

Abstract
In the title compound, C14H13NO, the two rings show significant deviation from coplanarity, with a dihedral angle between the two planes of 49.40 (5)°. The hydroxy group is involved in an intermolecular O—H⋯N hydrogen bond, forming an extended one-dimensional zigzag chain along (001).
Related literature
For the applications of Schiff bases, see: Qian & Cui (2009 ▶). For related structures, see: Burgess et al. (1999 ▶); Kaitner & Pavlovic (1995 ▶); Li (2010 ▶); Li et al. (2008 ▶); Yeap et al. (1993 ▶); Zhang (2010 ▶). For bond geometry, see: Allen et al. (1987 ▶).
Experimental
Crystal data
C14H13NO
M r = 211.25
Orthorhombic,

a = 21.618 (1) Å
b = 11.0561 (6) Å
c = 9.3318 (5) Å
V = 2230.4 (2) Å3
Z = 8
Mo Kα radiation
μ = 0.08 mm−1
T = 296 K
0.30 × 0.20 × 0.20 mm
Data collection
Bruker Kappa APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 1999 ▶) T min = 0.977, T max = 0.984
11344 measured reflections
1961 independent reflections
1559 reflections with I > 2σ(I)
R int = 0.028
Refinement
R[F 2 > 2σ(F 2)] = 0.036
wR(F 2) = 0.100
S = 1.08
1961 reflections
148 parameters
H-atom parameters constrained
Δρmax = 0.19 e Å−3
Δρmin = −0.14 e Å−3
Data collection: APEX2 (Bruker, 2004 ▶); cell refinement: APEX2 and SAINT (Bruker, 2004 ▶); data reduction: SAINT and XPREP (Bruker, 2004 ▶); program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶); software used to prepare material for publication: PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812007635/zs2179sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812007635/zs2179Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812007635/zs2179Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1⋯N1i | 0.88 | 1.87 | 2.7397 (17) | 170 |
Symmetry code: (i) 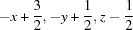 .
.
Acknowledgments
LJ thanks the Sophisticated Analytical Instrument Facility, IIT Madras, Chennai, for the single-crystal X-ray data collection.
supplementary crystallographic information
Comment
Schiff base compounds have attracted attention for the development of coordination chemistry related to catalysis and enzymatic reactions, magnetism and molecular architectures, e.g. (E)-2-methyl-N-[4-(methylsulfonyl)-benzylidene]aniline (Qian & Cui, 2009). As a part of our study on the coordination behaviour of ligands, an X-ray structural analysis of the title compound, C14H13NO (I) was carried out and the results are reported herein.
The molecule (I) (Fig. 1) may be described in terms of three planar subunits, namely two terminal benzene rings and their substituents bridged by a C═N imino moiety. The 4-hydroxybenzylidene system is nearly planar with r.m.s deviation of 0.0023 Å except for the hydroxy atom O1 which is 0.0183 Å out of the C9—C14 plane. The 4-methylbenzene system which is also essentially planar [r.m.s deviation, 0.0109 Å] except for the methyl atom C1 which is 0.0128Å out of the C2—C7 plane. The molecule has an E-configuration with respect to the C═N which is indicated by the torsion angle C9—C8—N1—C5 [-171.11 (13)°]. The twisting angles of the 4-hydroxybenzylidene and 4-methylbenzylidene groups with respect to the plane defined by the C5—N1—C8—C9 subunit [16.61 (15)° and 34.66 (10)°, respectively], are consistent with the general trend observed previously of aniline rings being more twisted than benzylidene rings, e.g. in 4-[(3-methoxyphenylimino)methyl]phenol [Yeap et al., 1993] and N-p-tolylvanillaldimine [Kaitner & Pavlovic, 1995] and in four N-(2-hydroxybenzylidene)aniline derivatives [Burgess et al., 1999]; 2-chloro-N-[4-(dimethylamino)benzylidene]aniline [Li et al., 2008); 4-bromo-N-[4-(diethylamino)benzylidene]aniline [Li, 2010]; (4-chloro-N-[4-(diethylamino)benzylidene]aniline [Zhang, 2010]. The C9—C8 and N1—C5 bond distances [1.451 (2) and 1.4221 (19) Å] confirm π-electron delocalization between the benzene rings, and the molecule can be regarded as a partially delocalized π-electron system as observed in related structures (Yeap et al., 1993; Kaitner & Pavlovic, 1995). In benzylideneaniline, where the phenyl ring has no substituents, the aromatic C—(Csp2), (Csp2) ═N and N—Car bond lengths of the azomethine portion are 1.496 (3), 1.237 (3) and 1.460 (3) Å, respectively (Kaitner & Pavlovic, 1995). If the terminal phenyl rings of benzylideneaniline have different substituents, the general pattern of two long and one short bond distance is not preserved. Contrary to this, the shortening of N—Car and aromatic C—(Csp2) [1.4221 (19) Å and 1.451 (2) Å. respectively] and the lengthening of N═(Csp2) [1.279 (2) Å] is observed in (I) and in similar structures (Yeap et al., 1993; Kaitner & Pavlovic, 1995). In (I), the two longer bonds are also shortened, while the shorter bond has lengthened, compared to the parent compound. The C2—C1 bond distance of 1.504 (2) Å is in good agreement with the aromatic C—(Csp3) bond lengths. Using a 3σ criterion, the lengths of O1—C12 [1.3496 (18) Å] is the same and fall into the range for the O—Car bond type. Expansion of the exocyclic angle O1—C12—C11 [123.45 (14)°] may be due to the steric interaction atoms H11 and H1 [H1···H1 = 2.3029 (1) Å]. The N1—C8—C9 [124.80 (14)°] is greater than the normal value of 120°. This might be a consequence of repulsion between the lone pair of electrons on N1 and H10 attached to C10 [N1···H10 = 2.6892 (1) Å]. All other bond lengths are within the expected ranges (Allen et al., 1987).
The crystal structure is stabilized by intermolecular hydroxy O—H···N hydrogen bonds (Table 1) linking the molecules into infinite one-dimensional chains extending along the c axis of the unit cell (Fig. 2).
Experimental
The title compound (I) was prepared by mixing equimolar quantities (10 mmol) of 4-hydroxybenzaldehyde and 4-methylaniline in ethanol (40 ml). The reaction mixture was refluxed for about 6 h and the resulting solution was allowed to slowly evaporate at room temperature. After three days colourless single crystals of the title compound, suitable for X-ray structure analysis were obtained.
Refinement
All of the H atoms were positioned geometrically and treated as riding on their parent atoms, with O—H = 0.88 Å, C—H = 0.93 Å (aromatic) or 0.96 Å (methyl), and refined using a riding model with Uiso(H) = 1.2Ueq(O or aromatic C) or 1.5Ueq(methyl C).
Figures
Fig. 1.

The molecular structure of the title compound showing atom numbering, with displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

A perspective view of the one-dimensional chain structure in the title compound showing O—H···N interactions as dashed lines. For symmetry code (i), see Table 1.
Crystal data
| C14H13NO | F(000) = 896 |
| Mr = 211.25 | Dx = 1.258 Mg m−3 |
| Orthorhombic, Pbcn | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2n 2ab | Cell parameters from 2333 reflections |
| a = 21.618 (1) Å | θ = 2.5–24.3° |
| b = 11.0561 (6) Å | µ = 0.08 mm−1 |
| c = 9.3318 (5) Å | T = 296 K |
| V = 2230.4 (2) Å3 | Needle, colourless |
| Z = 8 | 0.30 × 0.20 × 0.20 mm |
Data collection
| Bruker Kappa APEXII CCD diffractometer | 1961 independent reflections |
| Radiation source: fine-focus sealed tube | 1559 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.028 |
| ω and φ scans | θmax = 25.0°, θmin = 3.0° |
| Absorption correction: multi-scan (SADABS; Bruker, 1999) | h = −25→22 |
| Tmin = 0.977, Tmax = 0.984 | k = −13→13 |
| 11344 measured reflections | l = −9→11 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.036 | H-atom parameters constrained |
| wR(F2) = 0.100 | w = 1/[σ2(Fo2) + (0.0452P)2 + 0.5906P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.08 | (Δ/σ)max < 0.001 |
| 1961 reflections | Δρmax = 0.19 e Å−3 |
| 148 parameters | Δρmin = −0.14 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0028 (10) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| C1 | 0.98404 (9) | −0.3138 (2) | 0.5166 (2) | 0.0696 (6) | |
| H1A | 1.0178 | −0.2713 | 0.5605 | 0.104* | |
| H1B | 0.9999 | −0.3738 | 0.4519 | 0.104* | |
| H1C | 0.9597 | −0.3525 | 0.5894 | 0.104* | |
| C2 | 0.94434 (7) | −0.22573 (15) | 0.43514 (19) | 0.0463 (4) | |
| C3 | 0.93867 (8) | −0.10700 (16) | 0.47974 (19) | 0.0480 (4) | |
| H3 | 0.9599 | −0.0814 | 0.5610 | 0.058* | |
| C4 | 0.90213 (7) | −0.02566 (14) | 0.40601 (18) | 0.0424 (4) | |
| H4 | 0.9001 | 0.0544 | 0.4363 | 0.051* | |
| C5 | 0.86863 (7) | −0.06207 (13) | 0.28760 (16) | 0.0344 (4) | |
| C6 | 0.87510 (8) | −0.18013 (14) | 0.23975 (18) | 0.0425 (4) | |
| H6 | 0.8543 | −0.2055 | 0.1578 | 0.051* | |
| C7 | 0.91231 (8) | −0.25992 (15) | 0.31352 (19) | 0.0484 (5) | |
| H7 | 0.9160 | −0.3390 | 0.2805 | 0.058* | |
| C8 | 0.78057 (7) | −0.01218 (13) | 0.15734 (16) | 0.0364 (4) | |
| H8 | 0.7683 | −0.0915 | 0.1744 | 0.044* | |
| C9 | 0.74169 (7) | 0.06129 (13) | 0.06518 (16) | 0.0343 (4) | |
| C10 | 0.76120 (7) | 0.17174 (13) | 0.00844 (16) | 0.0348 (4) | |
| H10 | 0.7993 | 0.2033 | 0.0359 | 0.042* | |
| C11 | 0.72512 (7) | 0.23468 (12) | −0.08724 (16) | 0.0350 (4) | |
| H11 | 0.7389 | 0.3083 | −0.1234 | 0.042* | |
| C12 | 0.66836 (7) | 0.18910 (13) | −0.13011 (16) | 0.0344 (4) | |
| C13 | 0.64808 (7) | 0.07966 (13) | −0.07395 (18) | 0.0407 (4) | |
| H13 | 0.6099 | 0.0486 | −0.1011 | 0.049* | |
| C14 | 0.68428 (7) | 0.01724 (13) | 0.02146 (18) | 0.0402 (4) | |
| H14 | 0.6702 | −0.0561 | 0.0578 | 0.048* | |
| N1 | 0.83053 (6) | 0.02412 (11) | 0.21682 (13) | 0.0350 (3) | |
| O1 | 0.63156 (5) | 0.24423 (10) | −0.22698 (13) | 0.0457 (3) | |
| H1 | 0.6475 | 0.3149 | −0.2500 | 0.055* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0601 (12) | 0.0750 (14) | 0.0738 (14) | 0.0191 (11) | −0.0089 (11) | 0.0190 (11) |
| C2 | 0.0391 (9) | 0.0503 (10) | 0.0494 (10) | 0.0064 (7) | 0.0030 (8) | 0.0115 (8) |
| C3 | 0.0432 (9) | 0.0570 (11) | 0.0438 (10) | −0.0041 (8) | −0.0069 (8) | 0.0037 (8) |
| C4 | 0.0452 (9) | 0.0386 (9) | 0.0434 (9) | −0.0012 (7) | 0.0005 (8) | −0.0019 (7) |
| C5 | 0.0370 (8) | 0.0334 (8) | 0.0330 (8) | 0.0014 (6) | 0.0036 (7) | 0.0037 (6) |
| C6 | 0.0498 (9) | 0.0379 (9) | 0.0398 (9) | 0.0047 (7) | −0.0027 (7) | −0.0021 (7) |
| C7 | 0.0541 (10) | 0.0376 (9) | 0.0535 (11) | 0.0108 (8) | 0.0018 (9) | 0.0014 (8) |
| C8 | 0.0428 (8) | 0.0282 (8) | 0.0382 (9) | 0.0015 (6) | 0.0069 (7) | 0.0025 (6) |
| C9 | 0.0389 (8) | 0.0289 (7) | 0.0350 (8) | 0.0042 (6) | 0.0044 (7) | −0.0007 (6) |
| C10 | 0.0367 (8) | 0.0317 (8) | 0.0360 (9) | −0.0007 (6) | 0.0015 (7) | −0.0011 (6) |
| C11 | 0.0424 (8) | 0.0260 (7) | 0.0365 (9) | −0.0006 (6) | 0.0034 (7) | 0.0015 (6) |
| C12 | 0.0387 (8) | 0.0301 (8) | 0.0345 (9) | 0.0066 (6) | 0.0013 (7) | −0.0032 (6) |
| C13 | 0.0361 (8) | 0.0322 (8) | 0.0536 (11) | −0.0024 (7) | −0.0016 (8) | 0.0000 (7) |
| C14 | 0.0424 (9) | 0.0278 (8) | 0.0503 (10) | −0.0011 (6) | 0.0041 (8) | 0.0045 (7) |
| N1 | 0.0403 (7) | 0.0316 (7) | 0.0332 (7) | 0.0040 (5) | 0.0017 (6) | 0.0007 (5) |
| O1 | 0.0480 (7) | 0.0368 (6) | 0.0522 (7) | −0.0011 (5) | −0.0115 (6) | 0.0080 (5) |
Geometric parameters (Å, º)
| C1—C2 | 1.504 (2) | C8—N1 | 1.279 (2) |
| C1—H1A | 0.9600 | C8—C9 | 1.451 (2) |
| C1—H1B | 0.9600 | C8—H8 | 0.9300 |
| C1—H1C | 0.9600 | C9—C14 | 1.394 (2) |
| C2—C7 | 1.382 (2) | C9—C10 | 1.396 (2) |
| C2—C3 | 1.383 (2) | C10—C11 | 1.375 (2) |
| C3—C4 | 1.381 (2) | C10—H10 | 0.9300 |
| C3—H3 | 0.9300 | C11—C12 | 1.386 (2) |
| C4—C5 | 1.381 (2) | C11—H11 | 0.9300 |
| C4—H4 | 0.9300 | C12—O1 | 1.3496 (18) |
| C5—C6 | 1.387 (2) | C12—C13 | 1.390 (2) |
| C5—N1 | 1.4221 (19) | C13—C14 | 1.372 (2) |
| C6—C7 | 1.378 (2) | C13—H13 | 0.9300 |
| C6—H6 | 0.9300 | C14—H14 | 0.9300 |
| C7—H7 | 0.9300 | O1—H1 | 0.8811 |
| C2—C1—H1A | 109.5 | N1—C8—C9 | 124.80 (14) |
| C2—C1—H1B | 109.5 | N1—C8—H8 | 117.6 |
| H1A—C1—H1B | 109.5 | C9—C8—H8 | 117.6 |
| C2—C1—H1C | 109.5 | C14—C9—C10 | 117.61 (14) |
| H1A—C1—H1C | 109.5 | C14—C9—C8 | 119.57 (13) |
| H1B—C1—H1C | 109.5 | C10—C9—C8 | 122.63 (13) |
| C7—C2—C3 | 117.54 (15) | C11—C10—C9 | 121.17 (13) |
| C7—C2—C1 | 121.59 (17) | C11—C10—H10 | 119.4 |
| C3—C2—C1 | 120.87 (17) | C9—C10—H10 | 119.4 |
| C4—C3—C2 | 121.28 (16) | C10—C11—C12 | 120.39 (14) |
| C4—C3—H3 | 119.4 | C10—C11—H11 | 119.8 |
| C2—C3—H3 | 119.4 | C12—C11—H11 | 119.8 |
| C3—C4—C5 | 120.58 (15) | O1—C12—C11 | 123.45 (14) |
| C3—C4—H4 | 119.7 | O1—C12—C13 | 117.38 (13) |
| C5—C4—H4 | 119.7 | C11—C12—C13 | 119.16 (14) |
| C4—C5—C6 | 118.63 (14) | C14—C13—C12 | 120.19 (14) |
| C4—C5—N1 | 118.68 (13) | C14—C13—H13 | 119.9 |
| C6—C5—N1 | 122.66 (14) | C12—C13—H13 | 119.9 |
| C7—C6—C5 | 120.04 (16) | C13—C14—C9 | 121.47 (14) |
| C7—C6—H6 | 120.0 | C13—C14—H14 | 119.3 |
| C5—C6—H6 | 120.0 | C9—C14—H14 | 119.3 |
| C6—C7—C2 | 121.84 (16) | C8—N1—C5 | 118.71 (13) |
| C6—C7—H7 | 119.1 | C12—O1—H1 | 109.5 |
| C2—C7—H7 | 119.1 | ||
| C7—C2—C3—C4 | 0.3 (3) | C8—C9—C10—C11 | 174.95 (14) |
| C1—C2—C3—C4 | −179.72 (16) | C9—C10—C11—C12 | −0.3 (2) |
| C2—C3—C4—C5 | 2.0 (2) | C10—C11—C12—O1 | −177.82 (13) |
| C3—C4—C5—C6 | −3.5 (2) | C10—C11—C12—C13 | 0.7 (2) |
| C3—C4—C5—N1 | 178.62 (14) | O1—C12—C13—C14 | 177.90 (14) |
| C4—C5—C6—C7 | 2.7 (2) | C11—C12—C13—C14 | −0.7 (2) |
| N1—C5—C6—C7 | −179.48 (14) | C12—C13—C14—C9 | 0.3 (2) |
| C5—C6—C7—C2 | −0.4 (3) | C10—C9—C14—C13 | 0.1 (2) |
| C3—C2—C7—C6 | −1.1 (3) | C8—C9—C14—C13 | −175.12 (14) |
| C1—C2—C7—C6 | 178.95 (17) | C9—C8—N1—C5 | −171.11 (13) |
| N1—C8—C9—C14 | −171.69 (15) | C4—C5—N1—C8 | −147.79 (14) |
| N1—C8—C9—C10 | 13.4 (2) | C6—C5—N1—C8 | 34.4 (2) |
| C14—C9—C10—C11 | −0.1 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1···N1i | 0.88 | 1.87 | 2.7397 (17) | 170 |
Symmetry code: (i) −x+3/2, −y+1/2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: ZS2179).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L. & Orpen, A. G. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Bruker (1999). SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2004). APEX2, SAINT and XPREP Bruker AXS Inc., Madison, Wisconsin, USA.
- Burgess, J., Fawcett, J., Russell, D. R., Gilani, S. R. & Palma, V. (1999). Acta Cryst. C55, 1707–1710.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Kaitner, B. & Pavlovic, G. (1995). Acta Cryst. C51, 1875–1878.
- Li, X.-F. (2010). Acta Cryst. E66, o2417. [DOI] [PMC free article] [PubMed]
- Li, J., Liang, Z.-P. & Tai, X.-S. (2008). Acta Cryst. E64, o2319. [DOI] [PMC free article] [PubMed]
- Qian, S.-S. & Cui, H.-Y. (2009). Acta Cryst. E65, o3072. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Yeap, G.-Y., Teo, S.-B., Fun, H.-K. & Teoh, S.-G. (1993). Acta Cryst. C49, 1396–1398.
- Zhang, F.-G. (2010). Acta Cryst. E66, o382. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812007635/zs2179sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812007635/zs2179Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812007635/zs2179Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report