

Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare human genetic disease in which de novo osteogenesis – a developmental process occurring during embryonic skeletal formation – is induced aberrantly and progressively beginning during early childhood in soft connective tissues. Episodic initiation of spontaneous bone forming lesions occurs over time, affecting a generally predictable sequence of body locations following a pattern similar to that of the developing embryonic skeleton. The heterotopic (extra-skeletal) bone formation in FOP can also be induced by connective tissue injury. At the tissue level, an initial tissue degradation phase is followed by a tissue formation phase during which soft connective tissues are replaced by bone tissue through endochondral osteogenesis. This extra-skeletal bone is physiologically normal and develops through the same series of tissue differentiation events that occurs during normal embryonic skeletal development. The underlying genetic mutation in FOP alters the signals that regulate induction of cell differentiation leading to bone formation. In addition to post-natal heterotopic ossification, FOP patients show specific malformations of skeletal elements indicating effects on bone formation during embryonic development as well. Nearly all cases of FOP are caused by the identical mutation in the ACVR1 gene that causes a single amino acid substitution, R206H, in the bone morphogenetic protein (BMP) type I receptor ACVR1 (formerly known as ALK2). This mutation causes mild constitutive activation of the BMP signaling pathway and identifies ACVR1 as a key regulator of cell fate decisions and bone formation, providing opportunities to investigate previously unrecognized functions for this receptor during tissue development and homeostasis.
Keywords: Fibrodysplasia ossificans progressiva, FOP, heterotopic ossification, ACVR1, ALK2, BMP signaling, BMP receptors, bone formation, skeletal development, osteogenesis, chondrogenesis
Introduction
Bone formation, or osteogenesis, is normally restricted to the skeleton during embryonic development during the initial formation of the elements of the skeletal system and to fracture healing during repair of damage to the skeletal bone. Alterations in the regulatory processes that direct where and when osteogenesis occurs can cause disease. Heterotopic (extra-skeletal) ossification (HO) is a pathological condition in which qualitatively normal bone tissue forms in non-skeletal tissues in response to abnormal induction of osteogenesis.1, 2 Acquired, or non-hereditary, HO is usually associated with severe soft tissue trauma, and is a common secondary complication that is induced in response to central nervous system injury, total hip arthroplasty, deep tissue burns, and multiple other forms of trauma (including war-related injuries).1–6 HO has significant consequences for the health of affected individuals, causing restricted joint range of motion, severe pain, and limitations to prosthetic use. The onset of HO cannot be readily predicted, resulting in limited understanding of the inductive events and cellular mechanisms that lead to this ectopic bone formation.
In addition to acquired forms of HO, rare inherited disorders of extensive and progressive HO have been identified7 and provide opportunities to identify important components of the cellular mechanisms and pathways that regulate cell differentiation. Heterotopic bone can form through endochondral ossification, in which cartilage forms and then is replaced by bone – the same process through which most skeletal long bones develop, or through intramembranous ossification, in which bone forms directly – the process through which the skull forms. Fracture repair can occur through either endochondral or intramembranous bone formation.8 In fibrodysplasia ossificans progressiva (FOP), a genetic disease of extensive and progressive bone formation within soft connective tissues such as skeletal muscles, heterotopic bone forms through endochondral ossification as a result of mutations in one of the bone morphogenetic protein (BMP) type I receptors, identifying the BMP signaling pathway and a specific BMP receptor, ACVR1, as critical components regulating cell fate decisions and bone tissue formation.
Sidebar: Progressive osseous heteroplasia (POH)
Like FOP, progressive osseous heteroplasia (POH) is a human inherited disorder of heterotopic ossification (MIM #166350; http://www.omim.org/entry/166350?search=poh&highlight=poh).7 Both FOP and POH are monogenic autosomal dominant genetic disorders, identifying their disease-causing genes as critical components of the regulatory mechanisms that direct cell fate decisions and bone tissue and/or organ formation.
However, while activating mutations in ACVR1, a BMP type I receptor, induces heterotopic endochondral ossification in FOP, heterotopic ossification forms mainly intramembranous bone in response to inactivating mutations in the GNAS gene, a transcriptionally complex locus that includes multiple promoters and is regulated in part through genomic imprinting.7, 9 The most abundant protein product of this gene is GS alpha (Gsα), a heterotrimeric G-protein alpha subunit form that activates adenylyl cyclases and cyclic AMP and is ubiquitously expressed.
Although many cases of POH have been clinically misdiagnosed as FOP, POH can be distinguished from FOP by several clinical criteria. Unlike FOP, POH is characterized by ossification that initially develops within the dermis, often in association with adipose tissue. POH heterotopic ossification progresses from the dermis to the underlying deep connective tissues, with bone formation within skeletal muscle and in some cases fusing with skeletal bone. Also distinct from FOP, POH is not associated with inflammation, predictable regional patterns of HO, or FOP-like great toe malformations. Heterotopic bone formation in POH forms in an asymmetric mosaic distribution and shows a predominance of intramembranous bone formation.
DEVELOPMENTAL AND CLINICAL ASPECTS OF FIBRODYSPLASIA OSSIFICANS PROGRESSIVA (FOP)
Characteristic Features of FOP
Overview
Fibrodysplasia ossificans progressiva (FOP; MIM 135100; (http://www.omim.org/entry/135100?search=FOP&highlight=fop)) is a human disease of bone formation. The main characteristic and clinically significant feature of FOP is the formation of extra-skeletal (heterotopic) bone. However, specific skeletal bone malformations also occur, indicating that the underlying gene mutation influences the shape and structure of the skeletal elements during embryonic development in addition to regulating the cell fate decisions of progenitor cells in non-embryonic tissues.10, 11 FOP is a rare condition, occurring at a population frequency of about one per 2 million, and can be inherited by autosomal dominant inheritance, although most cases are sporadic new mutations in the affected person.12
A child carrying the FOP-causing mutation is usually born following an uneventful pregnancy. He/she appears healthy, although bent great toes are observed at birth. In many cases, the early childhood years are typical and active, although impaired neck movement is frequently noted. At about five years of age, the child begins to experience painful swellings that develop into hard bumps containing bone. These bone-forming events first appear in the upper back and neck, and then episodically progress through the trunk of the body and to the arms and legs. Over a time course of several years, this extra-skeletal bone fuses the skeleton and spans joints to form an extensive network of extra-skeletal bone that causes a near total immobilization of the body.
Skeletal malformations
In combination with heterotopic ossification, great toe dysmorphology is a diagnostic criteria for the standard, or “classic”, clinical presentation of FOP (Figure 1). Congenital malformation (hallux deformity) of the great toes is usually the earliest phenotypic evidence that a person has FOP. The proximal phalanx of the first digit of the foot is abnormally shaped and fused with the terminal first phalanx and an abnormal first metatarsal is frequently observed.13 Fusions of the middle and terminal phalanges of the second through fifth toes may also be present. These malformations are often bilaterally symmetrical. (A depiction of the normal skeletal structure of the foot can be viewed: http://www.britannica.com/EBchecked/media/101314/Bones-of-the-foot-showing-the-calcaneus-talus-and-other.)
Figure 1.

Clinical features of fibrodysplasia ossificans progressiva (FOP). The classic clinical phenotype of FOP is characterized by two features:
(a) The extensive heterotopic bone formation typical of FOP is seen in a three-dimensional reconstructed computed tomography (CT) scan of the back of a twelve-year-old child. Ribbons, sheets, and plates of bone form in extra-skeletal connective tissue and fuse the joints of the axial and appendicular skeleton. For comparison, an image of a normal skeleton (back view) can be viewed at: http://www.shutterstock.com/pic.mhtml?id=11313556.
(b) Radiograph of the feet of a three-year-old child shows symmetrical great toe malformations (a circle indicates the left foot malformation) of metatarsals and proximal phalanges along with microdactyly, fused interphalangeal joints, and hallux valgus deviations at the metatarsophalangeal joints.
[Reprinted from Shore et al., Nature Genetics 2006, 38, 525–527.]
Great toe dysmorphology is the most characteristic and consistent skeletal malformation of FOP, although other skeletal changes also commonly, but more variably, are present.10, 14 Skeletal dysmorphologies affecting the digits of the hands also occur but are less frequent than foot abnormalities. Early developmental malformations are frequently observed in the cervical spine. Other FOP-associated skeletal changes include short broad femoral necks and proximal medial tibial osteochondromas. Patient observations support that the craniofacial bone structure is also altered in FOP, with some effects possibly being secondary to disease progression affecting craniofacial bones, while others resulting from altered development or growth. Impaired conductive hearing is also a frequent feature of FOP.
Heterotopic ossification
HO in FOP begins early in life with a median age of onset of five years, although in some cases ossification has occurred as early as the first year. Subsequent gradual and progressive formation of extra-skeletal bone occurs within connective tissues such as skeletal muscle, tendon, ligament, fascia, and aponeuroses; smooth muscle and cardiac muscle do not become involved.10, 15
Episodes of HO generally occur in a characteristic anatomic and temporal pattern, with the first events frequently occurring along the upper back and neck. Bone formation typically occurs first in the dorsal, axial, cranial, and proximal regions of the body and later in the ventral, appendicular, caudal, and distal regions. This typical progression pattern can be altered in response to soft tissue injury which can induce an episode of heterotopic bone formation.16–20
By standard histology, biochemistry, metabolism, radiology, and biomechanics evaluations, the properties of the extra-skeletal bone that forms in FOP are consistent with those of the normal skeleton.10, 15 Marrow cavities and bone marrow cells can form within the mature heterotopic bone. The ectopic bone shows evidence of normal bone remodeling and responds to fracture with a normal healing process. 10, 15 The consequence is a bone tissue system that has developed within the soft tissues of the body.
Atypical forms of FOP
A small number of patients with FOP-type progressive HO show significant variation from the classic defining features of FOP (Table 1). These patients are described as FOP variants.14 FOP variants most often differ from classic FOP in the degree of severity of great toe malformations. Several FOP variants have more extensive developmental effects on the skeleton, with some cases showing severe reduction deficits or absence of toes and/or thumbs. Other very mild FOP variant cases have little or no developmental changes in the skeletal elements.
Table 1.
Clinical Features of Classic FOP, FOP-Plus, and FOP Variantsa
| Number of cases evaluated |
Classic FOP n>100 |
FOP-plus n=8 |
FOP Variants n=12 |
|---|---|---|---|
| ACVR1 mutation: codon change | all R206H | most R206H | in GS or kinase domains; no R206H |
| Age of HO onset (years) | 1–10 | <1–14 | <1–2 or 8–26 |
|
| |||
| Classic/Defining FOP Features: | |||
| Characteristic malformations of great toe (hallux valgus, malformed 1st metatarsal, and/or monophalangism) | 100% | 100% | 0% |
| Progressive heterotopic ossification in characteristic anatomic patterns | 100% | 100% | ~100% |
|
| |||
| Common Variable FOP Features: | |||
| Conductive hearing impairment | >50% | >50% | <50% |
| Cervical spine malformations | >80% | >80% | <80% |
| Proximal medial tibial osteochondromas | >90% | >90% | <50% |
| Short broad femoral necks | >70% | >70% | <70% |
| Thumb malformations (short 1st metacarpal, +/− monophalangism) | ~50% | ~50% | ~50% |
|
| |||
| Atypical FOP Clinical Features: | |||
| Severe variable reduction deficits of digits | 0% | 0% | ~50% |
| Absent finger/toe nails in digits with severe reduction deficits | 0% | 0% | ~50% |
| Normal or minimal changes in great toes | 0% | 0% | ~50% |
| Intra-articular synovial osteochondromatosis of hips and DJD of hips | 0% | 0% | ~25% |
| Sparse, thin scalp hair (more prominent in 2nd decade) | 0% | n=1 | ~50% |
| Mild cognitive impairment | 0% | n=1 | ~50% |
| Severe growth retardation | 0% | n=2 | 0% |
| Cataracts | 0% | n=1 | 0% |
| Retinal detachment | 0% | n=1 | 0% |
| Childhood glaucoma | 0% | n=2 | 0% |
| Craniopharygioma | 0% | n=1 | 0% |
| Persistence of primary teeth in adulthood | 0% | n=1 | 0% |
| Anatomic abnormalities of cerebellum | 0% | n=0 | n=2 |
| Diffuse cerebral dysfunction w seizures | 0% | n=1 | 0% |
| Polyostotic fibrous dysplasia | 0% | n=1 | 0% |
| Primary amenorrhea | 0% | n=1 | n=1 |
| Aplastic anemia | 0% | n=1 | 0% |
| Hypospadias | 0% | n=0 | n=1 |
| Cerebral cavernous malformations | 0% | n=0 | n=1 |
Patients reported in Kaplan et al., Human Mutation 2009, 30, 379–390 are summarized. Additional individual FOP variant cases have also been reported.
A second small group of patients have the standard characteristics of classic FOP (progressive HO and great toe malformations) but also have clinical features that are not commonly associated with FOP. These patients are described as FOP-plus.14 Atypical features that have been observed include sparse scalp hair, absence of finger/toe nails, cataracts, retinal detachment, childhood glaucoma, persistence of primary teeth in adulthood, mild cognitive impairment, and cerebellar abnormalities. It has not yet been established whether these features are influenced by the underlying genetic mutation or are due to independent causes.
GENETICS AND GENE MUTATIONS
ACVR1, the mutated gene in FOP
Genetic inheritance of FOP
In most cases, FOP is caused by a new mutational event in a child who has unaffected parents and no prior family history. FOP is a severely debilitating condition and inheritance from an affected parent to his/her children has been identified in only a small number of families, however when genetic transmission occurs, the inheritance pattern is autosomal dominant.12, 21
The gene mutation in FOP was identified through linkage analysis using five families with affected members in two generations.21 Linkage to chromosome 2q23–24 was observed in all families and led to the identification of mutations in the ACVR1 gene which encodes ACVR1 (activin A receptor, type I, which was formerly known as ALK2, activin-like kinase 2), a bone morphogenetic protein (BMP) type I receptor (http://www.genecards.org/cgi-bin/carddisp.pl?gene=ACVR1).
The bone morphogenetic protein (BMP) signaling pathway
Bone morphogenetic proteins (BMPs) are members of the transforming growth factor beta (TGFB) family of extra-cellular signaling factors that activate heterotetramer receptor complexes of two type I and two type II serine-threonine kinase receptors (Figure 2). In response to ligand binding, type II receptors phosphorylate the glycine-serine (GS) cytoplasmic domain of the type I receptors to activate the receptor complex.22–25 Activated type I receptors induce the BMP signaling pathway and transcriptional regulation through phosphorylation of BMP pathway-specific SMAD proteins (SMADs 1, 5, 8) and through MAP kinase pathways.22, 23 In addition to ACVR1, the BMP type I receptor mutated in FOP, BMP signal transduction can be mediated through the BMPR1A (formerly known as ALK3), BMPRIB (formerly known as ALK6), and ACVRL1 (formerly known as ALK1) type I receptors.
Figure 2.

Generalized schematic of the BMP signaling pathway.
Type I and type II BMP receptors span the cell membrane and bind extracellular BMP ligand. Ligand binding to receptor complexes activates signaling through type II receptor phosphorylation of the type I receptor at the GS domain. Type I receptor phsophorylation is accompanied by reduced GS binding by proteins, such as FKBP1A, that regulate receptor signaling in the absence of ligand binding. Activated type I receptor phosphorylates cytoplasmic signal transduction proteins such as R-SMADs and MAPKs, which in turn, directly or indirectly regulate transcription of target genes in the nucleus.
[Reprinted from Shore and Kaplan, Nature Reviews Rheumatology 2010, 6,518–527.]
FOP mutations
The classic R206H ACVR1 mutation
All individuals with classic clinical features of FOP (sporadic and inherited cases) contain the identical heterozygous single nucleotide substitution (c.617G>A) that changes amino acid 206 from arginine to histidine (R206H).14, 26 Codon 206 is highly conserved and occurs within the glycine-serine (GS) region of the cytoplasmic domain of ACVR1.
Mutations in Atypical Forms of FOP
As found in patients with a classic clinical presentation of FOP, all examined atypical patients with FOP-type heterotopic ossification also have heterozygous ACVR1 missense mutations in conserved amino acids.14 FOP-plus patients have the classic defining FOP features of progressive HO and great toe malformations in combination with clinical characteristics that are uncommon in classic FOP patients. The c.617G>A; R206H mutation that occurs in all cases of classic FOP occurs in most cases of FOP-plus. FOP variants present with significant differences in the standard clinical presentation of one or both of the two classic defining features of FOP, most commonly milder or more severe malformation of the great toes. A large patient series as well as individual case reports identified novel (non-R206H) ACVR1 mutations in each of the FOP variant cases and in two patients with FOP-plus.14, 27
ACVR1 Mutations
FOP patients are heterozygous for ACVR1 mutations, indicating that one mutant allele is sufficient to cause the disease. This is consistent with the observed autosomal dominant mode of inheritance. Homozygous mutations have not been found and are thought to be embryonic lethal.
ACVR1 mutations in FOP are fully penetrant. None of the identified ACVR1 mutations have been identified in unaffected individuals. DNA sequencing of patient genomic DNA supports that there is no locus heterogeneity in FOP; all patients with FOP-type heterotopic ossification thus far examined have a mutation in the ACVR1 gene.
All mutations identified in classic, atypical, and variant FOP patients are single nucleotide substitutions that cause missense mutations, with the exception of a FOP variant case who has a 3-nucleotide deletion that replaces two amino acids with a single codon.14 No frameshift or nonsense mutations have been identified, supporting that in each case a mutant protein with altered function is produced.
Genotype-phenotype correlations
Although the rate of progression and the severity of HO vary greatly among individuals with classic and atypical FOP forms, progressive postnatal HO is the common feature shared by all classic FOP, FOP-plus, and FOP variant patients. Among the identified ACVR1 mutations in these patients, there is limited correlation between the severity of heterotopic ossification and specific mutations, supporting that all of the identified ACVR1 missense mutations influence the promiscuous post-natal induction of cartilage and bone cell differentiation in similar ways on a cellular level. However, given that the identified mutations occur in more than one functional domain of the ACVR1 protein, some variation in specific molecular mechanisms that are altered by the different mutations likely occur.
A strong influence of environmental factors in triggering episodes of heterotopic bone formation in the context of ACVR1 mutation is supported by clinical observations of three pairs of identical twins with FOP28 who show wide variability in disease progression within each twin pair. However, skeletal formation, as reflected by toe malformations, is remarkably similar between members of each twin pair suggesting a significant genetic influence on embryonic development.
Among patients with classic FOP, FOP-plus, and FOP variants, a wide range of variability in the malformations of the great toes is observed, and genotype-phenotype correlations may explain at least some of this variation. However, given the rarity of FOP-plus and FOP variant patients, some of the ACVR1 mutations have been identified in only a single patient, therefore such correlations are difficult to determine through currently available clinical information.
Effects of FOP mutations on BMP signaling
All ACVR1 mutations in classic FOP, FOP-plus, and FOP-variants reside in or adjacent to the glycine-serine (GS) activation domain or within the active site of the kinase, regions of the receptor that participate in downstream signal transduction. Each of these mutated amino acids is highly conserved evolutionarily, further supporting their functional significance. Protein structure homology modeling predicts that each of the amino acid substitutions activates the ACVR1 receptor and enhances signaling through both BMP-responsive and BMP-independent mechanisms.14, 27, 29
Structure modeling predictions of the consequences of ACVR1 mutations are supported by experimental studies examining the effects of the recurrent R206H FOP mutation on BMP signaling. In vitro signaling assays demonstrated that ACVR1 R206H induces mild constitutive activation of the BMP signaling cascade.30–33 These data are consistent with studies in cells from FOP patients that demonstrated altered signal transduction through the BMP pathway with increased phosphorylation of BMP pathway signaling mediators (BMP-specific SMADs and MAPK14, formerly known as p38MAPK) and increased expression of BMP transcriptional targets in the absence of BMP ligand as well as the ability to further increase BMP signaling in response to ligand.34–37
The zebrafish (Danio rerio) is an important in vivo model for studying BMP signaling activity during development and has been used to show that BMPs induce the formation of ventral tissues during early embryonic development. Acvr1 (formerly known as Alk8), the zebrafish functional orthologue of human ACVR1, acts as a BMP2/4/7 receptor, upstream of Smad5. The zebrafish acvr1 mutant model (lost-a-fin; laf) develops dorsalization of the embryonic axis, indicating insufficient BMP signaling.38, 39 By contrast, over-activation of BMP signaling induces embryonic ventralization.40 The dorsalized phenotype of zebrafish acvr1 null (laf) embryos was rescued by injection with wild-type human ACVR1 mRNAs, however mutant R206H ACVR1 mRNA over-compensated for the lack of Acvr1 in laf embryos, causing a highly ventralized phenotype.31 That this ventralized phenotype occurs even in embryos that are genetically null for all BMP ligands, indicates that the R206H mutation confers ligand-independent hyper-activation of BMP signaling. Further analyses demonstrated that downstream signaling by the mutant receptor is mediated through the BMP-SMAD signaling pathway.30, 31, 33
Binding and activation of the SMAD signal transduction proteins occur through interaction with the GS region of type I TGFB/BMP receptors, the location of the FOP R206H mutation in ACVR1. In the absence of ligand-receptor binding, the GS domain of the type I receptors binds FKBP1A (formerly known as FKBP12), a highly conserved inhibitory protein that prevents leaky activation of type I receptors in the absence of ligand. In response to ligand binding, FKBP1A dissociates from the receptor and SMADs are activated.22, 24, 25 The ACVR1 R206H receptor has reduced interaction with FKBP1A,31–33, 41 suggesting that impaired FKBP1A-ACVR1 interaction contributes to BMP-independent BMP pathway signaling. The ACVR1 R206H mutation does not fully prevent FKBP1A interaction suggesting that FKBP1A-mutant ACVR1 interactions are less stable and/or of shorter duration, leading to mild constitutive receptor activation. FKBP1A-type I receptor binding has been suggested to act as a gradient reader for morphogenetic activities of the inducing ligands, such as BMPs and TGFB, with reduced FKBP1A binding leading to changes in the strength or timing of downstream signaling and dysregulation of differentiation and other cellular processes.41
BMP signaling regulates a diverse range of cellular activities including differentiation, proliferation, apoptosis, migration, positional information, and stem cell renewal.29, 45–49 Signaling mediated through subsets of BMPs and their cell surface receptors is important during embryonic development to establish dorsal-ventral polarity in the developing embryo, specifying the initial differentiation of ectoderm- and mesoderm-derived tissues and organs.46 Mouse Acvr1 knockout or activation models have shown that BMP signal transduction through ACVR1 contributes to tissue/organ development including embryonic patterning during gastrulation, and the migration of neural crest cells that contribute to cardiac and craniofacial development, eye development, and formation of germ cells.50–55 Clinical observations support that ACVR1 FOP activating mutations affect the development and function of a range of tissues and organs.14
CELLULAR/TISSUE EFFECTS - FORMATION OF HETEROTOPIC LESIONS
Endochondral ossification during embryonic development and fracture healing occur through the same stages of cellular differentiation to form cartilage and bone. These processes differ, however, in that fracture repair initiates with a robust immune and inflammatory response, while embryonic skeletal development does not. Histological evaluation of FOP patient biopsies,56, 57 supports that inflammation and an immune response contribute to the early stages of heterotopic bone formation as occurs during fracture repair.
Initiation of heterotopic ossification
Episodes of HO in FOP can be induced by soft tissue injury, such as surgery, muscle fatigue, intramuscular injections, pre-school immunizations, and viral illnesses.10, 15 These events are accompanied by robust inflammation through the innate immune wound-repair response. In the absence of injury, episodes of soft connective swelling leading to heterotopic bone formation (or ‘flare-ups’) appear to initiate spontaneously with no overt induction by tissue trauma.
However, even non-injury-associated HO events, as seen by analysis of spontaneous lesions formed in an FOP knock-in mouse model (our unpublished observations), an immune response is the first histological evidence of an HO lesion (Figure 3). The initiating stages of early FOP lesions are recognized by the presence of mononuclear cells, monocytes, macrophages, mast cells, and B- and T-lymphocytes within skeletal muscle and other connective tissues.56 This immune response is similar to, but appears greater in magnitude than, a normal tissue response to injury56, 58, 59 and accompanies the destruction and turnover of the surrounding connective tissues.
Figure 3.
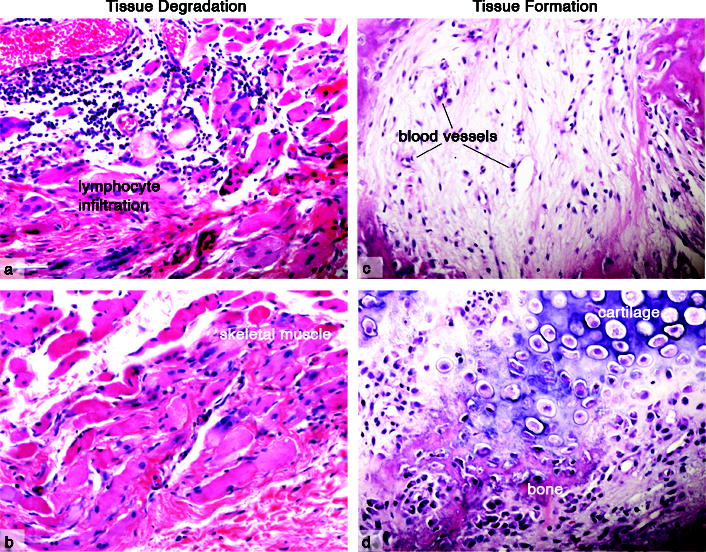
Heterotopic ossification in FOP.
The initial stages of FOP lesion formation involves inflammation and the destruction of connective tissues (a, b), followed by tissue replacement (c, d). Inflammatory and immune cell infiltration (a) is followed by tissue degradation and loss of the skeletal muscle structure (b). Subsequent tissue formation includes fibroproliferation and angiogenesis (c), followed by chondrogenesis and osteogenesis (d) and the formation of heterotopic endochondral bone. (Images are from patient biopsies obtained prior to a diagnosis of FOP. Tissue trauma, including biopsies, can induce FOP lesion formation and therefore are not obtained following diagnosis.)
[Figure courtesy of Salin Chakkalakal, PhD.]
A normal tissue repair response includes a hypoxic microenvironment.60 Protein structure homology modeling of the mutant R206H ACVR1 receptor predicts that an aberrant salt bridge would promote over-activity of the FOP receptor under hypoxic conditions and lower pH,61 and a hypoxic tissue environment has been associated with induction of HO in a mouse model.62 Our preliminary studies63 tested the hypothesis that a hypoxic microenvironment enhances signaling through the mutant ACVR1 receptor and demonstrated that signaling was enhanced and of longer duration in the presence of the ACVR1 R206H mutation under hypoxic conditions.
In the absence of detectable injury, low levels of mutant ALK2 activity have been hypothesized7 to prime tissues to aberrantly trigger a tissue repair response and stimulate immune cell migration to sites of an altered tissue microenvironment. Continued and/or enhanced levels of BMP signaling mediated through mutant ACVR1 during this process could exceed the signaling threshold that normally contributes to tissue repair and instead lead to induction of cell differentiation programs that direct endochondral bone formation. The described pattern of episodes of heterotopic ossification in the absence of overt trauma is an observation for which we have no explanation.
Aberrant cellular differentiation
The initial phase of tissue turnover in FOP lesion formation is followed by a phase of tissue formation (Figure 3) that is first indicated by angiogenesis and fibroproliferation, then is followed by production of newly formed cartilage and bone through a normal sequence of endochondral ossification stages.64, 65 In essence, one tissue is replaced by another.
BMPs were initially recognized for their bone forming activity, and many proteins within the BMP family have specific abilities to induce chondrogenesis and osteogenesis and promote endochondral bone formation.29, 42 Acvr1 is expressed in chondrocytes and osteoblasts and a constitutively active form (caACVR1; Q207D) enhances chondrogenesis and induces HO.43, 44 BMPs and BMP receptors are expressed in many adult tissues including skeletal muscles and chondrocytes.
Comparison of ACVR1 R206H to the strongly constitutively active and ligand independent ACVR1 Q207D mutation (caACVR1) confirmed that the FOP R206H mutation is only mildly constitutively activating.31–33 Chick limb bud micromass cultures were used for chondrogenesis assays and demonstrated that the ACVR1 R206H receptor has a milder stimulation of cartilage cell differentiation compared to caACVR1 Q207D.31
In recent years, there has been a growing interest in identifying the osteoprogenitor cells that participate in heterotopic bone formation. A number of sources of putative progenitor cells have been suggested,66–69 but few studies have investigated the identity of the specific cell lineages responsible for the chondro-osseous anlagen BMP-associated heterotopic ossification.
Early FOP lesions are highly angiogenic64 and recent investigations of FOP patients and in vivo mouse models of FOP-like heterotopic ossification demonstrated that connective tissue progenitor cells of endothelial origin contribute to multiple stages in the development of the heterotopic anlagen.70, 71 While data support that endothelial lineage cells are not the only source of chondro/osseous progenitor cells that contribute to heterotopic ossification, such cells may have a significant role in this process.
Embryonic Skeletal Development
Malformations of the great toes are the most consistent and readily detected skeletal anomaly caused by the ACVR1 R206H mutation. The malformation occurs during embryonic development and is observed in newborn children with FOP. More detailed analyses are required to explain the mechanisms through which this and other more subtle skeletal effects of the mutation occur. However, it has been established that BMP signaling plays key roles in the formation and differentiation of the mesenchymal cell condensations that are precursors of the skeletal elements.72 Interestingly, the great toe is one of the final skeletal elements to form during embryogenesis, raising speculation that the FOP ACVR1 mutation alters levels of signaling that direct skeletal patterning, perhaps by perturbing a signaling gradient, and this effect is more pronounced as skeletal development is completed. Mutations in BMP ligands, receptors, and antagonists have been identified in a number of specific skeletal diseases including multiple forms of brachydactyly (shortening of fingers/toes) that are associated with impaired BMP signaling as well as joint fusions that are associated with loss-of-function Noggin (a BMP antagonist) mutations.73, 74 BMP signaling is also an important contributor to the growth 0f skeletal elements with ACVR1 expressed primarily in resting and proliferating chondrocytes in the growth plates and contributing to bone growth.75, 76 It seems likely that ACVR1 mutations and altered BMP signaling may affect skeletal development at multiple stages.
Conclusion
Multipotent progenitor cells are precisely regulated and directed to coordinately differentiate to specialized cells in tissues and organs during embryonic development and to repair damage in adult tissues under the guidance of signals such as morphogen gradients and cell shape change-induced biomechanical stress. Cells interpret these complex signals through signal transduction pathways that direct transcriptional activation and repression, which in turn determine cell fate. FOP is a human genetic disease that alters the regulation of progenitor cell differentiation as a consequence of mutations that activate the BMP type I receptor ACVR1 and downstream signaling. While the identification of dysregulated BMP signaling through a specific BMP receptor was a significant advance in our understanding of this disease, much remains to be understood regarding the developmental and other phenotypic consequences of the mutation and the cellular mechanisms that mediates these effects.
Most patients with FOP share the identical ACVR1 mutation, a single nucleotide substitution within a region of the ACVR1 receptor that is critical for downstream signal transduction. The R206H mutation causes mild constitutive activation of the BMP signaling pathway in the absence of receptor activation by ligand and shows enhanced signaling in response to ligand. As a consequence of this mutation, at birth, patients are noted to have short, malformed great toes, as well as other variable skeletal effects. After birth, usually during early childhood, progenitor cells are mobilized to aberrantly differentiate to cartilage and bone, forming extensive ectopic bone tissue in skeletal muscles, tendons, ligaments.
Given the multiple roles of the BMP signaling pathway in many stages of development and in a wide range of cells and tissues, it would not be unexpected that the enhanced BMP signaling induced by the FOP ACVR1 mutation contributes to multiple stages of heterotopic bone formation, including an aberrant response to tissue injury (and/or tissue changes that are perceived as tissue damage) that initiates lesion formation, an induced or enhanced tissue environment that supports and promotes bone formation, recruitment of progenitor cells with potential for chondrogenesis and osteogenesis, and directing cell differentiation to cartilage and bone fates.
In addition to heterotopic osteogenesis after birth. malformations of the skeletal elements in FOP demonstrate the participation of BMP signaling through the ACVR1 receptor during embryonic bone development. Further, given the multiple roles of the BMP signaling pathway in many stages of development and in a wide ranges of cells and tissues, it is highly likely that the FOP mutation affects the development and functions of a range of tissues and organs outside of the musculoskeletal system.
FOP provides a unique view of the importance of a precisely regulated ACVR1-BMP pathway. As we have learned more about this disorder, new insight has generated many new questions about developmental biology, signaling mechanisms, and determination of cell fate including – What are the roles of ACVR1 during embryonic development? Why are specific FOP phenotypic effects exhibited in the great toe? What are the molecular mechanisms that permit leaky activation and enhanced ligand response from the mutant receptor? What are the critical downstream BMP/ACVR1 signaling targets that direct HO and normal skeletogenesis? What are the sources of chondro/osseous progenitor cells in adult tissues? Does the mutation have a cell autonomous or non-autonomous effect on the precursor cells, or both? How does the mutation initiate the earliest pre-osteogenic events that support bone formation? These issues remain open questions, however with emerging genetically-engineered mouse models containing the Acvr1 R206H mutation along with other models of heterotopic ossification, answers to these questions can be addressed.
For people who have FOP and their families, the most significant question to be answered is the identity of the specific and effective treatments that will prevent the action of the ACVR1 mutation to form cartilage and bone. Understanding the developmental biology of HO and endochondral osteogenesis give us opportunities to identify strategies for therapeutic interventions that rescue the effects of a perturbed signaling pathway to prevent unwanted bone formation as well as additional opportunities to modify the pathway to enhance signaling and engineer replacement cartilage and bone tissue.
Acknowledgments
This work reflects contributions from many collaborators and research group members. I especially acknowledge Dr. Fred Kaplan. This work was supported through the Center for Research in FOP and Related Disorders, the International FOP Association (IFOPA), the Ian Cali Endowment, the Weldon Family Endowment, the Rita Allen Foundation, the NIH/NIAMS-supported Penn Center for Musculoskeletal Disorders (AR050950), and by grants from the National Institutes of Health (R01-AR41916 and R01-AR046831).
References
- 1.McCarthy EF, Sundaram M. Heterotopic ossification: a review. Skeletal Radiology. 2005;34:609–619. doi: 10.1007/s00256-005-0958-z. [DOI] [PubMed] [Google Scholar]
- 2.Pignolo RJ, Foley KL. Nonhereditary heterotopic ossification. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:261–266. [Google Scholar]
- 3.Forsberg JA, Pepek JM, Wagner S, Wilson K, Flint J, Andersen RC, Tadaki D, Gage FA, Stojadinovic A, Elster EA. Heterotopic ossification in high-energy wartime extremity injuries: prevalence and risk factors. Journal of Bone & Joint Surgery - American Volume. 2009;91:1084–1091. doi: 10.2106/JBJS.H.00792. [DOI] [PubMed] [Google Scholar]
- 4.Mohler ER, 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 5.Neal B, Gray H, MacMahon S, Dunn L. Incidence of heterotopic bone formation after major hip surgery. ANZ Journal of Surgery. 2002;72:808–821. doi: 10.1046/j.1445-2197.2002.02549.x. [DOI] [PubMed] [Google Scholar]
- 6.van Kuijk AA, Geurts ACH, van Kuppevelt HJM. Neurogenic heterotopic ossification in spinal cord injury. Spinal Cord. 2002;40:313–326. doi: 10.1038/sj.sc.3101309. [DOI] [PubMed] [Google Scholar]
- 7.Shore EM, Kaplan FS. Inherited human diseases of heterotopic bone formation. Nature Reviews Rheumatology. 2010;6:518–527. doi: 10.1038/nrrheum.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schindeler A, McDonald MM, Bokko P, Little DG. Bone remodeling during fracture repair: The cellular picture. Seminars in Cell & Developmental Biology. 2008;19:459–466. doi: 10.1016/j.semcdb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Shore EM, Ahn J, de Beur SJ, Li M, Xu MQ, Gardner RJM, Zasloff MA, Whyte MP, Levine MA, Kaplan FS. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. New England Journal of Medicine. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan FS, Glaser DL, Shore EM, Deirmengian G, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, et al. The phenotype of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:183–188. [Google Scholar]
- 11.Kaplan FS, Groppe JC, Seeman P, Pignolo RJ, Shore EM. Fibrodysplasia Ossificans Progressiva: Developmental Implications of a Novel Metamorphogene. In: Bronner F, Farach-Carson MC, Roach HI, editors. Bone and Development. London: Springer-Verlag; 2010. pp. 233–249. [Google Scholar]
- 12.Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:201–204. [Google Scholar]
- 13.Schroeder HW, Zasloff M. The hand and foot malformations in fibrodysplasia ossificans progressiva. Johns Hopkins Medical Journal. 1980;147:73–78. [PubMed] [Google Scholar]
- 14.Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, Delai P, Fastnacht-Urban E, Forman SJ, Gillessen-Kaesbach G, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Human Mutation. 2009;30:379–390. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM. Fibrodysplasia ossificans progressiva. Best Practice and Research Clinical Rheumatology. 2008;22:191–205. doi: 10.1016/j.berh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaser DL, Rocke DM, Kaplan FS. Catastrophic falls in patients who have fibrodysplasia ossificans progressiva. Clinical Orthopaedics & Related Research. 1998;346:110–116. [PubMed] [Google Scholar]
- 17.Janoff HB, Zasloff MA, Kaplan FS. Submandibular swelling in patients with fibrodysplasia ossificans progressiva. Otolaryngology - Head & Neck Surgery. 1996;114:599–604. doi: 10.1016/S0194-59989670253-X. [DOI] [PubMed] [Google Scholar]
- 18.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. Journal of Pediatrics. 1995;126:762–764. doi: 10.1016/s0022-3476(95)70408-6. [DOI] [PubMed] [Google Scholar]
- 19.Luchetti W, Cohen RB, Hahn GV, Rocke DM, Helpin M, Zasloff M, Kaplan FS. Severe restriction in jaw movement after routine injection of local anesthetic in patients who have fibrodysplasia ossificans progressiva. Oral Surgery Oral Medicine Oral Pathology Oral Radiology & Endodontics. 1996;81:21–25. doi: 10.1016/s1079-2104(96)80141-7. [DOI] [PubMed] [Google Scholar]
- 20.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 2004;423:275–279. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 21.Shore EM, Xu MQ, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 22.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 23.Guo X, Wang X-F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Research. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nature Reviews Molecular Cell Biology. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 26.Shore EM, Xu M, Connor JM, Kaplan FS. Mutations in the BMP type I receptor ACVR1 in patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2006;21:S75–S75. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 27.Katagiri T. Heterotopic bone formation induced by bone morphogenetic protein signaling: Fibrodysplasia ossificans progressiva. J Oral Biosci. 2010;52:33–41. [Google Scholar]
- 28.Hebela N, Shore EM, Kaplan FS. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:205–208. [Google Scholar]
- 29.Bragdon B, Moseychuk O, Saldanha S, King D, Julian J, Nohe A. Bone morphogenetic proteins: a critical review. Cellular Signaling. 2011;23:609–620. doi: 10.1016/j.cellsig.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, Noguchi Y, Iwakiri K, Kondo T, Kurose J, et al. Constitutively Activated ALK2 and Increased SMAD1/5 Cooperatively Induce Bone Morphogenetic Protein Signaling in Fibrodysplasia Ossificans Progressiva. Journal of Biological Chemistry. 2009;284:7149–7156. doi: 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, Mundlos S, Seemann P, Kaplan FS, Mullins MC, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. Journal of Clinical Investigation. 2009;119:3462–3472. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song GA, Kim HJ, Woo KM, Baek JH, Kim GS, Choi JY, Ryoo HM. Molecular Consequences of the ACVR1(R206H) Mutation of Fibrodysplasia Ossificans Progressiva. Journal of Biological Chemistry. 2010;285:22542–22553. doi: 10.1074/jbc.M109.094557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Dinther M, Visser N, de Gorter DJJ, Doorn J, Goumans MJ, de Boer J, ten Dijke P. ALK2 R206H Mutation Linked to Fibrodysplasia Ossificans Progressiva Confers Constitutive Activity to the BMP Type I Receptor and Sensitizes Mesenchymal Cells to BMP-Induced Osteoblast Differentiation and Bone Formation. Journal of Bone and Mineral Research. 2010;25:1208–1215. doi: 10.1359/jbmr.091110. [DOI] [PubMed] [Google Scholar]
- 34.Billings PC, Fiori JL, Bentwood JL, O’Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2008;23:305–313. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiori JL, Billings PC, Serrano de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 36.Serrano de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. Journal of Bone and Mineral Research. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 37.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. New England Journal of Medicine. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 38.Bauer H, Lele Z, Rauch GJ, Geisler R, Hammerschmidt M. The type I serine/threonine kinase receptor Alk8/Lost-a-fin is required for Bmp2b/7 signal transduction during dorsoventral patterning of the zebrafish embryo. Development. 2001;128:849–858. doi: 10.1242/dev.128.6.849. [DOI] [PubMed] [Google Scholar]
- 39.Mintzer KA, Lee MA, Runke G, Trout J, Whitman M, Mullins MC. Lost-a-fin encodes a type I BMP receptor, Alk8, acting maternally and zygotically in dorsoventral pattern formation. Development. 2001;128:859–869. doi: 10.1242/dev.128.6.859. [DOI] [PubMed] [Google Scholar]
- 40.Little SC, Mullins MC. BMP heterodimers assemble hetero-type I receptor complexes that pattern the DV axis. Nature Cell Biology. 2009;11:637–643. doi: 10.1038/ncb1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groppe JC, Wu J, Shore EM, Kaplan FS. In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs. 2011 doi: 10.1159/000324230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 43.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, et al. BMP type I receptor inhibition reduces heterotopic ossification. Nature Medicine. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. Journal of Bone & Mineral Research. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 45.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nature Cell Biology. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 46.Eivers E, Fuentealba LC, De Robertis EM. Integrating positional information at the level of Smad1/5/8. Current Opinion in Genetics & Development. 2008;18:304–310. doi: 10.1016/j.gde.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watabe T, Miyazono K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Research. 2009;19:103–115. doi: 10.1038/cr.2008.323. [DOI] [PubMed] [Google Scholar]
- 48.Wu MY, Hill CS. Tgf-beta superfamily signaling in embryonic development and homeostasis. Developmental Cell. 2009;16:329–343. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 49.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Research. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chuva de Sousa Lopes SM, Roelen BAJ, Monteiro RM, Emmens R, Lin HY, Li E, Lawson KA, Mummery CL. BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes & Development. 2004;18:1838–1849. doi: 10.1101/gad.294004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mechanisms of Development. 2004;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- 53.Komatsu Y, Scott G, Nagy A, Kaartinen V, Mishina Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Developmental Dynamics. 2007;236:512–517. doi: 10.1002/dvdy.21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rajagopal R, Dattilo LK, Kaartinen V, Deng C-X, Umans L, Zwijsen A, Roberts AB, Bottinger EP, Beebe DC. Functions of the type 1 BMP receptor Acvr1 (Alk2) in lens development: cell proliferation, terminal differentiation, and survival. Investigative Ophthalmology & Visual Science. 2008;49:4953–4960. doi: 10.1167/iovs.08-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajagopal R, Huang J, Dattilo LK, Kaartinen V, Mishina Y, Deng CX, Umans L, Zwijsen A, Roberts AB, Beebe DC. The type I BMP receptors, Bmpr1a and Acvr1, activate multiple signaling pathways to regulate lens formation. Developmental Biology. 2009;335:305–316. doi: 10.1016/j.ydbio.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 1998;346:19–25. [PubMed] [Google Scholar]
- 57.Glaser DL, Economides AN, Wang LL, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. Journal of Bone and Joint Surgery-American Volume. 2003;85A:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 58.Hegyi L, Gannon FH, Glaser DL, Shore EM, Kaplan FS, Shanahan CM. Stromal cells of fibrodysplasia ossificans progressiva lesions express smooth muscle lineage markers and the osteogenic transcription factor Runx2/Cbfa-1: clues to a vascular origin of heterotopic ossification? Journal of Pathology. 2003;201:141–148. doi: 10.1002/path.1413. [DOI] [PubMed] [Google Scholar]
- 59.Kaplan FS, Shore EM, Gupta R, Billings PC, Glaser DL, Pignolo RJDG, Kamoun M. Immunological features of fibrodysplasia ossificans progessiva and the dysregulated BMP4 pathway. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:189–193. [Google Scholar]
- 60.Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P. Oxygen in acute and chronic wound healing. British Journal of Dermatology. 2010;163:257–268. doi: 10.1111/j.1365-2133.2010.09804.x. [DOI] [PubMed] [Google Scholar]
- 61.Groppe JC, Shore EM, Kaplan FS. Functional Modeling of the ACVR1 (R206H) mutation in FOP. Clinical Orthopaedics and Related Research. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- 62.Olmsted-Davis E, Gannon FH, Ozen M, Ittmann MM, Gugala Z, Hipp JA, Moran KM, Fouletier-Dilling CM, Schumara-Martin S, Lindsey RW, et al. Hypoxic adipocytes pattern early heterotopic bone formation. Am J Pathol. 2007;170:620–632. doi: 10.2353/ajpath.2007.060692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang H, Shore EM, Kaplan FS, Groppe J, Pignolo RJ. Hypoxia Promotes Ligand- independent Activation of the ACVR1 (R206H) Mutant Receptor in C2C12 Cells. Journal of Bone and Mineral Research. 2008;23:S433–S433. [Google Scholar]
- 64.Kaplan FS, Tabas JA, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. Journal of Bone & Joint Surgery - American Volume. 1993;75:220–230. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 65.Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:195–200. [Google Scholar]
- 66.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–319. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joe AWB, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FMV. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biology. 2010;12:153–U144. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leblanc E, Trensz F, Haroun S, Drouin G, Bergeron E, Penton CM, Montanaro F, Roux S, Faucheux N, Grenier G. BMP9-induced muscle heterotopic ossification requires changes to the skeletal muscle microenvironment. Journal of Bone and Mineral Research. 2011 doi: 10.1002/jbmr.311. [DOI] [PubMed] [Google Scholar]
- 69.Zheng B, Cao BH, Li GH, Huard J. Mouse adipose-derived stem cells undergo multilineage differentiation in vitro but primarily osteogenic and chondrogenic differentiation in vivo. Tissue Engineering. 2006;12:1891–1901. doi: 10.1089/ten.2006.12.1891. [DOI] [PubMed] [Google Scholar]
- 70.Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment ADA, Shore EM, Glaser DL, Goldhamer DJ, Kaplan FS. Identification of progenitor cells that contribute to heterotopic skeletogenesis. Journal of Bone & Joint Surgery - American Volume. 2009;91:652–663. doi: 10.2106/JBJS.H.01177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nature Medicine. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22:138–147. doi: 10.1002/(SICI)1521-1878(200002)22:2<138::AID-BIES5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 73.Kornak U, Mundlos S. Genetic disorders of the skeleton: A developmental approach. American Journal of Human Genetics. 2003;73:447–474. doi: 10.1086/377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seemann P, Mundlos S, Lehmann K. Alterations of BMP signaling pathway(s) in skeletal diseases. In: Vukicevic S, Sampath KT, editors. Bone Morphogenetic Proteins: From Local to System Therapeutics. Basel/Switzerland: Birkhauser Verlag; 2008. pp. 141–159. [Google Scholar]
- 75.Minina E, Schneider S, Rosowski M, Lauster R, Vortkamp A. Expression of Fgf and Tgfbeta signaling related genes during embryonic endochondral ossification. Gene Expression Patterns. 2005;6:102–109. doi: 10.1016/j.modgep.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 76.Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128:4523–4534. doi: 10.1242/dev.128.22.4523. [DOI] [PubMed] [Google Scholar]