhill JV. Chem Soc Rev. 1977;6:277–294.and references cited therein.
- 34.Ramírez A, García-Rubio S. Curr Med Chem. 2003;10:1891–1915. doi: 10.2174/0929867033457016. [DOI] [PubMed] [Google Scholar]
- 35.Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704–724. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- 36.Wong FM, Wang J, Hengge AC, Wu W. Org Lett. 2007;9:1663–1665. doi: 10.1021/ol070345n. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura E, Yoshikai N, Yamanaka M. J Am Chem Soc. 2002;124:7181–7192. doi: 10.1021/ja017823o. [DOI] [PubMed] [Google Scholar]
- 38.For a similar type of cyclobutanone formation by C-H activation, see: Sonawane HR, Bellur NS, Ahuja JR, Kulkarni DG. J Org Chem. 1991;56:1434–1439.Hashimoto S, Watanabe N, Ikegami S. Tetrahedron Lett. 1992;33:2709–2712.Cane DE, Thomas PJ. J Am Chem Soc. 1984;106:5295–5303.
- 39.For the synthesis of an akuammiline alkaloid via CuOTf catalyzed diazo decomposition, see: Shen L, Zhang M, Wu Y, Qin Y. Angew Chem Int Ed. 2008;47:3618–3621. doi: 10.1002/anie.200800566.
- 40.Danheiser RL, Millar RF, Brisbois RG, Park SZ. J Org Chem. 1990;55:1959–1964. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.