

Abstract
Therapeutic use and function of recombinant molecules can be studied by the expression of foreign genes in mice. In this study, we have expressed human Fcgamma receptor –Ig fusion molecules (FcγR-Igs) in mice by administering FcγR-Ig plasmid DNAs hydrodynamically and compared their effectiveness to purified molecules in blocking immune-complex (IC) mediated inflammation in mice. The concentration of hydrodynamically expressed FcγR-Igs (CD16AF-Ig, CD32AR-Ig and CD32AH-Ig) reached a maximum of 130 μg/ml of blood within 24 h after plasmid DNA administration. The in vivo half-life of FcγR-Igs was found to be 9-16 days and Western blot analysis showed that the FcγR-Igs were expressed as a homodimer. The hydrodynamically expressed FcγR-Igs blocked 50-80% of IC-mediated inflammation up to 3 days in a reverse passive Arthus reaction model. Comparative analysis with purified molecules showed that hydrodynamically expressed FcγR-Igs are more efficient than purified molecules in blocking IC-mediated inflammation and had a higher half-life. In summary, these results suggest that the administration of a plasmid vector with a FcγR-Ig gene can be used to study the consequences of blocking IC-binding to FcγRs during the development of inflammatory diseases. This approach may have potential therapeutic value in treating IC-mediated inflammatory autoimmune diseases such as lupus, arthritis and autoimmune vasculitis.
Keywords: Hydrodynamic delivery, Plasmid DNA, Fcgamma receptors, Inflammation
INTRODUCTION
The in vivo expression of a foreign gene in animal models is of great interest because it not only provides an opportunity to study the structure/function of a protein, but also has therapeutic efficacy in treating various pathological disorders. In vivo transgene expression requires efficient delivery of specific genes into the cells. Currently, viral and non-viral vectors are the two predominant gene delivery systems being used. Recently, it has been shown that a significant amount of protein can be expressed in vivo by rapidly injecting plasmid DNA in a large volume through the tail vein by a process called hydrodynamic-based gene delivery 1-4. Though the mechanism of this hydrodynamic-based in vivo gene expression is not clearly understood, it has been suggested that the rapid injection of a large volume of plasmid DNA solution causes shearing forces on the hepatocytes. These forces induce transient pore formation in the plasma membrane facilitating the direct entry of the plasmid DNA into the hepatocytes’ cytosol resulting in a high level of transient in vivo gene expression 1,2,5-7. Since the discovery of this hydrodynamic gene delivery technique in the 1990s, it has been reported as an effective method of gene delivery in experimental animal models. The introduction of exogenous genes by the hydrodynamic method using plasmid DNA has many advantages such as ease of preparation of large quantities of DNA in a short period of time and stability. Recently, several studies have shown the hydrodynamic-based in vivo expression of several protein molecules (CTLA4-Ig, IL22-Ig, IL10-Ig, CD40-Ig, fetal liver kinase-1, DNA cancer vaccine, hFlex-TRAIL) and their role in various disease conditions such as experimental autoimmune myocarditis, allergic encephalomyelitis, systemic lupus erythematosus, collagen-induced arthritis, nephritis, and cancer 8-17.
In this report, we have investigated whether hydrodynamic-based delivery of FcγR genes results in a functional product that can block immune-complex (IC)-mediated inflammation. The receptors (FcγR) for the Fc domain of IgG molecules play a vital role in IC-mediated autoimmune diseases. Inflammatory cells, such as neutrophils, monocytes, and NK cells, express three types of FcγR 18-22. FcγRI (CD64) is a high affinity receptor for monomeric IgG whereas FcγRII (CD32) and FcγRIII (CD16) are low affinity receptors for monomeric IgG; however, all three bind stably to ICs. Both in vivo and in vitro studies from various laboratories have shown that interaction of FcγRs expressed on inflammatory cells with antibody-coated target cells/tissues is a key event in the destruction of antibody coated tissues through antibody dependent cellular cytotoxicity (ADCC) and phagocytosis23-31, which leads to the development of various autoimmune diseases 28,32-37. During the development of autoimmune diseases such as arthritis, systemic lupus erythematosus and autoimmune vasculitis, autoantibodies bind to the antigen expressed on cells and form ICs. These ICs bind to inflammatory cells through FcγRs leading to chronic inflammation and destruction of the target cells. Therefore, blocking the interaction of pathogenic ICs with the cell surface FcγRs expressed on inflammatory cells using recombinant FcγR-Igs could be a potential therapeutic approach. We have previously demonstrated that the administration of a purified dimeric form of a low affinity FcγR (CD16A-Ig) can be successfully employed to treat IC-mediated acute inflammation in mice 38. .In this report, we have hydrodynamically expressed human low affinity FcγR-Igs in vivo and studied their effectiveness in blocking IC-mediated inflammation in a murine model. We show that in vivo expressed recombinant FcγR-Ig molecules are secreted in high concentrations and are sustained for longer periods of time in circulation compared to the administered purified FcγR-Ig. These molecules are also as effective as purified FcγR-Igs in blocking the interaction of ICs with inflammatory cells, thus preventing inflammation in vivo for a relatively longer period of time.
RESULTS
Purified human FcγR-Igs block IC-binding to mouse FcγRs in vitro and antibody-mediated inflammation in vivo in mice
Previously, we have shown that the purified human CD16A-Ig molecule, when administered in mice, is capable of blocking IC-induced inflammation in a reverse passive Arthus (RPA) model 38. In this study we have made additional human FcγR-Ig molecules. Prior to testing the anti-inflammatory effect of human FcγR-Ig molecules in mice, we assessed the ability of purified human FcγR-Ig molecules (CD16AV-Ig, CD16AF-Ig, CD32AR-Ig and CD32AH-Ig) to compete and cross-block IC-binding to mouse FcγRs in vitro. We carried out a soluble IC (sIC)-binding assay using the mouse macrophage cell line (P388D1) as described earlier 38. P388D1 cells express all four types of mouse FcγRs: CD16A, CD32B, CD64 and FcγRIV. As shown in Figure 1a, all of the hFcγR-Ig molecules, except hCD32AR-Ig, inhibited sIC binding to mouse FcγRs expressed on P388D1 cells in a dose dependent fashion. More than 70% inhibition was achieved when P388D1 cells were co-incubated with 25 μg/ml of hCD16AF-Ig, hCD16AV-Ig and hCD32AH-Ig, whereas, hCD32AR-Ig was not able to compete with mouse FcγRs at the same concentration. The results with purified hCD16AF-Ig, and hCD32AR-Ig are consistent with our previous studies 38.
Figure 1. Purified human FcγR-Igs compete with cell surface mouse FcγRs and block IC binding both in vitro and in vivo.
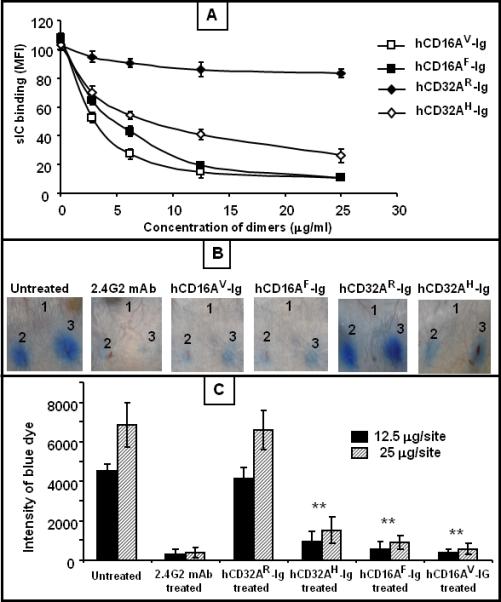
(A) Human FcγR-Igs blocked the binding of sICs to the mouse macrophage cell line P388D1 in a dose dependent manner. P388D1 cells were incubated with FITC-IC (sIC) in the presence and absence of various concentrations of hFcγR-Ig dimers, and the cells were analyzed for binding of FITC-IC using flow cytometry. (B) Systemic administration of purified hFcγR-Ig dimers efficiently blocked the RPA reaction in vivo in mice. Groups of mice (n=3/group) were injected with hFcγR-Ig intravenously (100 μg/mouse). After 1 h, the mice were injected intradermally with PBS (site 1) or anti-Ova (12.5 μg in site 2 and 25 μg in site 3). RPA was initiated by injecting Ova with 1% Evan's blue intravenously through the tail vein. The anti-FcγR antibody (2.4G2) treated control mice were injected with 25 μg/ml of blood (40 μg/mice) mAb. The PBS injected mice served as the untreated positive control. After 3 h the mice were euthanized, and the dorsal side of the skin was photographed for analysis. The figure shows three representative mice. (C) Quantitative analysis of RPA. The dermal lesion, seen blue in the photographs, was quantified using ImageJ and KaleidaGraph softwares for groups with or without FcγR-Ig dimer treatment. Data are presented as the mean ± SD from three experiments. *p<0.01, **p<0.001.
The in vivo efficacy of purified human FcγR-Ig molecules was analyzed using RPA an acute antibody-mediated inflammation in a murine model 38. In this model, inflammation is initiated by the formation of antigen-antibody complexes at the antibody-injected site. To induce inflammation, rabbit anti-chicken ovalbumin (anti-Ova) IgG was injected intradermally on the dorsal side of the mouse skin and chicken ovalbumin (Ova) along with 1% Evan's blue (blue dye) was injected intravenously. Extravasation of inflammatory cells and vascular permeability during the inflammation at the antibody-injected site can be visualized upon leakage of the blue dye 38,39. Before the induction of RPA, hFcγR-Ig molecules (50 μg/ml of mouse blood) were administered intravenously into a separate group of mice (n=3) to study the effect of purified hFcγR-Ig molecules. As shown in Figure 1b, hCD16AF-Ig, hCD16AV-Ig and hCD32AH-Ig inhibited more than 70% inflammation as measured by the intensity of the Evan's blue extravasation (Fig. 1c); whereas under similar conditions, hCD32AR-Ig did not inhibit inflammation (Fig. 1b). The lack of inhibition by purified hCD32AR-Ig may be due to the difference in affinity of human FcγR-Igs towards rabbit IgG binding or because hCD32AR-Ig might have partially denatured during the purification process. As specificity controls, mice treated with 2.4G2 antibody, a mAb to mouse FcγRs (CD16/32), did not cause RPA (Fig. 1b).
In vivo expression of human FcγR-Ig molecules in mice by the hydrodynamic-based method has a longer half-life
Next, we determined the level and kinetics of expression of human FcγR-Ig molecules in vivo. As shown in Figure 2, all of the hydrodynamically expressed hFcγR-Ig molecules in mice reached maximum expression by 24h. The concentration of hFcγR-Ig in circulation was found to be around 130 μg/ml of blood. The in vivo half-life was 9 days for hCD16AF-Ig; whereas for the hCD32A-Ig molecules, the half-life was estimated to be 16 days. hCD16AF-Ig was detected up to 17 days in circulation, while, hCD32A-Ig molecules were detected until day 24. We reported previously that the in vivo half-life of the hFcγR-Ig molecules is 5 days when administered as purified molecules 38. These results suggest that the in vivo expression of a foreign gene in mice by the hydrodynamic-based method increases the in vivo half-life of the foreign protein when compared to administration of purified proteins. At present the reason for increased half-life of hydrodynamically expressed FcγR-Igs is not clear. However it is possible that the purification process may have made the molecules more susceptible to in vivo degradation.
Figure 2. Hydrodynamic based expression of human FcγR-Ig molecules in mice.
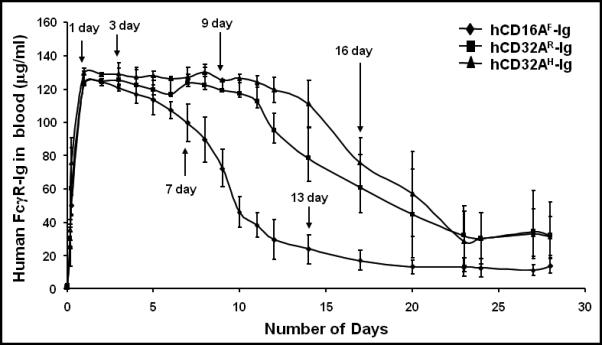
Human FcγR-Ig genes in plasmid DNA (10 μg) were diluted in 1.6 ml of sterile saline and injected within 5 seconds into each group of mice through the tail vein. Subsequently, 5 μl blood samples were collected at different time points and diluted in PBS/EDTA. Plasma was separated to detect FcγR-Ig dimers using ELISA as described under Materials and Methods. The plasma from a group of normal mice injected with PBS served as a specificity control. Purified hCD16A-Ig and hCD32A-Ig were used as positive controls, while BSA coated wells served as negative controls. Purified hCD32A-Ig was used as a standard to quantify the level of dimers in the blood. Experiments are representative of three individual experiments. Data are mean ± SD of triplicates.
Hydrodynamically expressed human FcγR-Ig molecules block antibody-mediated inflammation in mice
We then determined whether hydrodynamically expressed hFcγR-Ig can block antibody-mediated inflammation in a murine RPA model. In these experiments, a group of mice (n=12 for each molecule) were injected with plasmid DNA of hFcγR-Ig as described under Materials and Methods. DNA was injected on day 0 and at different time intervals, and a group of mice (n=3) were used to conduct the RPA for each molecule. We have chosen different time points for each molecule to conduct the RPA since the hydrodynamically expressed hCD16A-Ig and hCD32A-Ig molecules differ in their in vivo half-life. For hCD16AF-Ig, the RPA was carried out on days 1, 3, 7 and 13. As shown in Figure 3a, in vivo expressed hCD16AF-Ig was able to significantly (P=0.001) reduce antibody-mediated inflammation up to 3 days. As measured by the intensity of the Evan's blue extravasation, hCD16AF-Ig was able to block inflammation by 83% and 77% on days 1 and 3, respectively, while on day 7 it was only able to block 20% (Fig. 3a; left panel). These results suggest that in vivo expressed hCD16AF-Ig can block antibody-mediated inflammation up to 3 days and becomes ineffective by day 7 (Fig. 3a; right panel bar graph). For hCD32AR-Ig and hCD32AH-Ig DNA injected animals, the RPA was conducted on days 1, 3, 9 and 16. Interestingly, as shown in Figure 3b, the in vivo expressed hCD32AR-Ig was able to block antibody mediated inflammation completely on day 1, but the blocking efficiency as measured by the intensity of the blue dye began to decline by day 3 and was completely ineffective by day 9 (Fig. 3b; right panel). Most notably, hydrodynamically expressed hCD32AR-Ig blocks IC-mediated inflammation in mice, while the purified molecule cannot (compare Fig. 1 and Fig. 3). The in vivo expressed hCD32AH-Ig was able to block antibody-mediated inflammation completely up to 9 days (Fig. 3c). About 50% of RPA was blocked by hCD32AH-Ig even on day 9 and became ineffective on day 16 as indicated by the decrease in the intensity of the blue dye (Fig. 3c; right panel). As a specificity control, mice were treated with 2.4G2 mAb, which completely blocked RPA on day 3. The untreated group of mice (n=3) injected with rabbit anti-Ova antibody alone served as the positive control. These results suggest that hydrodynamically expressed hFcγR-Ig molecules are capable of competing with mFcγRs and can effectively block IC binding.
Figure 3. Hydrodynamically expressed human FcγR-Ig molecules block antibody mediated inflammation in mice.
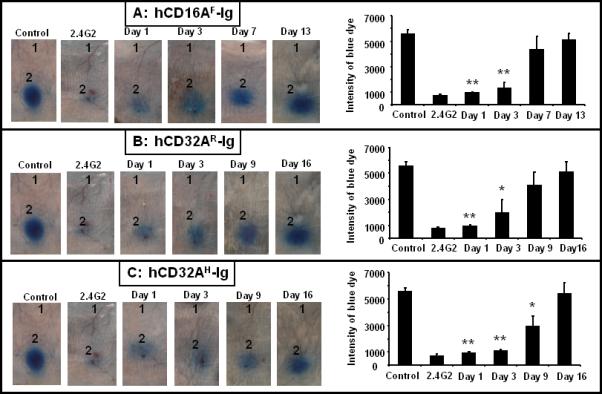
Mice (n=12) were injected with 10 μg of plasmid vector containing human FcγR-Ig cDNA (Panel A: hCD16AF-Ig, Panel B; hCD32AR-Ig, Panel C; hCD32AH-Ig) intravenously as described under Material and Methods. RPA was carried out using three mice at each time point. Mice were injected intradermally with PBS (site 1) and 25 μg (site 2) of anti-Ova per site. RPA was initiated by injecting Ova with 1% Evan's blue intravenously through the tail vein. The antibody (2.4G2) treated control mice were injected with 25 μg/ml of blood (40 μg/mouse) mAb. The PBS injected mice served as the untreated positive control. After 3 h the mice were euthanized, and the dorsal side of the skin was photographed for analysis. The figure is representative of three individual mice. Bar graph on the right side represents the quantitative analysis of RPA. The dermal lesion, seen blue in the photographs, was quantified using ImageJ and KaleidaGraph softwares for groups with or without FcγR dimer treatment. Data are presented as the mean ± SD from three experiments. *p<0.01, **p<0.001.
Functionality of hydrodynamically expressed hFcγR-Ig molecules
To determine whether the level of expression correlates with the ability to block RPA, plasma was collected from the mice used for RPA experiments before induction of RPA, and the concentration of human FcγR-Igs in the blood of the mice was determined using a sandwich ELISA. As shown in Figure 4 (upper panel), hCD16AF-Ig was expressed at about 120 μg/ml of blood up to day 3. About 50% of the hCD16AF-Ig was cleared from circulation by day 7 and was not detectable in circulation on day 13. hCD32AR-Ig and hCD32AH-Ig were expressed at 160 and 120 μg/ml of blood, respectively, within 24 h of hydrodynamic injection, (Fig. 4, middle and lower panel). The same concentration was maintained until day 9, and about 50% was cleared from circulation by day 16. Although the concentration of FcγR-Ig molecules present within the mice should be sufficient to block RPA, CD16AF-Ig and CD32AR-Ig were not able to block RPA after three and nine days of expression, respectively. This suggests that the level expression of hFcγR-Ig does not correlate with the ability to block RPA when the molecules are in circulation more than 3 days. Therefore, we determined whether the hFcγR-Ig molecules present in circulation are capable of binding IgG in in vitro assays.
Figure 4. Expression of human FcγR-Ig molecules in mice used to conduct RPA.
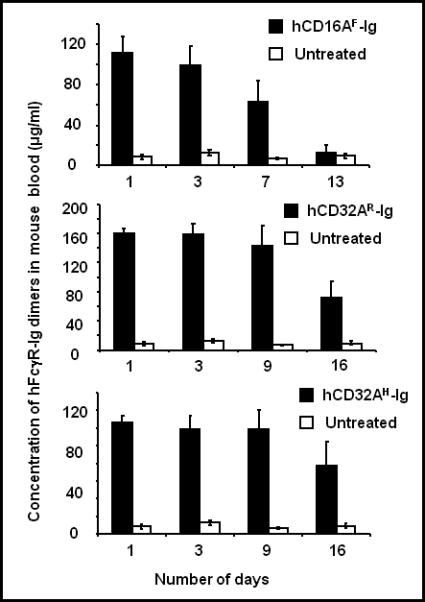
Before the initiation of RPA (presented in Fig. 3), blood was collected (5 μl) and diluted in 500 μl of PBS/EDTA and plasma was used to detect the expression of human FcγR-Ig molecules by sandwich ELISA as described under the figure 3 legend. Experiments are an average of three individual mice.
ELISA plates were coated with 10 μg/ml of rabbit IgG, and then 100 μl of 1:100 diluted plasma was added to the wells. The plates were washed, and the bound hFcγR-Ig was detected using HRP-conjugated anti-human Fc specific antibody. The mAb coated wells (CLB-Fcgran-1 for to human CD16A and IV.3 for CD32A) and the plasma from naïve mice served as specificity controls. As shown in Figure 5 (upper panel), in vivo expressed hCD16AF-Ig bound to rabbit IgG efficiently on day 1 of its expression. On day 3, its binding efficiency to rabbit IgG was reduced by 50% and by day 7 it was no longer able to bind to rabbit IgG. The in vivo expressed hCD32AR-Ig (Fig. 5: middle panel) bound to rabbit IgG poorly on day 1 of expression, and its binding efficiency was reduced by 50% on day 3. On day 9 it was not able to bind to rabbit IgG. The in vivo expressed hCD32AH-Ig (Fig. 5: lower panel) bound efficiently to rabbit IgG on day 1 and retained up to 50% of its binding efficiency until day 16. Correlation of the level of expression (Fig. 4) and functional activity (Fig. 5 and Fig. 3) shows that hCD32AH-Ig and hCD16AF-Ig bind more efficiently to rabbit IgG when compared to hCD32AR-Ig. These data suggest that dimeric FcγR-Ig molecules may become inactive or degraded partially after being present in circulation for several days, and the lack of functionality may vary from molecule to molecule. At present, the reason for the ineffectiveness of CD16AF-Ig and CD32AR-Ig after being in circulation for a few days is not clear.
Figure 5. Binding of hydrodynamically expressed human FcγR-Igs to rabbit IgG.
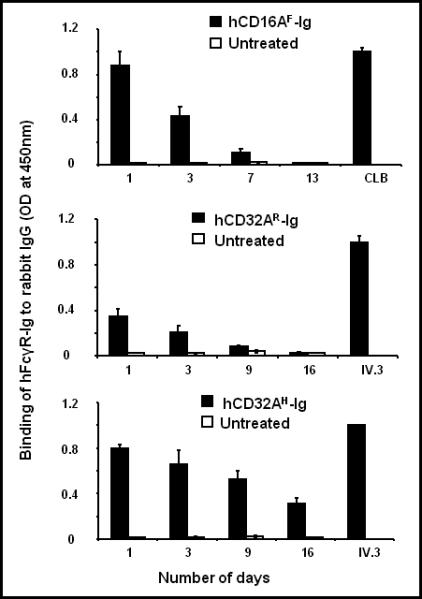
Plasma collected from the mice used to conduct the RPA in figure 3 was used to determine whether the in vivo expressed hFcγR-Igs bind to rabbit IgG using a sandwich ELISA. Rabbit IgG (100 μl of 10 μg/ml) was coated on ELISA plates overnight, and the binding of hFcγR-Ig molecules to rabbit IgG was determined as described under Materials and Methods. The hFcγR-Ig alleles that bound to BSA-coated wells were taken as non-specific binding. Experiments are an average of three individual mice. Data are mean ± SD of triplicates.
Hydrodynamically expressed hFcγR-Ig molecules are secreted as a homodimer
To determine the molecular nature and kinetics of degradation of circulating hFcγR-Ig, we analyzed the plasma by Western blot. Previously we have reported that recombinant hFcγR-Igs secreted by CHO-K1 cell transfectants are disulfide-linked homodimers 38,40,41. As shown in Figure 6a, on day 1, hCD16AF-Ig is secreted as a disulfide-linked homodimer as evidenced by the presence of 120 kDa band under non-reducing conditions and the 70 kDa band under reducing conditions. However, we also observed a band around 200 kDa in CD16A-Ig blots, while on day 3 this band had mostly disappeared. The reason for the secretion of this high molecular weight band is not clear; however it may be due to the aggregation of hCD16AF-Ig. We did not observe this high molecular weight band when we purified the hCD16AF-Ig molecule in vitro 40. On day 3, the molecules began to degrade, as suggested by the appearance of low molecular weight bands, and was completely degraded by day 13. These results are consistent with the detectable rabbit IgG binding activity (Fig. 5). The hCD32AR-Ig (Fig. 6b) and hCD32AH-Ig (Fig. 6c) molecules are also secreted as disulfide linked homodimers as seen by the 110 kDa band under non-reducing conditions and a 60 kDa band under reducing conditions. The intensity of the bands under reducing conditions was lower which may be due to the decreased reactivity of the antibodies with denatured molecules. Interestingly, hCD32A-Ig molecules are more stable in vivo than hCD16A-Ig (compare Fig. 6a, b and c) as suggested by its dimeric nature up to 16 days. In addition to the major bands, we observed minor low molecular weight bands, which may be due to different glycosylation or degradation products of the hCD32A-Ig molecules. These results suggest that the progressive loss of ability to block inflammation in RPA with time by the in vivo expressed FcγR-Ig molecules correlates with the in vitro rabbit IgG binding activity and is partly due to the degradation of the molecules in circulation.
Figure 6. Western blot analysis of hydrodynamically expressed human FcγR-Igs in mice.
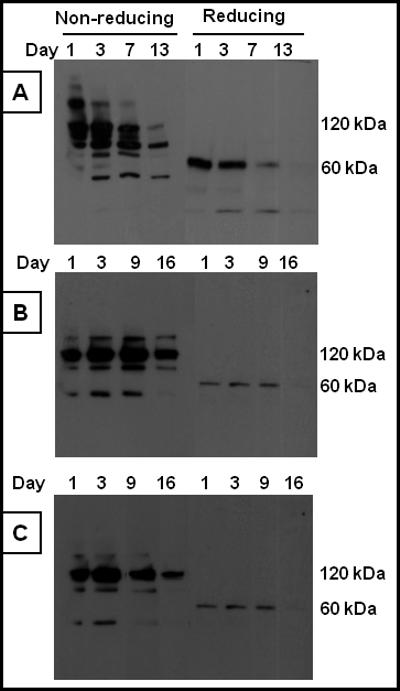
To determine the dimeric nature of hydrodynamically expressed hFcγR-Igs, the plasma samples collected from the mice, used to conduct the RPA in figure 3, were subjected to Western blot analysis. 20 μl of plasma was mixed with SDS-PAGE sample buffer under both reducing and nonreducing conditions. After electroblotting the protein onto the PVDF membrane, the proteins were detected using the HRP-conjugated goat anti-human Fc specific antibody. (a) hCD16AF-Ig, (b) hCD32AR-Ig, (c) hCD32AH-Ig.
Hepatocytes and antigen presenting cells (APCs) such as macrophages in the spleen are responsible for the hydrodynamic expression of hFcγR-Ig
To investigate the specific organ and cell type involved in the high expression of hFcγR-Ig molecules, a group of mice (n=3) were hydrodynamically injected with hCD32AH-Ig plasmid DNA. Various organs were then harvested at different time points post DNA injection and subjected to immunohistochemistry. The expression of hCD32AH-Ig was detected using the F(ab)’2 fragment of goat anti-human Fc specific antibody conjugated with Cy-5. Consistent with previous reports 1,42, our results show that hepatocytes are the major cell type responsible for the expression of hFcγR-Igs and maintained expression for up to 48h after plasmid DNA administration (Fig 7A). Interestingly, APCs such as macrophages in the red pulp region of the spleen also expressed the hCD32AH-Ig molecules, but not the B- or T-cell zones in the white pulp region (Fig 7B). Maximum expression was reached after 24h and declined thereafter (Fig 7B). In contrast, expression of hCD32AH-Ig was not found in other organs tested such as kidney, lung or heart (figure not shown). These results suggest that in addition to hepatocytes in the liver, APCs such as macrophages present in the red pulp area of the spleen are also responsible for the expression of hFcγR-Ig molecules following the hydrodynamic delivery of plasmid DNA.
Figure 7. Expression of hFcγR-Ig in liver and spleen cells after hydrodynamic administration of plasmid DNAs.
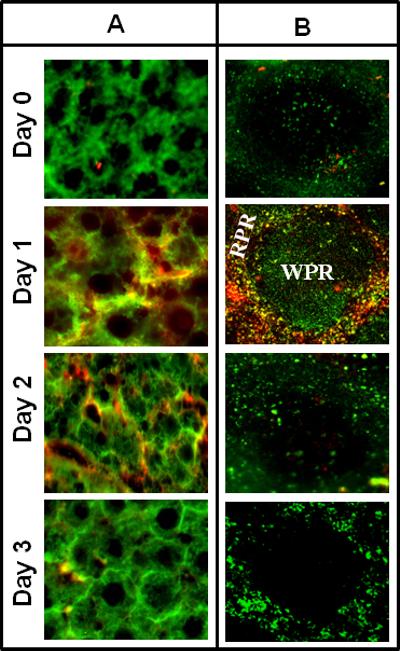
To determine the specific organ and cell type involved in the expression of hFcγR-Ig molecules, a group of mice (n=3) were hydrodynamically injected with hCD32AH-Ig plasmid DNA (10 μg) and then various organs at different time points were collected and immunohistochemistry was carried out as described under Materials and Methods. A: Liver sections were stained with a liver mitochondrial marker specific rabbit antibody to detect hepatocytes followed by FITC conjugated goat anti-rabbit as secondary antibody (green). Liver sections are also counterstained with Cy-5 conjugated goat anti-human Fc specific antibody to detect the presence of hCD32AH-Ig expression (red). The merger of green and red color (yellow) represents the expression of hCD32AH-Ig by hepatocytes. Photographs were taken at 40X magnification. B: Spleen sections were stained with FITC conjugated macrophage specific rat antibody to detect macrophages in the spleen (green) and counter stained with Cy-5 conjugated goat anti-human Fc specific antibody to detect the presence of hCD32AH-Ig expression (red). The yellow color, which is a result of merger of green and red color indicates, the expression of hCD32AH-Ig by macrophages. Photographs were taken at 20X magnification. WRP represents white pulp region where T and B-cells are present. RPR represents red pulp region around WRP where the hCD32AH-Ig molecules were expressed by macrophages.
DISCUSSION
In the recent past, the significance of low affinity FcγRs in autoimmune/inflammatory disease conditions has been well-documented 43-47. The interaction of ICs with FcγRs on inflammatory cells is a key event in the progression of IC-mediated diseases. Previously, we have shown that inflammation caused by the interaction of ICs with inflammatory cells can be blocked by administering purified FcγR-Ig molecules 38,48,49. In this report, we demonstrate that the hydrodynamic administration of plasmid DNAs encoding human low-affinity FcγR-Igs is capable of blocking IC-mediated inflammation in a murine RPA model. The hFcγR-Ig molecules were secreted in high concentrations in circulation and sustained for longer periods of time after hydrodynamic administration when compared to the administered purified molecules. Recently, we have reported that the purified hCD32AR-Ig molecules, despite being active in vitro 41, failed to block inflammation induced by RPA in mice 38. The reason for the ineffectiveness of the purified hCD32AR-Ig molecule in vivo is not clear. However, it is possible that the purified molecules may not be able to compete with mouse FcγRs for IC binding 38 or may have partially denatured during the purification process, thus leading to its ineffectiveness. Interestingly, in this report we observed that hydrodynamically expressed hCD32AR-Ig molecules are effective in blocking antibody-induced inflammation in mice, suggesting that the hCD32AR-Ig molecules may have been partially denatured during the purification process. These results also suggest that in vivo expressed molecules are more active when compared to purified molecules, indicating that the in vivo expression of a transgene by the hydrodynamic-based method is a more desirable method to study the function of a particular gene product in vivo. It has been shown that after hydrodynamic injection of plasmid DNA, a high expression of recombinant luciferase was found in the liver whereas a very low level of expression was observed in the spleen, kidney, lung and heart 1. In agreement with this, we observed that the hepatocytes are the major cell types responsible for the high level of expression of recombinant molecules. Interestingly, we also found that APCs present in the red pulp region of the spleen are also responsible for the expression of recombinant molecules and not T or B-cells present in the white pulp region (fig 7). In contrast, we did not observe the expression of recombinant molecules in the other organs tested such as kidney, lung and heart (data not shown). Taken together, these results suggest that the hepatocytes in the liver and APCs in the spleen are responsible for the expression of recombinant molecules when plasmid DNA is hydrodynamically injected in mice.
Recently, hydrodynamic-based in vivo expression of several Ig fusion protein molecules such as interleukins and cell adhesion molecules (CTLA4-Ig, IL22-Ig, IL10-Ig, Flex/TRIAl-Ig) were studied, and their role in various disease conditions was established 8-10,15-17. For instance, hydrodynamically expressed CTLA4-Ig dramatically reduced experimental autoimmune myocarditis by blocking the costimulatory signal from the T cells. The in vivo expression of CTLA4-Ig was approximately 2 μg/ml of blood and was detectable up to 16 days 10. In this report, for the first time, we have expressed hFcγR-Ig molecules by injecting naked plasmid DNA in mice and showed that hydrodynamically expressed hFcγR-Ig molecules have a longer half life compared to the purified molecules, remain as dimers in circulation, and are effective in blocking IC-mediated inflammation in a mouse model for an extended period of time. It has been shown that the in vivo half-life of purified monomeric hCD32A was only 30 min in mice 25. Whereas, recently we have shown that the in vivo half-life of purified, dimeric forms of Fc fusion hCD16AF-Ig and hCD32AR-Ig was 120h when administered intravenously to mice 38. In this report we observed that the hydrodynamically expressed FcγR-Ig molecules exhibited a longer half-life when compared to purified FcγR-Ig molecules. The reason for this prolonged half-life is not clear, however, this may be due to continuous production of the FcγR-Ig molecules from hepatocytes and macrophages present in the spleen, which took up the DNA during hydrodynamic injection. Moreover it has been shown that the in vivo half-life of Fc fusion proteins is due to the presence of the Fc domain. The Fc domain binds to neonatal Fc receptors (FcRn) and protects the IgG from degradation by the endocytic machinery of cells 50. The Fc domain used in the present investigation is mutated to abolish the FcγR binding activity 51-53. Since both FcγRs and FcRn bind to different sites of the Fc domain 54,55, the mutation that we introduced in the Fc domain of the FcγR-Ig molecules will not affect the FcRn binding activity, therefore potentially aiding in extending the in vivo half-life of hFcγR-Ig molecules. It has been shown that hydrodynamically injected plasmid DNA can stay in the hepatocytes for upto 3 days after administration 15,42. Alternatively, hydrodynamically expressed FcγR-Igs may be in a less denatured state in circulation due to lack of exposure to harsh acidic conditions that are used for affinity purification of FcγR-Igs.
In conclusion, we have demonstrated that the hydrodynamic delivery of plasmid DNA encoding hFcγR-Igs inhibited antibody mediated inflammation in mice. The hFcγR-Ig molecules are expressed at high levels, intact and dimeric in nature, and sustained in circulation for an extended period of time. The data presented herein will be useful in studying the consequences of uncoupling IC binding to FcγRs expressed on inflammatory cells during IC-mediated inflammatory/autoimmune diseases such as arthritis, systemic lupus erythematosus, and autoimmune vasculitis. This approach also obviates the laborious purification process of recombinant molecules to delineate the specific role of a particular gene of interest in vivo within a short period of time and suggests that FcγR-Ig molecules can be expressed in vivo. Further, these hydrodynamically expressed molecules are useful in evaluating the roles and therapeutic use of FcγRs during the course of various inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Reagents
HRP-conjugated anti-human Fc antibody and ovalbumin (OVA) were purchased from Sigma (St. Louis, MO). The Ovalbumin-FITC, rabbit anti-Ovalbumin IgG and rabbit IgG were from Roche Molecular Biochemicals (Indianapolis, IN). HRP-substrate and SDS-PAGE gels were from BioRad (Hercules, CA). DNA preparation kits were from QIAGEN (Valencia, CA). DH5α bacteria, CHO serum free media and lipofectamin were from Invitrogen (Carlsbad, CA). The Micro BCA-protein assay kit was from Pierce (Rockford, IL). Quick-change II site-directed mutagenesis kit was from Stratagene (La Jolla, CA). Cell culture reagents were from Life Technologies (Gaithersburg, MD). Site directed mutagenesis kit was from Stratagene (La Jolla, CA). 2.4G2 (anti- mouse mAb CD16/32) mAb was from BD biosciences (San Jose, CA). Anti-human CD16 and CD32A mAb were referenced previously 40,41. Human Fc specific Cy5 conjugated F(ab)’2 fragment of goat antibody was from Jackson Immunoresearch Laboratories (West groove, PA). Mouse macrophage specific FITC conjugated rat monoclonal (clone#BM8) antibody F4/80, rabbit polyclonal mouse liver mitochondrial marker antibody (CSP1) and FITC conjugated, affinity purified goat polyclonal antibody against rabbit IgG were purchased from Abcam (San Francisco, CA). C57BL/6 (8-10 weeks old) female mice were from Jackson laboratory (Bar Harbor, Maine). The animal experiments were conducted according to the Emory University IACUC protocol.
Construction of human FcγR-Ig plasmids
The construction of the dimeric form of hCD32AR-Ig and hCD16AF-Ig was carried out by ligating the extracellular domain of hCD32AR or CD16AF to the mutated Fc domain of the human IgG1 heavy chain as described earlier 38,40,41. The mutations in the Fc domain were shown to abolish the binding of FcγRs 51-53. The hCD32AH-Ig and hCD16AV-Ig alleles were constructed by Quick-change II site directed mutagenesis kit using hCD32AR-Ig and hCD16AF-Ig DNA as a template. cDNA encoding the FcγR-Ig were then subcloned into the pcDNA3.1 expression vector with neomycin selection marker 40,41. The plasmid DNAs of hFcγR-Ig were transfected into CHO-K1 cells and culture supernatant was collected and used to purify the FcγR-Ig molecules using a Protein-G column as previously described 40,41.
Soluble IC binding assay
Soluble immune complex (sIC) was prepared as described 38,41. Briefly FITC-Ova was mixed with rabbit anti-Ova IgG (1:1 molar ratio) and incubated for 4 h at 4°C. The complex was centrifuged using a microcentrifuge at 15,000 rpm for 30 min at 4°C and the supernatant was used for the FITC-IC binding assay. The P388D1 mouse macrophage cells (50 μl of 5×106) were preincubated with recombinant receptors or mAbs for 30 min at 4°C and then incubated with FITC-IC (20 μg/ml) in binding buffer (PBS/EDTA with 1% BSA, pH 7.4) for 1 h at 4°C. The cells were then washed and analyzed by flow cytometry.
Hydrodynamic-based injection of FcγR-Ig in mice
Hydrodynamic-based injection of plasmid DNA was carried out as described 1,56. Briefly, 10 μg of pcDNA3.1 plasmid vector containing human FcγR-Ig cDNAs (hCD16AF-Ig, hCD16AV-Ig, hCD32AR-Ig or hCD32AH-Ig) was diluted in 1.6 ml of sterile saline (0.9% NaCl) and injected into each group of mice (n=5 per group) through the tail vein within 5-7 sec, using a 27.5-gauge needle. Subsequently, 5 μl blood samples were collected at several time points and diluted to 500 μl in PBS/5 mM EDTA, and the plasma was separated by centrifugation. The plasma samples were frozen immediately at -80° C for future analysis.
Estimation of human FcγR-Ig levels in mice blood
Plasma collected at different time points was used to detect FcγR-Ig dimers by sandwich ELISA. ELISA plates were coated with 100 μl of 10 μg/ml anti-hCD16A (CLBFcgran-1) and anti-hCD32A (IV.3) mAbs overnight at 4°C. The wells were then blocked with PBS/5 mM EDTA /1% BSA. After washing, 100 μl of the plasma samples were added into the wells and incubated for 1 h. The wells were washed and then incubated for another 1 h after adding 100 μl of HRP-conjugated goat anti-human IgG Fc specific antibody. HRP substrate was added to the washed wells and read at 450 nm. The plasma from mice injected with PBS served as a specificity control. To quantify the level of FcγR-Ig dimers in the blood, purified hCD16AF-Ig and hCD32AR-Ig were used as standards, while BSA coated wells served as negative controls.
Reverse passive Arthus reaction (RPA)
RPA was carried out as described 38. Briefly, the mice (n=12) were hydrodynamically injected with 10 μg of human FcγR-Ig plasmids in 1.6 ml of saline. Then RPA was carried out at different time points. For each time point, a group of mice (n=3) was used. Before the initiation of RPA, blood was collected (5 μl), diluted to 500 μl with PBS/EDTA, and centrifuged to collect plasma. The diluted plasma was used to analyze the hFcγR-Ig molecules in the circulation. At each time point, a group of mice (n=3) was injected with 12.5 or 25 μg per site of anti-Ova intradermally in a total volume of 25 μl of PBS at different sites. After 5 min following the injection of anti-Ova antibody, RPA was initiated by injecting 100 μl PBS containing 500 μg of ovalbumin and 1% Evan's blue through the tail vein. A group of mice (n=3) injected with PBS served as a control at each time point. Both the control and experimental mice were sacrificed after 3 h and the injection sites on the reverse side of the skin were examined for extravasation of the blue dye. Photographs were taken immediately and used to quantitate the degree of inflammation elicited by RPA reaction. The intensity of each dermal lesion, seen blue in the photographs, was quantified using ImageJ (National Institute of Health, Bethesda) and KaleidaGraph (Synergy Software, Reading, PA).
Western blot analysis
The plasma samples collected at various time points after the hydrodynamic injection of FcγR-Ig plasmid DNA were subjected to Western blot analysis. Briefly, 20 μl of plasma was mixed with SDS-PAGE sample buffer of both reducing and nonreducing conditions. Samples were boiled for 5 min and subjected to SDS-PAGE under reducing or non-reducing conditions. After blotting the protein onto a PVDF membrane, the proteins were detected using a HRP-conjugated goat anti-human Fc specific antibody.
Analyses of ligand binding ability of circulating FcγR-Ig by ELISA
The plasma sample collected at various time points were used to detect the ligand binding activity of hydrodynamically expressed human FcγR-Igs. Briefly, ELISA plates were coated overnight with rabbit IgG (100 μl of 10 μg/ml) and blocked for 1 h with a binding buffer. The plasma samples from mice injected with human FcγR-Ig (100 μl of 1:100 diluted) were then added to the plates, and the incubation continued for 1h at 4°C. The FcγR-Ig bound to the rabbit IgG was detected as described in a previous section. The hFcγR-Igs bound to BSA-coated wells were taken as non-specific binding. Experiments are representative of three individual experiments. Data are mean ± SD of triplicates.
Immunohistochemistry
Expression of FcγRs-Ig molecules in different organs was determined by immunohistochemistry. Briefly, 10 μg of pcDNA3.1 plasmid vector containing human hCD32AH-Ig was injected hydrodynamically into a group of mice (n=3 per group) as described above. Subsequently, different organs (liver, spleen, kidney, lung and heart) were collected at several time points and snap frozen immediately using liquid nitrogen. 5μm cryo sections were taken and stained for the hCD32AH-Ig, hepatocytes and macrophages using antibodies. The specific antibodies are: F4/80: a mouse macrophage specific FITC conjugated rat monoclonal antibody, CSP1: rabbit polyclonal mouse liver mitochondrial marker antibody, and a Cy5 conjugated F(ab)’2 fragment of goat antibody specific to human Fc domain.
Statistical analysis
A statistical comparison of the control and treated samples was performed using student T-test; p<0.01 (*) considered as significant and p<0.001 (**) considered as highly significant.
ACKOWLDEMENTS
This study was supported by NIH grants R21HL09126802 and R21AR05682101 to P.S and AHA grant 11SDG5710004 to R.S. We also thank Ms. Archana Boopathy, Ms. Sumi Selvaraj and Ms. Danielle Daniels, for critical reading of the article.
Footnotes
Conflict of interest: The authors declare no competing financial interests.
REFERENCES
- 1.Liu F, Song YK, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 2.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 3.Suda T, Liu D. Hydrodynamic gene delivery: Its principles and applications. Mol Ther. 2007;15:2063–2069. doi: 10.1038/sj.mt.6300314. [DOI] [PubMed] [Google Scholar]
- 4.Herweijer H, Wolff JA. Gene therapy progress and prospects: Hydrodynamic gene delivery. Gene Ther. 2007;14:99–107. doi: 10.1038/sj.gt.3302891. [DOI] [PubMed] [Google Scholar]
- 5.Budker VG, Subbotin VM, Budker T, Sebestyen MG, Zhang G, Wolff JA. Mechanism of plasmid delivery by hydrodynamic tail vein injection. II. Morphological studies. J Gene Med. 2006;8:874–888. doi: 10.1002/jgm.920. [DOI] [PubMed] [Google Scholar]
- 6.Andrianaivo F, Lecocq M, Wattiaux-De Conick S, Wattiaux R, Jadot M. Hydrodynamics-based transfection of the liver: entrance into hepatocytes of DNA that causes expression takes place very early after injection. J Gene Med. 2004;6:877–883. doi: 10.1002/jgm.574. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi N, Kuramoto T, Yamaoka K, Hashida M, Takakura Y. Hepatic uptake and gene expression mechanisms following intravenous administration of plasmid DNA by conventional and hydrodynamics-based procedures. J Pharm exper thera. 2001;297:853–860. [PubMed] [Google Scholar]
- 8.Chang HE, Hanawa H, Liu H, Yoshida T, Hayashi M, Watanabe R, Abe S, Toba K, Yoshida K, Elnagger R, Minagawa S, Okura Y, Kato K, Kodama M, Maruyama H, Miyazaki J, Aizawa Y. Hydrodynamic-based delivery of an interleukin-22-Ig fusion gene ameliorates experimental autoimmune myocarditis in rats. J Immunol. 2006;177:3635–3643. doi: 10.4049/jimmunol.177.6.3635. [DOI] [PubMed] [Google Scholar]
- 9.Chang HE, Hanawa H, Yoshida T, Hayashi M, Liu H, Ding L, Otaki K, Hao K, Yoshida K, Kato K, Toba K, Kodama M, Maruyama H, Miyazaki J, Aizawa Y. Alteration of IL-17 related expressions in experimental autoimmune myocarditis and inhibition of Il-17 by IL-10-Ig fusion gene transfer. Circ J. 2008;72:813–819. doi: 10.1253/circj.72.813. [DOI] [PubMed] [Google Scholar]
- 10.Abe S, Hanawa H, Hayashi M, Yoshida T, Komura S, Watanabe R, Lie H, Chang H, Kato K, Kodama M, Maruyama H, Nakazawa M, Miyazaki J, Aizawa Y. Prevention of experimental autoimmune myocarditis by hydrodynamics-based naked plasmid DNA encoding CTLA4-Ig gene delivery. J Card Fail. 2005;11:557–564. doi: 10.1016/j.cardfail.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Matsui Y, Inobe M, Okamoto H, Chiba S, Shimizu T, Kitabatake A. Blockade of T cell costimulatory signals using adenovirus vectors prevents both the induction and the progression of experimental autoimmune myocarditis. J Mol Cell Cardiol. 2002;34:279–295. doi: 10.1006/jmcc.2001.1511. [DOI] [PubMed] [Google Scholar]
- 12.Kawaguchi Y. A gene therapy or purified CTLA4IgG treatment of experimental allergic encephalomyelitis. Igaku Zasshi. 1999;74:467–475. [PubMed] [Google Scholar]
- 13.Mihara M, Tan I, Chuzhin Y, Reddy B, Budhai L, Holzer A, et al. CTLA4Ig inhibits T cell -dependent B-cell maturation in murine systemic lupus erythematosus. J Clin Invest. 2000;106:91–101. doi: 10.1172/JCI9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quattrocchi E, Dallman MJ, Feldmann M. Adenovirus-mediated gene transfer of CTLA-4Ig fusion protein in the suppression of experimental autoimmune arthritis. Arthritis Rheum. 2000;43:1688–1697. doi: 10.1002/1529-0131(200008)43:8<1688::AID-ANR4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, He Y, Falo L, Jr, Hui K, Huang L. Regression of human mammary adenocarcinoma by systemic administration of a recombinant gene encoding the hFlex-TRAIL Fusion Protein. Mol Ther. 2001;3:368–374. doi: 10.1006/mthe.2001.0280. [DOI] [PubMed] [Google Scholar]
- 16.Yazawa H, Murakami T, Li H-M, Back T, Kurosaka K, Suzuki Y, Shorts L, Akiyama Y, Maruyama K, Parsoneault E, Wiltrout RH, Watanabe M. Hydrodynamics-based gene delivery of naked DNA encoding fetal liver kinase-1 gene effectively suppresses the growth of pre-existing tumors. Cancer Gene Therapy. 2006;13:993–1001. doi: 10.1038/sj.cgt.7700970. [DOI] [PubMed] [Google Scholar]
- 17.Neal ZC, Bates MK, Albertini MR, Herweijer H. Hydrodynamics limb vein delivery of a xenogeneic DNA cancer vaccine effectively induces antitumor immunity. Mol Ther. 2007;15:422–430. doi: 10.1038/sj.mt.6300046. [DOI] [PubMed] [Google Scholar]
- 18.Hulett MD, Hogarth PM. Molecular basis of Fc receptor function. Adv Immunol. 1994;57:1–127. doi: 10.1016/s0065-2776(08)60671-9. [DOI] [PubMed] [Google Scholar]
- 19.Kimberly RP, Salmon JE, Edberg JC. Receptors for immunoglobulin G. Molecular diversity and implications for disease. Arthritis Rheum. 1995;38:306–314. doi: 10.1002/art.1780380303. [DOI] [PubMed] [Google Scholar]
- 20.Ravetch JV. Fc receptors. Curr Opin Immunol. 1997;9:121–125. doi: 10.1016/s0952-7915(97)80168-9. [DOI] [PubMed] [Google Scholar]
- 21.Unkeless JC, Scigliano E, Freedman VH. Structure and function of human and murine receptors for IgG. Annu Rev Immunol. 1988;6:251–281. doi: 10.1146/annurev.iy.06.040188.001343. [DOI] [PubMed] [Google Scholar]
- 22.Selvaraj P, Fifadara N, Nagarajan S, Cimino A, Wang G. Functional regulation of human neutrophil Fc gamma receptors. Immunol Res. 2004;29:219–230. doi: 10.1385/IR:29:1-3:219. [DOI] [PubMed] [Google Scholar]
- 23.Anderson CL, Looney RJ. Human leukocyte IgG Fc receptors. Immunol Today. 1986;7:264–266. doi: 10.1016/0167-5699(86)90007-1. [DOI] [PubMed] [Google Scholar]
- 24.Fanger MW, Shen L, Graziano RF, Guyre PM. Cytotoxicity mediated by human Fc receptors for IgG. Immunol Today. 1989;10:92–99. doi: 10.1016/0167-5699(89)90234-X. [DOI] [PubMed] [Google Scholar]
- 25.Hogarth PM. Fc receptors are major mediators of antibody-based inflammation in autoimmunity. Curr Opin Immunol. 2002;14:798–802. doi: 10.1016/s0952-7915(02)00409-0. [DOI] [PubMed] [Google Scholar]
- 26.Takai T. Fc receptors and their role in immune regulation and autoimmunity. J Clin Immunol. 2005;25:1–18. doi: 10.1007/s10875-005-0353-8. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 28.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotropic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 29.Ravetch JV, Clynes RA. Divergent roles for Fc receptors and complement in vivo. Annu Rev Immunol. 1998;16:421–432. doi: 10.1146/annurev.immunol.16.1.421. [DOI] [PubMed] [Google Scholar]
- 30.Clynes R, Dumitru C, Ravetch JV. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science. 1998;279:1052–1054. doi: 10.1126/science.279.5353.1052. [DOI] [PubMed] [Google Scholar]
- 31.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, Van de Winkel JGJ, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in FcγRIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 32.Clarkson SB, Bussel JB, Kimberly RP, Valinsky JE, Nachman RL, Unkeless JC. Treatment of refractory immune thrombocytopenic purpura with anti-Fcγ receptor antibody. New Engl J Med. 1986;314:1236–1239. doi: 10.1056/NEJM198605083141907. [DOI] [PubMed] [Google Scholar]
- 33.Ravetch JV, Bolland S. IgG Fc Receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 34.Takai T, Nakamura A, Akiyama K. Fc receptors as potential targets for the treatment of allergy, autoimmune disease and cancer. Curr Drug Targets Immune Endocr metabol Disord. 2003;3:187–197. doi: 10.2174/1568008033340180. [DOI] [PubMed] [Google Scholar]
- 35.Cohen-Solal JF, Cassard L, Fridman WH, Sautes-Fridman C. Fc gamma receptors. Immunol Lett. 2004;92:199–205. doi: 10.1016/j.imlet.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 36.Tarzi RM, CooK HT. Role of Fc gamma receptors in glomerulonephritis. Nephron Exp Nephrol. 2003;95:e7–12. doi: 10.1159/000073018. [DOI] [PubMed] [Google Scholar]
- 37.Nakamura A, Takai T. A role of FcgammaRIIB in the development of collagen-induced arthritis. Biomed Pharmacother. 2004;58:292–298. doi: 10.1016/j.biopha.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 38.Shashidharamurthy R, Hennigar RA, Fuchs S, Palaniswami P, Sherman M, Selvaraj P. Extravasations and emigration of neutrophils to the inflammatory site depend on the interaction of immune-complex with Fcγ receptors and can be effectively blocked by decoy Fcγ receptors. Blood. 2008;111:894–904. doi: 10.1182/blood-2007-04-085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arthus M. Injections repetees de serum de cheval chez le lapin. C R Soc Biol. 1903;55:817. [Google Scholar]
- 40.Li P, Nagarajan S, Zhu C, Selvaraj P. Recombinant CD16A-Ig forms a homodimer and cross-blocks the ligand binding functions of neutrophil and monocyte Fcγ receptors. Mol Immunol. 2002;38:527–538. doi: 10.1016/s0161-5890(01)00088-8. [DOI] [PubMed] [Google Scholar]
- 41.Shashidharamurthy R, Zhang F, Amano A, Kamat A, Panchanathan R, Ezekwudo D, Zhu C, Selvaraj P. Dynamics of the interaction of human IgG subtypes immune complexes with cells expressing R and H allelic forms of a low affinity Fc□ receptor CD32A. J Immunol. 2009;183:8216–8224. doi: 10.4049/jimmunol.0902550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ignatovich AI, Dizhes EB, Palvotskayan AV, Akifiev BN, Burov SV, Orlov SV, Perevozchikov AP. Complexes of plasmid DNA with basic domain 47-57 of the HIV-1 tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem. 2003;278:42625–42636. doi: 10.1074/jbc.M301431200. [DOI] [PubMed] [Google Scholar]
- 43.van Sorge NM, van der Pol W-L, Van de Winkel JGJ. FcγR Polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tis Ant. 2003;61:189–202. doi: 10.1034/j.1399-0039.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 44.Dijstelbloem HM, Bijl M, Fijnheer R, Scheepers RH, Oost WW, Jansen MD, Sluiter WJ, Limburg PC, Derksen RH, van de Winkel JG. Fcγ receptor polymorphisms in systemic lupus erythematosus: association with disease and in vivo clearance of immune complexes. Arthritis and Rheum. 2000;43:2793–2800. doi: 10.1002/1529-0131(200012)43:12<2793::AID-ANR20>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 45.Ludo van der Pol W, van de winkel GJ. IgG receptor polymorphism: risk factor for disease. Immunogen. 1998;48:222–232. doi: 10.1007/s002510050426. [DOI] [PubMed] [Google Scholar]
- 46.Tan SY. FcγRIIa polymorphism in systemic lupus erythematosus. Kidney Blood Press Res. 2000;23:138–142. doi: 10.1159/000025967. [DOI] [PubMed] [Google Scholar]
- 47.Manger K, Repp R, Spriewald BM, Rascu A, Geiger A, Wassmuth R, Westerdaal NA, Wentz B, Manger B, Kalden JR, van de Winkel JG. Fcγ receptor IIa polymorphism in caucasian patients with systemic lupus erythromatosus: association with clinical symptoms. Arthritis Rheum. 1998;41:1181–1189. doi: 10.1002/1529-0131(199807)41:7<1181::AID-ART6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 48.Ierino FL, Powell MS, McKenzie IFC, Hogarth PM. Recombinant soluble human Fc-gamma-RII: Production, characterization, and inhibition of the Arthus reaction. J Exp Med. 1993;178:1617–1628. doi: 10.1084/jem.178.5.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ellsworth JL, Hamacher N, Harder B, Bannink K, Bukowski TR, Byrnes-Blake K, Underwood S, Oliver C, Waggie KS, Noriega C, Hebb L, Rixon MW, Lewis KE. Recombinant soluble human FcγR1A (CD64A) reduces inflammation in murine collagen induced arthritis. J Immunol. 2009;182:7272–7279. doi: 10.4049/jimmunol.0803497. [DOI] [PubMed] [Google Scholar]
- 50.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the β2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci USA. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chappel MS, Isenman DE, Everett M, Xu YY, Dorrington KJ, Klein MH. Identification of the Fcγ receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc Natl Acad Sci USA. 1991;88:9036–9040. doi: 10.1073/pnas.88.20.9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chappel MS, Isenman DE, Oomen R, Xu YY, Klein MH. Identification of a secondary FcγRI binding site within a genetically engineered human IgG antibody. J Biol Chem. 1993;268:25124–25131. [PubMed] [Google Scholar]
- 53.Ledbetter JA, Gilliand LK, Hayden MS, Linsley PS, Bajorath J, Fell HP. Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in mammalian cells. United States Patent. 2000:6132992. [Google Scholar]
- 54.Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989;143:2595–2601. [PubMed] [Google Scholar]
- 55.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 56.He Y, Pimenov AA, Nayak JV, Plowey J, Falo LD, Huang L. Intravenous injection of naked DNA encoding secreted flt3 ligand dramatically increases the number of dendritic cells and natural killer cells in vivo. Hum Gene Ther. 2000;11:547. doi: 10.1089/10430340050015734. [DOI] [PubMed] [Google Scholar]