

Abstract
The amide synthase of the geldanamycin producer, Streptomyces hygroscopicus, shows a broader chemoselectivity than the corresponding amide synthase present in Actinnosynnema pretiosum, the producer of the highly cytotoxic ansamycin antibiotics, the ansamitocins. This was demonstrated when blocked mutants of both strains incapable of biosynthesizing 3-amino-5-hydroxybenzoic acid (AHBA), the polyketide synthase starter unit of both natural products, were supplemented with 3-amino-5-hydroxymethylbenzoic acid instead. Unlike the ansamitocin producer A. pretiosum, S. hygroscopicus processed this modified starter unit not only to the expected 19-membered macrolactams, but also to ring enlarged 20-membered macrolactones. The former mutaproducts revealed the sequence of transformations catalyzed by the post-PKS tailoring enzymes in geldanamycin biosynthesis. The unprecedented formation of the macrolactones together with molecular modeling studies shed light on the mode of action of the amide synthase responsible for macrocyclization. Obviously, the 3-hydroxymethyl substituent shows similar reactivity and accessibility towards C-1 of the seco-acid as the arylamino group, while phenolic hydroxyl groups lack this propensity to act as nucleophiles in the macrocyclization. The promiscuity of the amide synthase of S. hygroscopicus was further demonstrated by successful feeding of four other m-hydroxymethylbenzoic acids leading to formation of the expected 20-membered macrocycles. Good to moderate antiproliferative activity were encountered for three of the five new geldanamycin derivatives which matched well with a competition assay for Hsp90α.
Keywords: ansamitocins, amide synthases, geldanamycin, molecular modeling, mutasynthesis
INTRODUCTION
Geldanamycin 4 is a highly potent antitumor drug1 which binds to the N-terminal ATP-binding domain of heat shock protein 90 (Hsp90) and inhibits its ATP-dependent chaperone activities.2 It was demonstrated that geldanamycin could prevent restructuring of mutated cancer-relevant proteins such as p53.3 Most geldanamycin derivatives reported to date are 17-aminated compounds such as 5a and 5b and were obtained by semisynthesis while total synthesis has not provided essential information for structure-activity relationship studies (SAR).4 17-Dimethylaminoethylamino demethoxygeldanamycin hydrochloride (17-DMAG, 5a), also known as alvespimicin, shows improved water solubility, undergoes only limited metabolism compared to 4 and has reached clinical trials as anticancer agent against several tumors. Related to geldanamycin (4) is reblastatin (6) which instead of the quinone moiety contains a phenolic group at C-18. Importantly, it shows lower cytotoxicity than geldanamycin while having a higher affinity for Hsp90.5
The producing microorganism Streptomyces hygroscopicus var. geldanus NRRL 3602 generates geldanamycin through a biosynthetic machinery based on a polyketide synthase (PKS) and additional post-PKS enzymes (Scheme 1), encoded by the gdm biosynthetic gene cluster.6 The biosynthesis of 4 is primed by the starter unit, 3-amino-5-hydroxybenzoic acid (AHBA 1).6 Chain extension by the PKS using one malonate, four methylmalonate and two methoxymalonate extender units gives seco-progeldanamycin (2), which is cyclized and released from the PKS by an amide synthase, a key enzyme. The resulting progeldanamycin 3 is then further modified by a set of tailoring enzymes.
Scheme 1.
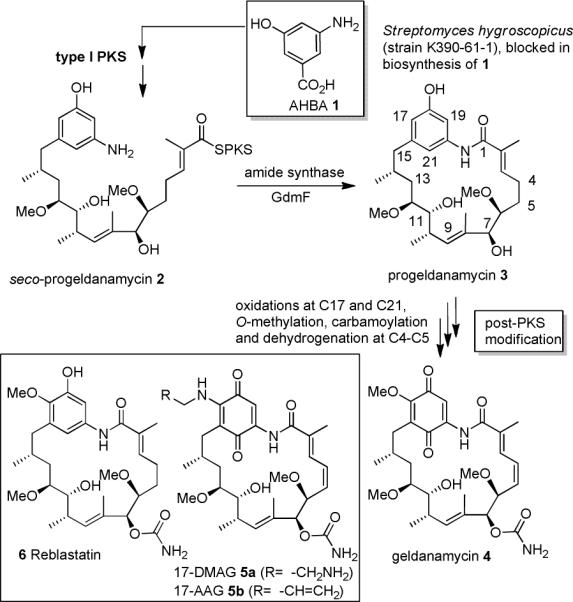
Biosynthesis starting from 3-amino-5-hydroxybenzoic acid (AHBA 1) of geldanamycin (4) via seco-(2) and progeldanamycin (3).
It was proposed that this sequence starts with the oxidation of C-21 and C-17, followed by O-methylation at C-17, introduction of the carbamoyl moiety and finalized by dehydrogenation across C-4,5.7a,b The oxidation of the hydroquinone moiety to the quinone only takes place after oxidation at C-21.7c Surprisingly, it still remains unclear at which stage during the biosynthesis bioactive geldanamycin analogues are formed or what the simplest biosynthetic intermediate is that inhibits Hsp90 activity.
We and two other groups8–10 have utilized mutants of the geldanamycin producer blocked in AHBA formation [AHBA-(−) mutants] to prepare several new geldanamycin derivatives in a mutasynthetic approach,11,12 a synthetic method that is inspired by the detailed biosynthetic knowledge of geldanamycin formation.7,13
In this report, we disclose the first mutasynthetic preparation of a metabolic series of geldanamycin derivatives using an AHBA-(−) mutant of S. hygroscopicus (strain K390-61-1), which reveal the order of the post-PKS modifications in geldanamycin biosynthesis and shed light on structure-activity relationships among the post-PKS modification products. We also disclose the unprecedented finding that ring-enlarged macrolactones instead of macrolactams can be generated by the amide synthase present in S. hygroscopicus. We finally discuss the possible molecular basis of this unusual behaviour of the amide synthase involved.
RESULTS AND DISCUSSION
Feeding experiments with mutasynthon 7 and biosynthetic consequences
In our previous mutasynthetic studies we found that when feeding aminobenzoic acid analogs to a growing culture of an AHBA-(−) mutant of Streptomyces hygroscopicus only one or two mutaproducts were formed in sufficient amounts for full structural analysis and preliminary biological evaluation. One aspect of the present report covers the unprecedented observation that the mutasynthon hydroxymethyl-aminobenzoic acid 7 yields a large variety of new geldanamycin derivatives when fed to fermentations (1.6 L) of Streptomyces hygroscopicus (strain K390-61-1). We first collected five new geldanamycin derivatives 8−12 in amounts that allowed full characterization (Scheme 2). The formation of this set of modified advanced biosynthetic intermediates is particularly interesting from two aspects: a) The new products can be arranged in a linear biosynthetic sequence of postketide transformations which slightly differs from the one proposed in the literature7 and b) SAR studies can be conducted on structurally closely related derivatives along a postketide biosynthetic sequence.
Scheme 2.
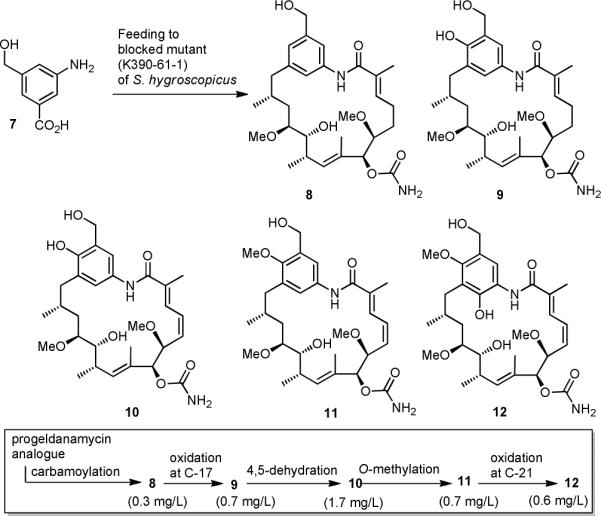
Mutasynthesis with S. hygroscopicus K390-61-1 using 3-aminobenzoic acid (7) and our proposed sequence of postketide transformations.
With respect to point a) we propose that the post-PKS modifications are initiated by carbamoylation at C-7 to yield mutaproduct 814 which is then oxidized at C-17 to yield phenol 9, followed by dehydrogenation across C-4,5 which furnishes geldanamycin derivative 10. The next post-PKS transformation is the O-methylation at C-17 to yield benzyl alcohol 11. Finally, we also isolated the fully substituted mutaproduct 12 which is formed upon oxidation at C-21 of 11. This sequence of events differs from the proposed one7 which postulates that carbamoylation takes place at a later stage.
Remarkably, oxidation at C-21 takes place despite the fact that the para position is hydroxymethylated instead of being functionalized with a phenolic moiety. It is noteworthy, that benzoic acid 7 is the first mutasynthon that is fully processed by the geldanamycin producer S. hygroscopicus, except for the oxidation of the aromatic ring to the quinone form. Up to the present study, our mutasynthetic experiments had only afforded new geldanamycin derivatives that are processed to the level of functionalization found in derivative 10. Lee, Hong and coworkers9 as well as Menzella et al.10 disclosed related studies and also only described new geldanamycin mutaproducts that were incompletely processed.
However, the most striking result of these feeding studies is the formation of the additional 20-membered macrolactone 13 and its metabolites 14 and 15. Structural proof for the formation of these macrolactones which are expanded in ring size compared to 4 was gained from HRMS spectrometric data and 1H-NMR studies. The benzyl ether protons at C-22 undergo a downfield shift (δ= 5.07 and 4.95 ppm for 13 compared to δ= 4.57 ppm for 8) which is diagnostic for acylated benzyl alcohols. The macrolactone formation was also confirmed by long range proton carbon couplings in HMBC spectra. The additionally isolated compounds 14 and 15 result from further metabolism of 12 by, respectively, non-specific N-acetylation, a common biotransformation in Actinomycetes, and carbamoylation, presumably catalyzed by the carbamoyltransferase GdmN of the gdm gene cluster.7a
Obviously, the amide synthase, that is responsible for the macrolactamization is able to recognize and utilize alcohols for cyclization as long as they are sufficiently nucleophilic. This is not the case for the phenolic moiety in seco-progeldanamycin 2, but in marked contrast it is for its hydroxymethyl derivative A/B.
Molecular modeling of amide synthases
Studies on the formation of ring-enlarged macrolactones reveal that the amide synthase GdmF carries a non-native functional group specificity when dealing with seco acids as substrate. In the non-natural seco-progeldanamycin A/B the benzylic alcohol serves as a potential nucleophile for cyclization to about the same extent as the naturally occurring aryl amine. So far, our mutasynthetic experiments with the AHBA-(−) mutants of S. hygroscopicus and A. pretiosum, the ansamitocin producer,16,17 unequivocally demonstrated that both amide synthases involved in macrolactamization accept a wide range of seco acids modified in the aromatic moiety.5 But when feeding aminobenzoic acid 7 to a AHBA-(−) blocked mutant of A. pretiosum in our previous studies, we had only encountered formation of lactams and never lactones, when screening the fermentation broth by UPLC HRMS.16 Thus the amide synthase Asm9 catalyzing the cyclization of seco-ansamitocin 16a to proansamitocin 17a (Scheme 3) shows clearly a narrower reaction specificity compared to GdmF, which also catalyzes the conversion of 16b to 17b. This specificity difference led us to analyze the specific amino acid composition at and around the catalytic site of the two amide synthases. An initial sequence alignment between GdmF and Asm9, revealed 42 % identity and 58 % similarity when accounting for conservative mutations.
Scheme 3.
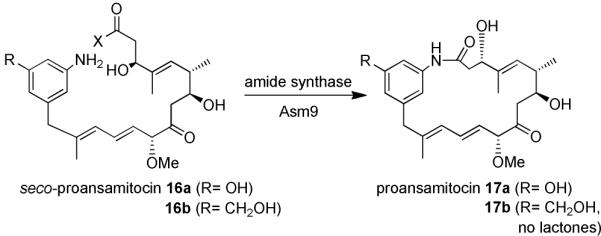
Macrolactamization of seco-proansamitocin 16a and derivative 16b to proansamitocin 17a and derivative 17b, respectively, by the amide synthase (Asm9) from Actinosynnema pretiosum (for X refer to legend of figure 1).15
Therefore, any of a large number of residues (105) could potentially be responsible for the different specificity of GdmF compared to Asm9 when handling the aromatic hydroxymethyl group in 6 (Figure 2A).18
Figure 2.
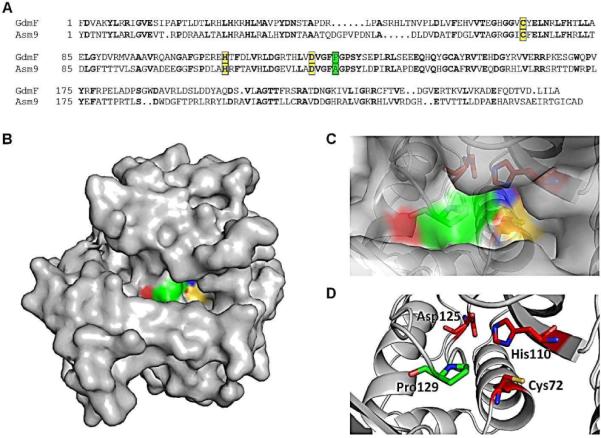
(A) Sequence alignment of GdmF and Asm9 using web-based resources (http://blast.ncbi.nlm.nih.gov). Amino acids in bold indicate a perfect match. Yellow ground displays the catalytic triad: Cys72, His110 and Asp125; Green ground, Pro/Ala129 (for GdmF and Asm9, respectively); (B) Surface representations of the homology model of GdmF using SWISS-MODEL.18–20 Note the tight channel leading to the catalytic triad (Cys72, His110 and Asp125 are colored, compare to C and D); (C, D) Zoom-in of Figure 2B showing the catalytic triad and Pro129.
For the amide synthase to perform the cyclization of seco-progeldanamycin 2 or the corresponding hydroxymethyl analogue 15, both ends of the PKS-bound polyketide have to access the catalytic pocket containing the catalytic triad, Cys72, His110 and Asp125 (Figure 2B–D). Hydrogen-bonding between those three residues enhances the nucleophilic power (partial charge) of the Cys72 sulphur atom, enabling it, as the first reaction step to covalently link to C-1 of an incoming precursor molecule via nucleophilic attack, displacing it from the PKS. In the second reaction step the aromatic ring enters the catalytic pocket through a tight channel. Once the aromatic ring is in close proximity to C-1 (covalently bound to Cys72), one of the two functional groups present (-NH2 vs. -OH or -NH2 vs. -CH2OH) at the aromatic ring can nucleophilically attack C-1. Cys72 then serves as a leaving group while the macrocycle is formed.
Restrained molecular dynamics (MD) simulations on a nanosecond time scale using explicit solvent allowed us to probe qualitatively the conformational space available to the aromatic ring while penetrating the catalytic pocket and thus the distance of the nucleophilic groups -NH2 vs. -OH/ -CH2OH, respectively, relative to C-1 (Figure 318). The dynamic trajectories obtained imply that the natural substrate 2 has some degree of mobility within the catalytic pocket, while the hydroxymethyl derivative is more statically trapped. Further detailed analysis of the trajectories pointed towards Pro129 being a key residue for the specific macrocyclization of seco-progeldanamycins via the amino group by introducing stiffness inside the catalytic pocket of GdmF (Figure 3). Intriguingly, the sequence alignment of GdmF and Asm9 shows that the latter amide synthase contains Ala129 instead of proline, which could explain the different substrate specificity of this enzyme. Indeed, our modeling indicates that a proline-to-alanine mutation at position 129 in GdmF (which mimics the catalytic pocket of Asm9) changes the dynamic penetration trajectory for the hydroxymethyl derivative of seco-geldanamycin substantially (see Figures 3 and 4)18.
Figure 3.
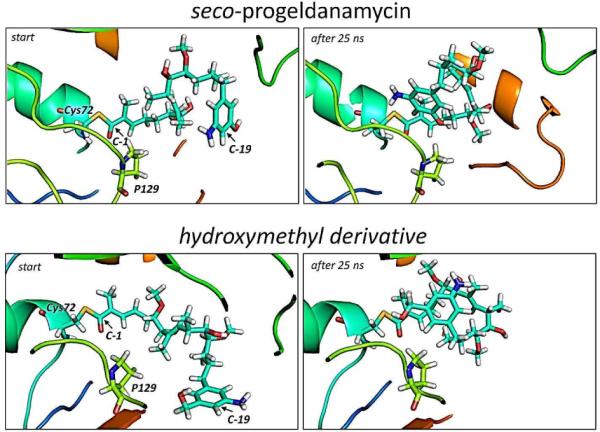
Starting and end trajectory after 25 nanoseconds of the restrained MD simulations of GdmF covalently linked to seco-progeldanamycin (top panel) and its hydroxymethyl derivative (bottom panel). The distance restraint (5 Å) between C-1 and C-19 contained a linearly increasing energy penalty during the simulation. Note, that for the hydroxymethyl derivative the aromatic ring moves out of plane in order to avoid clashes with P129.
Figure 4.
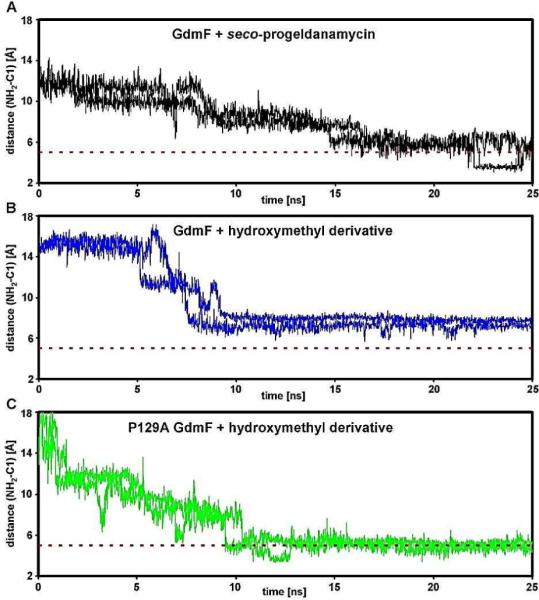
Restrained MD simulations of the aromatic ring whilst penetrating the catalytic site of GdmF (and its variant P129A) using a distance restraint (5 Å) with linearly increasing energy penalty between C-1 and C-19, the latter being located halfway between the nucleophilic groups (–NH2, –OH or –CH2OH) decorating the aromatic ring (Figure 3). Using this approach we were able to qualitatively probe the conformational space available to the aromatic ring and thus the distance of the nucleophilic groups to C-1. The dashed red line indicates close proximity of the NH2 nucleophile to C-1 after penetration as observed for the natural ligand seco-progeldanmycin (in black). In contrast, the approach of the NH2 nucleophile of the hydroxymethyl derivative of seco-geldanamycin towards C-1 appears hindered (in blue) which might explain observed slower lactam cyclization rates for this non-natural substrate. In fact, detailed analyses of the dynamic trajectories imply that the naturally occurring compound is still mobile within the catalytic pocket of GdmF, while the hydroxymethyl derivative shows static properties (compare Figure 4A and 4B and data not shown). The static ligand properties can be overcome once proline 129 is mutated to an alanine (P129A, in green) rationalizing the observed turnover specificity for Asm9 (compare Figure 4B and 4C). Note that P129A mimics the catalytic pocket of Asm9 (see Figures 2,3)).
It needs to be stressed that proline residues are the most rigid amino acids due to phi and psi torsion angle restrictions in the protein backbone. Thus, our results suggest that catalysis of seco-progeldanamycin 2 cyclization via the amino group by GdmF is fast due to smooth penetration into the catalytic channel as well as the ideal distance and flexibility of the amino group towards C-1. In contrast, several factors such as penetration hindrance, less mobility and a non-ideal distance of the amino group with respect to C-1 for the hydroxymethyl derivative of seco-progeldanamycin all reduce the efficiency of the nucleophilic attack dramatically. In addition to these bioinformatic considerations, the differences in nucleophilicity of the three different relevant functional groups can also account for the observed reactivity pattern as it is well established that anilines and benzyl alcohols are more reactive towards electrophiles than phenols.21 However, further computational and biochemical experimental data are needed to fully understand the molecular mechanisms underlying catalysis of macrocyclization of these enzymes.
Formation of other macrolactones
These unprecedented results along with our preliminary computational analyses paved the way to the design of alternative benzoic acids 18, 20, 22 and 24 that upon feeding to S. hygroscopicus (strain K390-61-1) under the typical fermentation conditions were expected to be transformed into lactones. In fact, the expected lactones 19, 21, 23 and 25 were isolated and characterized (Scheme 4). Again, post-PKS carbamoylation occurred in all cases. Notably, benzyl amine 26 was also processed to the corresponding 20-membered macrolactam 27, as judged by HRMS-MS analysis.22 In contrast, the ortho- and para-substituted benzoic acids 29 and 30, regioisomers of the meta-substituted mutasynthon 24 did not serve as substrates for the biosynthetic assembly line of the geldanamycin producer S. hygroscopicus (Figure 3). Obviously, only the meta architecture provides a geometric environment for successful PKS-loading and PKS-processing of unnatural starter units. One cannot exclude the possibility that charging onto the loading module does happen but that further processing is inefficient or is blocked. However, we were never able to detect any oligoketide intermediates in cases of unsuccessful mutasyntheses.
Scheme 4.

New 20-membered geldanamycin-derived macrolactones 19, 21, 23 and 25 (isolated yields) and 20-membered macrolactam 30.
The modelling analyses and the feeding experiments are also in line with the following observation. Benzoic acid 28 containing two meta-oriented phenolic hydroxyl groups does not act as a substrate for mutasynthetic formation of macrocyclized geldanamycin derivatives. This would require that one of the two phenolic groups has to act as nucleophile during amide synthasecatalyzed macrocyclization, but ring closure via a phenolic moiety has never been observed.
Biological evaluation
The new geldanamycin derivatives 8 – 12 were tested for their inhibitory effects on the proliferation of different mammalian cell lines. The cell lines tested are cancer derived, permanent - but not cancer derived, and primary cells. In general all derivatives show a lower activity than geldanamycin 4, the least active derivative being 10, the most active one 8 but still less active than 4 by a factor of 10 to 100.
Metabolites 9, 11, and 12 show medium antiproliferative activity. Clearly, the primary human umbilical vein endothelial cells (HUVEC) and the cancer derived KB-3-1 cells are the most sensitive to the geldanamycin derivatives while the permanent mouse cell line L-929 is 5 to 15 times more resistant. Surprisingly, there is no positive correlation between biological activity and the progress of the biosynthesis. Decrease of activity with each post-PKS transformation is particularly striking for the desaturation step from metabolite 9 to 10 which results in a basically inactive geldanamycin analog. The relative antiproliferative activities of mutaproducts 8 – 12 are unexpected because not each successive post-PKS modification leads to metabolites with greater activity. Starting from carbamoylated progeldanamycin 8 continuous decrease up to derivative 10 is observed before antiproliferative activity is partly restored in fully substituted homogeldanamycin derivative 12.
In order to verify these unexpected in vivo results we decided to conduct an in vitro competition assay which utilizes recombinant, purified full length human heatshock protein Hsp90α. The assay relies on the competition of fluorescently (fluorescein isothiocyanate (FITC)) labeled geldanamycin bound to Hsp90α with new geldanamycin derivatives. The geldanamycin-FITC conjugate was first incubated at different concentrations withHsp90α (Figure 5). Half saturation of binding was achieved at 11 nM as judged from the fluorescence polarization signal for bound geldanamycin-FITC. This value compares favorably to the sensitivity observed by Kim et al. for a similar geldanamycin conjugate (2.3 nM). This group had developed such an assay for high-throughput screening in search for novel Hsp90 inhibitors.23 The dose-response activities of geldanamycin derivatives 5b and 8 – 12 were tested in the presence of Hsp90α loaded with the geldanamycin-FITC conjugate (Table 2).18 The data favorably compare with the in vivo data collected and listed in table 1 with geldanamycin derivative 10 being the least active new metabolite and indicate, that this binding/competition assay well complements our in vivo results.
Figure 5.
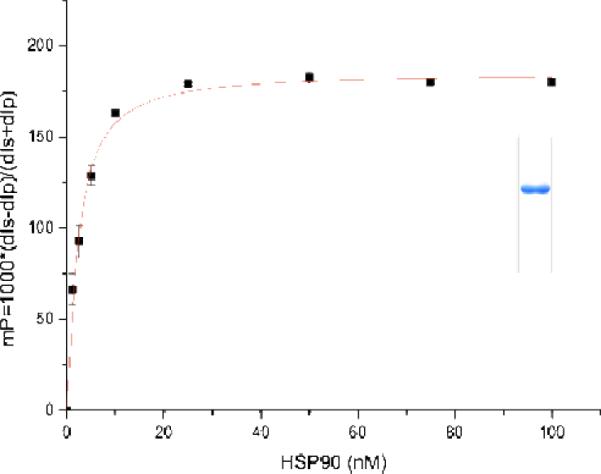
Dose-response curve for the binding of 10 nM geldanamycin-FITC conjugate to human Hsp90α. Different amounts of purified protein were incubated with geldanamycin-FITC at 4° C and fluorescence polarization was measured after 16 h. Data were imported into Origin 6.0 to determine the dissociation constant from a dose-response curve as a function of the Hsp90α concentration.22 The tracer had a Kd of 11 nM for HSP90α. The assay was performed under conditions at or above 130 mP (inset: purified full length Hsp90α).
Table 2.
Competition assay of geldanamycin-FITC conjugate (10 nM) bound to Hsp90α (50 nM) with geldanamycin derivatives 5b, 8 – 12. The IC50 values obtained from dose-response fitting curves as a function of the competitor concentration are given in the table.18
| Geldanamycin derivative | |||||||
|---|---|---|---|---|---|---|---|
| GMFICT | 5b | 8 | 9 | 10 | 11 | 12 | |
| Fluorescence polarization [nM] | 2.3±0.1 | 8±6 | 42±7 | 95±8 | 256±84 | 19±4 | 24±3 |
Table 1.
Antiproliferative activity (IC50 [ng/mL]), compared to geldanamycin 4, of new geldanamycin derivatives 8 – 12 against different cell lines: a permanent mouse line, four human cancer-derived lines, and a primary HUVEC (values shown are means of two parallel determinations).
| Cell line | ||||||
|---|---|---|---|---|---|---|
| L-929 Mouse cell line | KB-3-1 Cervix carc. | PC-3 Prostate carc. | SK-OV-3 Ovarian carc. | A-431 Epidermoid carc. | HUVEC Primary cells | |
| 4 | 5.0 | 2.7 | 21 | 4.1 | 2.0 | 2.1 |
| 8 | 470 | 22 | 130 | 60 | 30 | 38 |
| 9 | 3200 | 210 | 600 | 380 | 270 | 210 |
| 10 | >4000 | 640 | 2800 | 1900 | 1300 | 840 |
| 11 | 880 | 210 | 440 | 280 | 240 | 190 |
| 12 | 980 | 75 | 400 | 170 | 240 | 95 |
In contrast to the geldanamycin derivatives the ring enlarged 20-membered lactones 13 – 15 and 19, 21, and 23 did not show antiproliferative activity nor Hsp90 binding affinity.
Conclusions
In summary, mutasynthetic feeding studies with the AHBA-(−) mutant of the geldanamycin producer S. hygroscopicus using 3-amino-5-hydroxymethylbenzoic acid 7 provided five new geldanamycin derivatives differing in the degree of post-PKS modification. This mutasynthetic experiment allowed us to propose the sequence of post-PKS transformations by the tailoring enzymes in geldanamcin biosynthesis. The second set of mutaproducts, three 20-membered macrolactones, revealed an unexpected and unprecedented chemoselectivity of the amide synthase responsible for macrocyclization, which is not shown by the corresponding amide synthase operating in ansamitocin biosynthesis.15,16 This substrate flexibility with respect to the aryl moiety of the bound nucleophile could be extended using other m-hydroxymethylbenzoic acid starter units. Finally, new geldanamycin derivatives 8, 11 and 12 show good to moderate antiproliferative activities on different cell lines while partially post-PKS processed mutaproducts 9 and 10 have lost most of this biological property. The active mutaproducts, as the parent natural product geldanamycin 4, act on Hsp90.
To the best of our knowledge we describe here the first mutasynthetic experiment that yields macrocyclic metabolites with an altered type of ring closure and ring size.24 These results pave the way to utilize the amide synthase of S. hygroscopicus, after overexpression and mutagenetic optimization, as a macrocylization tool of chemically broader substrate flexibility.
Supplementary Material
Figure 1.
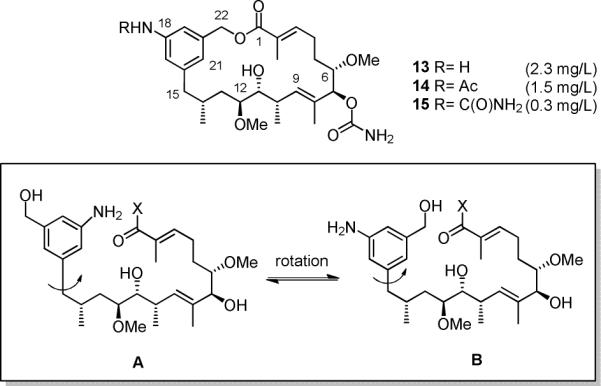
New 20-membered macrolactones 13–15 and precursors A/B responsible for macrolactamization and macrolactonization, respectively (X; The nature of the leaving group at C-1 is unknown, so that X principally could be -OH, -SCoA or –SPKS, the latter two being the more likely cases).15
ACKNOWLEDGEMENT
We thank the US National Institutes of Health (grant CA 76461), and the Deutsche Forschungsgemeinschaft (grant 13-1) and the Fonds der Chemischen Industrie. We thank Kosan Biosciences Incorporated for providing us with strain S. hygroscopicus (K390-61-1) and Wera Collisi (HZI) for performing the cell proliferation assays. We are grateful to David Agard (UCSF, USA) for a human HSP90α clone. We are grateful to Gerald Dräger for expert support in mass spectrometry analyses. We thank the referees for helpful suggestions.
Footnotes
Supporting Information. Details on the synthesis of benzoic acids and copies of 1H-, 13C-NMR and MS spectra of mutaproducts as well as details on the assay and modeling studies are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).a) Workman P. Curr. Cancer Drug Targets. 2003;3:297–300. doi: 10.2174/1568009033481868. [DOI] [PubMed] [Google Scholar]; b) Neckers L, Neckers K. Expert Opin. Emerg. Drugs. 2005;10:137–149. doi: 10.1517/14728214.10.1.137. [DOI] [PubMed] [Google Scholar]; c) Whitesell L, Lindquist SL. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]; d) Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- (2).Review: Biamonte MA, van de Water R, Arndt JW, Scannevin RH, Perret D, Lee W-C. J. Med. Chem. 2010;53:3–17. doi: 10.1021/jm9004708..
- (3).Blagosklonny MV, Toretsky J, Neckers L. Oncogene. 1995;11:933–939. [PubMed] [Google Scholar]
- (4).Janin L. J. Med. Chem. 2005;48:7503–7512. doi: 10.1021/jm050759r. [DOI] [PubMed] [Google Scholar]
- (5).a) Stead P, Latif S, Blackaby AP, Sidebottom PJ, Deakin A, Taylor NL, Life P, Spaull J, Burrell F, Jones R, Lewis J, Davidson I, Mander T. J. Antibiot. 2000;53:657–663. doi: 10.7164/antibiotics.53.657. [DOI] [PubMed] [Google Scholar]; b) Takatsu T, Ohtsuki M, Muramatsu A, Enokita R, Kurakata SI. J. Antibiot. 2000;53:1310–1312. doi: 10.7164/antibiotics.53.1310. [DOI] [PubMed] [Google Scholar]
- (6).a) Kim C-G, Yu TW, Fryhle CB, Handa S, Floss HG. J. Biol. Chem. 1998;273:6030–6040. doi: 10.1074/jbc.273.11.6030. [DOI] [PubMed] [Google Scholar]; b) Arakawa K, Müller R, Mahmud T, Yu T-W, Floss HG. J. Am. Chem. Soc. 2002;124:10644–10645. doi: 10.1021/ja0206339. [DOI] [PubMed] [Google Scholar]
- (7).a) Rascher A, Hu Z, Viswanathan N, Schirmer A, Reid R, Nierman WC, Lewis M, Hutchinson CR. FEMS Microbiol. Lett. 2003;218:223–230. doi: 10.1016/S0378-1097(02)01148-5. [DOI] [PubMed] [Google Scholar]; b) Hong Y-S, Lee D, Kim W, Jeong JK, Kim CG, Sohng JK, Lee JH, Paik SG, Lee JJ. J. Am. Chem. Soc. 2004;126:11142–11143. doi: 10.1021/ja047769m. [DOI] [PubMed] [Google Scholar]; c) Lee D, Lee K, Cai XF, Dat NT, Boovanahalli SK, Lee M, Shin JC, Kim W, Jeong JK, Lee JS, Lee CH, Lee JH, Hong Y-S, Lee JJ. ChemBioChem. 2006;7:246–248. doi: 10.1002/cbic.200500441. [DOI] [PubMed] [Google Scholar]
- (8).Eichner S, Floss HG, Sasse F, Kirschning A. ChemBioChem. 2009;10:1801–1805. doi: 10.1002/cbic.200900246. [DOI] [PubMed] [Google Scholar]
- (9).a) Kim W, Lee JS, Lee D, Cai XF, Shin JC, Lee K, Lee C-H, Ryu S, Paik S-G, Lee JJ, Hong Y-S. ChemBioChem. 2007;8:1491–1494. doi: 10.1002/cbic.200700196. [DOI] [PubMed] [Google Scholar]; b) Kim W, Lee D, Hong SS, Na Z, Shin JC, Roh SH, Wu C-Z, Choi O, Lee K, Shen Y-M, Paik S-G, Lee JJ, Hong Y-S. ChemBioChem. 2009;10:1243–1251. doi: 10.1002/cbic.200800763. [DOI] [PubMed] [Google Scholar]
- (10).a) PCT Int. Appl. 2008:53. Pub. No.: US 2008/0188450 A1. [Google Scholar]; b) Menzella HG, Tran T-T, Carney JR, Lau-Wee J, Galazzo J, Reeves CD, Carreras C, Mukadam S, Eng S, Zhong Z, Timmermans PBMWM, Murli S, Ashley GW. J. Med. Chem. 2009;52:1518–1521. doi: 10.1021/jm900012a. [DOI] [PubMed] [Google Scholar]
- (11).Shier WT, Rinehart KL, Jr., Gottlieb D. Proc. Nat. Acad. Sci. USA. 1969;63:198–204. doi: 10.1073/pnas.63.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rinehart L., Jr Jpn. J. Antibiot. 1979;32(Suppl):S32–46. [PubMed] [Google Scholar]
- (12).Reviews Weist S, Süssmuth RD. Appl. Microbiol. Biotechnol. 2005;68:141–150. doi: 10.1007/s00253-005-1891-8.. Kirschning A, Taft F, Knobloch T. Org. Biomol. Chem. 2007:3245–3259. doi: 10.1039/b709549j..
- (13).Rascher A, Hu Z, Buchanan GO, Reid R, Hutchinson CR. Appl. Environ. Microbiol. 2005;71:4862–4871. doi: 10.1128/AEM.71.8.4862-4871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).The carbamoyltransferase shows fairly broad promiscuity. Thus, it cannot be excluded that amidination takes place on any of the post-PKS intermediates and therefore it is not necessarily the first post-PKS modification.
- (15).Harmrolfs K, Brünjes M, Dräger G, Floss HG, Sasse F, Taft F, Kirschning A. ChemBioChem. 2010;11:2517–2520. doi: 10.1002/cbic.201000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Knobloch T, Harmrolfs K, Taft F, Thomaszewski B, Sasse F, Kirschning A. ChemBioChem. 2011;12:540–547. doi: 10.1002/cbic.201000608. [DOI] [PubMed] [Google Scholar]
- (17).a) Taft F, Brünjes M, Knobloch T, Floss HG, Kirschning A. J. Am. Chem. Soc. 2009;131:3812–3813. doi: 10.1021/ja8088923. [DOI] [PubMed] [Google Scholar]; b) Taft F, Brünjes M, Floss HG, Czempinski N, Grond S, Sasse F, Kirschning A. ChemBioChem. 2008;7:1057–1060. doi: 10.1002/cbic.200700742. [DOI] [PubMed] [Google Scholar]; c) Kubota T, Brünjes M, Frenzel T, Xu J, Kirschning A, Floss HG. ChemBioChem. 2006;7:1221–1225. doi: 10.1002/cbic.200500506. [DOI] [PubMed] [Google Scholar]
- (18).For details see supporting information.
- (19).Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- (20).a) Schwede T, Kopp J, Guex N, Peitsch MC. Nucl. Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guex N, Peitsch MC. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- (21).Pérez P, Domingo LR, Duque-Noreña M, Chamorro E. J. Mol. Struct.: THEOCHEM. 2009;895:86–91. [Google Scholar]
- (22).The production was too small to isolate an amount of 27 sufficient for complete NMR assignment.
- (23).Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G. J. Biomol. Screen. 2004;9:375–381. doi: 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]
- (24).Ring expanded 16-membered macrolactones derived from 6-deoxyerythronolide B were reported by Cane and Koshla when feeding a triketide to a blocked mutant of S. coelicolor. However, the type of ring closure was not altered: Kinoshita K, Williard PG, Khosla C, Cane DE. J. Am. Chem. Soc. 2001;123:2495–2502. doi: 10.1021/ja004139l..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.