
Fig. 2.
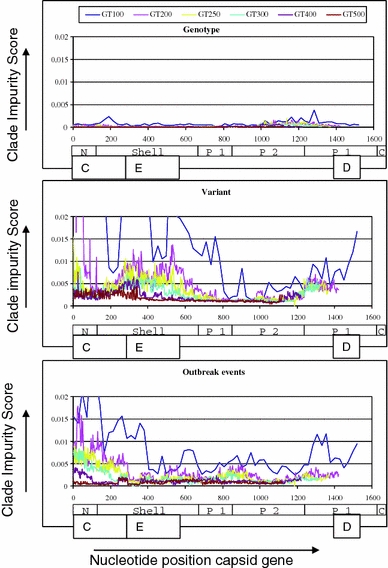
Summary of performance of phylogeny-based typing of norovirus capsid gene sequences. Clade impurity scores were calculated for each of 3,075 ML trees built in RAxML 7.0.4 [25] and presented per center position of the window along all nt positions of the full capsid gene of the reference sequence AB220921. A score of 0 is optimal and indicates that all clades of a specific level do not show invading sequences within this sub-alignment tree, for example, all genotypes are correctly positioned together while separate from others. Scores >0 indicate that some of the minimal differentiating clades within levels in the sub-alignment tree contain invading sequences. Scores were calculated for six fragment lengths, which are indicated as window-100 to window-500, and with each fragment length represented by a different color, and calculated separately for genotypes (upper panel), variants (mid panel), and outbreak events (lower panel). Scores for the full capsid alignment were 0 for genotypes, 0.000759 for variants, and 0 for outbreak events. The different domains in ORF2 are depicted: the N-terminal domain, the Shell domain, the Protruding domain split up into P1 and P2. The norovirus particle is built from 180 copies of the capsid protein (90 dimers)