

Abstract
Reverse Phase Protein Microarray analysis was used to identify cell-signaling derangements in squamous cell carcinoma (SCC) compared to actinic keratosis (AK) and upper inner arm (UIA). We analyzed 2 independent tissue sets with isolation and enrichment of epithelial cells by Laser Capture Microdissection. Set 1 served as a pilot and a means to identify protein pathway activation alterations that could be further validated in a second independent set. Set 1 was comprised of 4 AK, 13 SCC, and 20 UIA. Set 2 included 15 AK, 9 SCC and 20 UIA. Activation of 51 signaling proteins known to be involved in tumorigenesis were assessed for set 1 and demonstrated the MEK-ERK pathway was activated in SCC compared to AK and UIA, that EGFR and mTOR pathways were aberrantly activated in SCC. Unsupervised two-way hierarchical clustering revealed that AK and UIA shared a common signaling network activation architecture while SCC was dramatically different. Statistical analysis found that pro-survival signaling through phosphorylation of ASK and 4EBP1 as well as increased Bax and Bak expression was higher in AK compared to UIA. We expanded pathway network activation mapping in Set 2 to 101 key signaling proteins, which corroborated activation of MEK-ERK, EGFR, and mTOR pathways through discovery of a number of upstream, and downstream signaling molecules within these pathways to conclude that SCC is indeed a pathway activation-driven disease. Pathway activation mapping of SCC compared to AK revealed several interconnected networks that could be targeted with drug therapy for potential chemoprevention and therapeutic applications.
Keywords: protein microarrays, squamous cell carcinoma, actinic keratosis, melanoma/skin cancers, premalignant lesions, signal transduction, cell signaling, proteomics
Introduction
Skin cancer, which includes both melanoma and nonmelanoma (NMSC) cancers, represents a significant health problem. Skin cancer incidence is higher than all other cancers and the rate skin cancer is increasing in the United States and worldwide (1). NMSC represents the largest proportion of skin cancer diagnoses (~96%) in the US (2, 3) of which approximately 16% are squamous cell carcinomas (SCC). SCC involves the malignant transformation and proliferation of squamous cells, which are the most abundant cell type in the epidermis. SCC can demonstrate aggressive behavior as seen in advanced high risk lesions with increased risk for metastasis although this represents a small proportion (4, 5). The majority of SCCs occur on sun-exposed areas of the body and have been strongly associated with chronic sun exposure (6). Actinic keratosis (AK) is an intraepidermal malignancy that exists in a continuum with SCC although not all AK progress to SCC and the factors responsible for AK progression are largely unknown (2). The transformation rates of AK to SCC have been variably estimated to be between 0.025% and 16% per year (7, 8). In addition, AKs are an important risk factor for identifying those at increased risk of SCC (9). While mortality due to SCC is relatively low, it represents a significant public health burden making the study of SCC pathogenesis imperative (10). The 3-year cumulative risk of recurrence of SCC is at least ten times the rate compared with the incidence of first tumors of other types in a comparable general population (11, 12). From a cost and morbidity perspective, there is a need for a shift in current practices of applying primarily destructive methods for treatment of SCC and AK to interventions that can prevent or reverse these keratinocytic neoplasias (13).
The most significant molecular alteration found in AK and SCC is in the mutation of the p53 gene, which occurs early in the sequential development of chronically sun-damaged skin, AK, and SCC (14, 15). The subset of SCCs considered to be advanced have a higher rate of mortality and recurrence but there is a lack of understanding of the specific molecular mechanisms that lead to the development and progression of SCC (16).
Ultraviolet light (UV) has both acute and chronic effects on mouse and human skin (17). Although not all of these effects are known, it has been shown that acute UV irradiation leads to the activation of critical molecular targets and multiple signal transduction pathways that result in the induction of expression of specific genes that lead to skin cancer (18–20). Analysis of the functional activation protein network architecture altered by both acute and chronic exposure to ultraviolet irradiation represents a significant means of uncovering the signaling pathways responsible for progression to SCC. Molecular profiling using gene expression arrays provide evidence of altered gene expression at the messenger RNA level. However, gene expression studies lack the ability to show how these changes manifest at the protein level, and most critically how the post-translational modifications that drive and control cell signaling such as protein phosphorylation are modulated. Reverse Phase Protein Microarray (RPMA) is a technology platform that allows for the broad-scale and quantitative measurements of the activation/phosphorylation of dozens to hundreds of proteins at once from very small tissue samples. The elucidation and characterization of on-going protein signaling activation can complement and synergize with gene-based analysis to generate a better understanding of the underpinning biology of tumors (21).
This study uses RPMA to assess changes in phosphoprotein and total protein expression of these complex signaling pathways during the progression from normal skin to AK and SCC in a pilot and second independent sample set. We chose skin taken from the upper inner arm (UIA) since this region is likely exposed to substantially lower amounts of UV. Ultimately, identification of molecular targets essential to the solar UV light signal transduction pathways leading to SCC carcinogenesis will facilitate the evaluation of novel and more effective chemopreventive therapies to inhibit the development of cutaneous SCCs.
Materials and Methods
Patient Samples
Subjects, males and females aged 18 or older were recruited from the University of Arizona Medical Center Section of Dermatology, the Southern Arizona Veterans Affairs Health Care System (SAVAHCS), local dermatologist offices, and advertisements. Specimens were obtained with informed consent and the study was approved by the Institutional Review Board review of the University of Arizona and the SAVAHCS. All lesions were immediately divided and placed into 10% formalin or snap frozen in liquid nitrogen and frozen specimens were then maintained at −80° C until analysis. The diagnosis of AK and SCC was confirmed by study pathologists (AP and PS). Tissue samples were collected prospectively for studies related to the Skin Cancer Prevention program at the University of Arizona Cancer Center and approved for future use. Specimens were then retrospectively selected based on whether there was a frozen biopsy available with a prior histologic diagnosis of AK or SCC and a matched upper-inner arm sample (UIA). Advanced SCCs were categorized as having evidence of large size (≥2.0 cm) (16). The matched UIA was a 4 mm punch biopsy of the right upper inner arm and was handled in the same manner as described for the SCC and AK.
The first study set was comprised of 4 cases of AK, 6 cases of advanced SCC, 4 non-advanced low risk SCC and 20 UIA. The second independent study set included 15 AK, 5 advanced SCC, 4 non-advanced SCC and 20 UIA. Table 1 lists the demographic and pathologic characteristics of subjects and corresponding specimens. Table 1 also contains subject age, gender, tumor diameter, tumor depth, differentiation, site, presence of perineural invasion, whether the tumor was a primary or recurrent lesion, and sentinel lymph node status. The mean age for advanced SCC was 79.7 ± 3.2 years, for non-advanced SCC was 81.5 ± 4.4 years, and for AK was 69 ± 9.7 years for sample set 1. In sample set 2, the mean age was 77 ± 10.7 years, 67.8 ± 10.4 years, and 65 ± 7.44 years for advanced SCC, for non-advanced SCC, and AK, respectively. In sample set 1 males made up 83% (5/6) of advanced SCCs, 100% (4/4) non-advanced SCCs, and 100% (5/5) of subjects with AK. In sample set 2, 80% (4/5) of subjects with advanced SCCs, 100% (4/4) with non-advanced SCCs and 73% (11/15) of subjects with AK were males.
Table 1.
Patient Demographics
| A: Sample Set 1 SCCs | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Gender | Age | Diagnosis | Size (cm) | Depth Invasion (cm) | Differentiation State | Site | Perineural Invasion | Recurrent or Primary | Sentinel lymph node* | Adv/Not Adv |
| 3392 | F | 80 | Invasive SCC | 3.0 | 0.5 | Moderate – poor | Temple | No | Primary | No | Adv |
| 3552 | M | 79 | Invasive SCC | 3.0 | 1.9 | Poor | Scalp | Yes | Primary | 4/4 | Adv |
| 3393 | M | 79 | Invasive SCC | 2.5 | 0.8 | Well | Scalp | No | Recurrent | No | Adv |
| 3396 | M | 75 | Invasive SCC | 5.5 | 0.8 | Poor | Shoulder | Yes | Primary | No | Adv |
| 3395 | M | 85 | Invasive SCC | 3.0 | 1.2 | Moderate | Cheek | Yes | Recurrent | No | Adv |
| 3397 | M | 80 | Invasive SCC | 2.5 | 0.6 | Well | L Back of Hand | No | Primary | No | Adv |
| 3387 | M | 77 | SCC in situ | NA | NA | NA | NA | NA | NA | NA | Non-Adv |
| 3549 | M | 87 | Invasive SCC arising in an AK | 1.3 | 0.5 | Moderate | L Ear | No | Primary | No | Non-Adv |
| 3545 | M | 83 | Invasive SCC | 0.6 | 0.3 | Moderate | Chin | Yes | Primary | No | Non-Adv |
| 3402 | M | 79 | SCC in situ | NA | NA | Well | Forearm | No | NA | NA | Non-Adv* |
| B: Sample Set 2 SCCs | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Id | Gender | Age | Diagnosis | Size (cm) | Depth Invasion (cm) | Differentiation State | Site | Perineural Invasion | Recurrent or Primary | Sentinel lymph node* | Adv/Not Adv |
| 3408 | F | 86 | Invasive SCC | 3.0 | 0.6 | Moderate | R Thigh | No | Primary | No | Adv |
| 3544 | M | 79 | Invasive SCC | 2.2 | 0.8 | Well | L Forearm | Yes | Primary | No | Adv |
| 3542 | M | 59 | Invasive SCC | 3.5 | 0.7 | Moderate | Scalp | Yes | Primary | 0/1 | Adv |
| 3550 | M | 84 | Invasive SCC | 2.0 | 0.7 | Moderate – poor | Scalp | Yes | Primary | No | Adv |
| 3555 | M | 77 | Invasive SCC | 2.0 | 1.2 | Poor | R Scalp | No | Primary | 0/3 | Adv |
| 3383 | M | 61 | Invasive SCC | 1.7 | 0.4 | Well | Upper chest | No | Primary | No | Non-Adv |
| 3407 | M | 69 | Invasive SCC | 0.7 | 0.3 | Moderate | L Ear | No | Primary | No | Non-Adv |
| 3551 | M | 59 | Invasive SCC | 0.6 | 0.2 | Moderate | L Ear | No | Primary | No | Non-Adv |
| 3554 | M | 82 | Invasive SCC | 1 | 0.5 | Moderate | R Ear | Yes | Primary | No | Non-Adv |
No denotes that a sentinel lymph node was not done
Sample Preparation and Reverse Phase Protein Microarray
Highly enriched populations of approximately 6000 tumor epithelial cells were obtained from SCCs by Laser Capture Microdissection (LCM) using a PixCell IIa (Molecular Devices, Sunnyvale, CA). An equivalent amount of epithelium from AK and UIA samples was isolated by UV cutting laser microdissection using the Veritas (Molecular Devices, Sunnyvale, CA). Microdissected cells were lysed on the CapSure® Macro LCM Caps (Molecular Devices, Sunnyvale, CA) using with a 1:1 mixture of T-Per™ Tissue Protein Extraction Reagent (Pierce, Rockford, IL) and 2X Tris-Glycine SDS Sample Buffer (Invitrogen, Carlsbad, CA) containing 5% β-mercaptoethanol. Reverse Phase Protein Microarrays (RPMA) were prepared using an Aushon 2470 solid pin microarrayer (Aushon Biosystems, Billerca, MA). For sample set 1, the arrayer was outfitted with 350 μm pins, and for sample set 2, 185 μm pins were used. A series of positive and negative control lysates consisting of cell lines treated or untreated with compounds that cause broad phosphoprotein increases (e.g. pervanadate, calyculin) were also printed and slides were stored dessicated at −20°C prior to staining with antibody. For estimation of total protein amounts, selected arrays were stained with Sypro Ruby Protein Blot Stain (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and visualized on the NovaRay scanner (Alpha Innotech, San Leandro, CA). Printed slides were prepared for staining by treating with 1x Reblot (Chemicon, Temecula, CA) for 15 min, followed by 2×5 min washes with PBS. Slides were treated for at least 5 hours or overnight with blocking solution (1g I-block (Applied Biosystems, Bedford, MA), 0.5% Tween-20 in 500mL PBS) with constant rocking. Blocked arrays were stained with antibodies on an automated slide stainer (Dako North America, Inc., Carpinteria, CA) using the Catalyzed Signal Amplification System kit according to the manufacturer’s recommendation (CSA; Dako). Briefly, endogenous biotin was blocked for 10 minutes with the biotin blocking kit (Dako), followed by application of protein block for 5 minutes; primary antibodies were diluted in antibody diluent and incubated on slides for 30 minutes and biotinylated secondary antibodies were incubated for 15 minutes. Signal amplification involved incubation with a streptavidin-biotin-peroxidase complex provided in the CSA kit for 15 minutes, and amplification reagent (biotinyl-tyramide/hydrogen peroxide, streptavidin-peroxidase) for 15 minutes each. A signal was generated using streptavidin-conjugated IRDye680 (LI-COR Biosciences, Lincoln, NE). Slides were allowed to air dry following development. Slides containing sample set 1 were stained with a set of 51 antibodies against phospho-specific and total proteins, and slides containing sample set 2 were stained with a set of 101 antibodies. The proteins and phosphoproteins measured in the two sample sets are shown in a supplementary Table. All of sample set 1endpoints were repeated in set 2 with the exception of cleaved caspase 3 (D175), HIF-1α, pEGFR (Y992), pFOX01 (S256), pNFkB (S536), and pp90RSK (S380) due to antibody availability. All antibodies were subjected to extensive validation for single band, appropriate MW specificity by Western blot as well as phosphorylation specificity through the use of cell lysate controls (e.g. HeLa +/− pervanadate, Jurkat +/− Calyculin purchased from Cell Signaling as lysates).
Stained slides were scanned individually on the NovaRay scanner (Alpha Innotech) or the Vidar Scanner (Vidar Systems, Herndon, VA). The TIF images for antibody-stained slides and Sypro-stained slide images were analyzed using MicroVigene v2.9.9.9 (VigeneTech, Carlisle, MA, USA). Briefly, Microvigene performed spot finding, local background subtraction, replicate averaging and total protein normalization, producing a single value for each sample at each endpoint. All data was background subtracted (local and slide average), normalized to total protein, and all signal values produced for data analysis were at least 2 standard deviations above background.
Data and Statistical Analysis
Statistical analyses were performed on final RPMA intensity values obtained using SAS version 9 software (SAS Institute, Cary, NC). Initially, the distribution of variables was checked. If the distribution of variables for the analyzed groups was normal, a two-sample t-test was performed. If the variances of the groups were equal, two-sample t-test with a pooled variance procedure was used. Otherwise, two-sample t-test without a pooled variance procedure was adopted. For non-normally distributed variables, the Wilcoxon rank sum test was used. All significance levels were set at p < 0.05.
Pathway Mapping and Visualization
The “Pathways in Human Cancer” diagram (reproduced courtesy of Cell Signaling, Inc (46) along with the Google Maps Application Programming Interface (API) (47) to create a dynamic web application called Cscape (Cancer Landscape) was used for visualizing and navigating through RPMA generated data. Cell signaling analytes from the RPMA data were manually annotated to image coordinates using Adobe Dreamweaver. The application was written within a web application framework based off of the Perl CGI: Application module and hosted on a Red Hat Enterprise Linux Advanced Server using tan Apache HTTP Server. After data and parameters for the job (colors, values to use) were submitted to the server via a web form, JavaScript calls to the Google Maps API were returned in the result in order to place the appropriate markers on the image. The color legend and custom markers were generated using Perl’s Image:Magick module. The Perl modules mentioned are available on CPAN Comprehensive Perl Archive Network (48).
Results
Two independent sets of samples were analyzed by RPMA in this study. Table 1 provides the subjects age, gender, diameter of the tumor, tumor depth, differentiation state, anatomical site, presence of perineural invasion, whether the tumor was a primary or recurrent lesion, and sentinel lymph node status for sample sets 1 and 2. The original criteria for selecting and categorizing SCCs as advanced was tumor diameter, subsequent to this original selection the study pathologist (AP) evaluated each SCC for depth of invasion, level of differentiation, and perineural invasion all of which contribute to revised criteria for classifying an SCC as advanced (16). Sample set 1 was used to obtain pilot information and as a means to generate candidate pathway alterations that could be further validated in a second independent sample set with an expanded number of analytes. The 51 proteins and phosphoproteins were chosen based on knowledge of alterations seen in both mouse and human skin after UV irradiation and with SCC progression. Sample set 1 was comprised of 4 cases of AK, 5 cases of advanced SCC, 8 non-advanced SCC, and 20 UIAs, which included both matched and unmatched samples of UIA. In each case, snap frozen human skin biopsies were subjected to LCM to enrich for epidermal cells and to reduce contamination of surrounding normal material prior to RPMA analysis. This step was determined to be necessary as our group (22) found that in human breast cancer and colorectal cancer study sets LCM was necessary to determine accurate signaling data for tumor epithelium and stroma. The list of validated antibodies measured for both sample set 1 and 2 is shown in a supplementary Table. Multiplexed quantitative cell signaling analysis was performed using RPMA on the lysates of the captured material using these validated antibodies. Parametric and non-parametric statistical analysis of each signal pathway protein was performed between AK and advanced SCC samples from Study Set 1 revealing a number of significant differences (p <0.01). These statistically significant endpoints were then plotted using an unsupervised two-way hierarchical clustering heatmap (Figure 1). As seen in Figure 1, unsupervised two-way hierarchical clustering analysis revealed clear differences between advanced SCC (≥2 cm) and AK, and importantly, despite the fact that 51 endpoints were measured, those with statistically significant changes were all contained within a distinct EGFR-driven signaling network through both MAPK family signaling and the AKT-mTOR pathway. The signaling proteins shown in Figure 1 are cleaved caspase-9, (D330), phospho-ERK 1/2 (Thr202/Tyr204), phospho-EGFR (Tyr1173), phospho-MEK1/2 (Ser217/221), phospho-p38 MAP Kinase (Thr180/Tyr182), total MEK1/2, total PTEN, phospho-mTOR (Ser2448), phospho-mTOR (Ser2481), and phospho-NF-kappaB p65 (Ser536).
Figure 1.
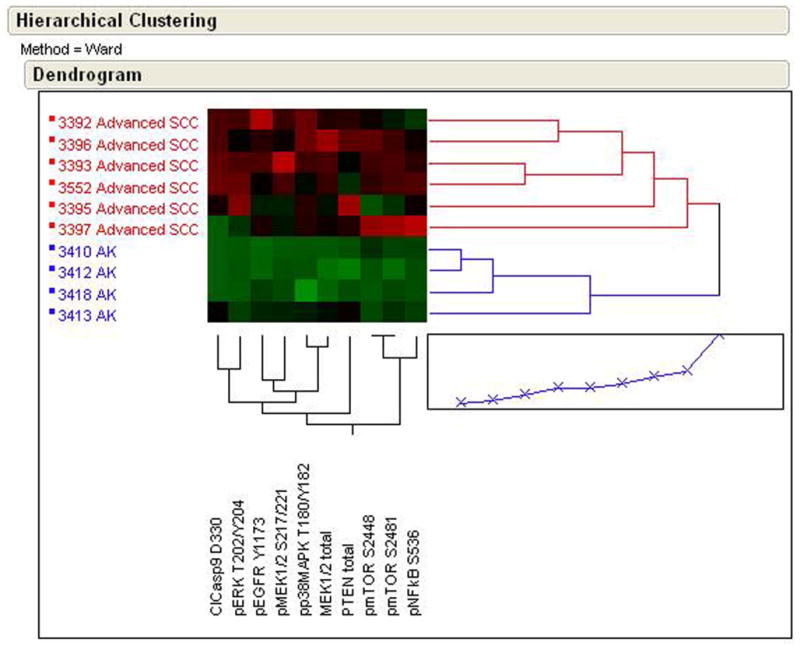
RPMA analysis of human skin biopsy samples containing AK or advanced SCC showing an unsupervised two-way hierarchical clustering heatmap. Samples with the highest relative level of phosphorylation are shown as red and lowest levels as green. Median values are black.
In a second comparison of sample set 1, unsupervised two-way hierarchical clustering analysis was performed on statistically significant endpoints between advanced SCC and non-advanced SCC (in situ SCC and superficially invasive), shown in Figure 2. Advanced SCC and non-advanced SCC could be discriminated using unsupervised two-way hierarchical clustering analysis for phospho-FAK (Tyr397), phospho-p38 MAPK (Thr202/Tyr204), HIF 1α, phospho-Chk1 (Ser345), phospho-Cofilin (Ser3), phospho-Bcl2 (Ser70), phospho-IRS1 (Ser612), and phospho-PDGFR (Tyr751). Again, despite the 51 endpoints measured, a significant number were part of the same EGFR signaling pathway. In a third comparison using unsupervised two-way hierarchical clustering analysis AK and UIA samples (data not shown) were found to be similar for those proteins and phosphoproteins measured.
Figure 2.

RPMA analysis of human skin biopsy samples containing advanced SCC or non-advanced SCC (in situ and lower risk SCCs) showing an unsupervised two-way hierarchical clustering heatmap. Samples with the highest relative level of phosphorylation are shown as red and lowest levels as green. Median values are black.
Study set 2 was used to both confirm and validate our pilot findings from study set 1 as well as to expand the number of signaling proteins to 101. This independent group of samples consisted of 15 AK, 5 advanced SCC, 4 non-advanced SCC and 20 UIA. Using this expanded group of proteins, unsupervised two-way hierarchical clustering of UIA, AK, non-advanced SCC, and advanced SCC (Figure 3) revealed significant pathway activation in all SCC samples compared to AK, as well as differences between non-advanced SCC and advanced SCC. Moreover, the clustering highlighted the overarching similarity in the signaling architecture of UIA and AK as demonstrated in the co-mingled clustering of AK and UIA in Figure 3. Again, despite the large number of analytes measured, we saw activation of the same key signaling proteins from sample set 1 in the advanced and non-advanced SCC that included phospho-p38, MEK1/2, PTEN, EGFR and phospho-mTOR. Statistical analysis of advanced and non-advanced SCC compared to AK (Table 2) revealed a rich archive of pathway activation differences. Finally, statistical analysis also revealed that pro-survival signaling through phosphorylation of ASK, and 4EBP1 as well as increased Bax and Bak expression were higher in AK compared to UIA (Table 2).
Figure 3.

Unsupervised clustering of RPMA pathway activation data from study set 2 showing AK, UIA, non-advanced SCC and advanced SCC samples. Samples with the highest relative level of phosphorylation are shown as red and lowest levels as green. Median values are black.
Table 2.
Statistically Significant Endpoints from Sample Set 2
| Increased in SCC p <0.01* | Increased in AK p<0.01* |
|---|---|
| Bak | EstrRec a total |
| Bax | p4EBP1 S65 |
| ClCasp3 D175 | pAkt S473 |
| ClCasp9 D330 | pAkt T308 |
| ErbB2 total | pALK Y1586 |
| MEK1/2 total | pcKit Y703 |
| p4EBP1 T70 | pcKit Y719 |
| pAdducin S662 | pBcl2 S70 |
| pAMPKa S485 | pErbB2 Y1248 |
| pAMPKb S108 | pFKHRL1 S253 |
| pARaf S299 | pIGF1R Y1135/36 - IR Y1150/51 |
| pATF2 T71 | pIkBa S32/36 |
| pATP/CitrateLyase S454 | pLKB1 S334 |
| pAuroraA/B/C T288/232/198 | pMSK1 S360 |
| pBad S112 | pPKCd T505 |
| pBad S136 | pProgRec S190 |
| pBad S155 | pStat1 Y701 |
| pBRaf S445 | pTyk2 Y1054/1055 |
| pcAbl T735 | |
| pcAbl Y245 | |
| pCatenin B S33/35/T41 | |
| pChk1 S345 | |
| pcRaf S338 | |
| pCREB S133 | |
| pCrkL Y207 | |
| pEGFR Y1068 | |
| pEGFR Y1173 | |
| peIF4G S1108 | |
| peNOS S113 | |
| peNOS S1177 | |
| pErbB3 total | |
| pErbB3 Y1289 | |
| pERK T202/Y204 | |
| pEzrin/Radixin/Moesin T567/564 | |
| pFADD S194 | |
| pFAK Y576/77 | |
| pFKHR/RL1 T24/32 | |
| pFKHRL1 S253 | |
| pGSK3a/B S21/9 | |
| pHistoneH3 S10 | |
| pMARCKs S152/156 | |
| pMEK1/2 S217/221 | |
| pMet Y1234/5 | |
| pmTOR S2448 | |
| pmTOR S2481 | |
| pp27 T187 | |
| pp38MAPK T180/Y182 | |
| pPDGFRb Y751 | |
| pPDK1 S241 | |
| pPKA C T197 | |
| pPKCa S657 | |
| pPKCth T538 | |
| pPKCz/l T410/403 | |
| pPLK1 T210 | |
| pPRAS40 T246 | |
| pPTEN S380 | |
| pRaf S259 | |
| pRasGRF1 S916 | |
| pS6RiboProt S235/236 | |
| pSmad2 S465/467 | |
| pSrc Y527 | |
| pStat3 S727 | |
| PTEN total | |
| pVEGFR2 Y996 | |
| XIAP total |
Compared to normal upper-inner arm (UIA)
We next utilized a novel pathway visualization tool called CScape, or Cancer Landscape, as a means to take RPMA data directly and map the expression/activation changes onto a network image so that pathway activation portraits can be easily generated, and network connections clearly revealed. As seen in Figure 4, CScape pathway activation mapping of those proteins showing the statistically significant differences between AK and all SCCs combined (advanced and non-advanced SCCs) from Study Set 2 revealed that the protein phosphorylation alterations found from biochemically linked networks indicate specific pathway activation changes between AK and all SCCs. The AMPK-AKT-mTOR and RAS-RAF-ERK pathways are highlighted as examples of systemic pathway activation in SCC. A Cancer Landscape (CScape) Protein Pathway Activation Map (Figure 4, Top left) is shown using population averages of RPMA data from individuals with AK and SCC. Higher fold differences in the SCC group are shown in increasing shades of red while higher fold differences for the AK group are shown in green. Each balloon pin is placed over the protein measured. Enlargements (middle top and bottom, Figure 4) of the AMPK-AKT-mTOR and RAS-RAF-ERK pathways are again highlighted with a green box and a red box, respectively. Linked members from each pathway are numbered and the individual protein data are plotted as scatter plots for the statistically significant endpoints. The scatter plots for the AMPK-AKT-mTOR pathway (far right of Figure 4) include phospho-AMPKα (Ser485), phospho-AMPKβ Ser108), phospho-S6Ribosomal Protein (Ser235/236), phospho-mTOR (Ser2448, 2481), phospho-4EBP1 (Thr70), phospho-eIF4G (Ser1108), and phospho-GSKα/β (Ser219) for both AK and all SCCs. Similarly, the RAS-RAF-ERK pathway scatter plots (Figure 4, bottom left) include phospho-BRAF (Ser445), phospho-MEK1/2 (Ser217/221), phospho-ERK (Tr202/Ty204), and phospho-Creb (Ser133) for AK compared to all SCCs.
Figure 4.

Protein Pathway Activation Map of SCC. A Cancer Landscape (CScape) Protein Pathway Activation Map (4 Top left) is shown using population averages of RPMA data from patients with AK and SCC. Higher fold differences in the SCC group are shown in increasing shades of red while higher fold differences in signaling in the AK group are shown in green. Each balloon pin is placed over the protein measured. Magnified views of the AMPK-mTOR pathway (green box, middle top) and RAS-RAF-ERK pathway (red box, middle bottom) are shown to reveal pathway detail. Linked pathway members from each pathway are numbered and the individual protein data are plotted as scatter plots for the statistically significant endpoints.
DISCUSSION
In this study, two independent sample sets of normal appearing skin, AK, non-advanced SCC, and advanced SCC were analyzed using RPMA as a means to interrogate the activation of selected kinase pathways in the progression of cutaneous SCC. Protein microarrays represent a rapidly emerging field and the RPMA format specifically was originated to provide for broad-scale mapping of signaling proteins from microscopic quantities of cells of interest. A direct analysis of the protein signaling architecture generates functional information about the protein pathways compared genomic or RNA expression profiling techniques (22) and can provide key insights into the activation state of key therapeutic targets (23).
While the signalling analysis performed in this study included a broad-scale analysis of the activation/phosphorylation state of key signalling proteins we did not measure the activation state of every protein. Indeed, like any immunoassay, the arrays are limited by the availability of well performing highly specific antibodies. However, based on our initial findings, we expect that even further coverage of the signal transduction machinery will yield new discoveries. We chose to focus our analysis on key signalling hubs known to integrate information about known tumorigenic processes such as inflammation, survival, energy metabolism, growth and differentiation. Furthermore, while signalling can be regulated by a number of post-translational modification driven events (e.g. glycosylation, acetylation, etc.), we chose to measure protein phosphorylation, the principal regulator of signal transduction and the key analyte endpoint for the recording of on-going cellular kinase activity, so that we could generate a direct knowledge snapshot of the on-going signalling cascades within the tumor cells.
The first sample set was used to obtain pilot information and as a means to generate candidate pathway alterations that could be further validated in a second independent sample set with an expanded number of analytes. In study set 2, we confirmed results from study set 1 that showed activation of a number of key proteins involved in MEK-ERK, EGFR, and mTOR in SCC compared to AK, by evaluating an expanded number of upstream and downstream signaling molecules within these pathways (Supplementary Table), as well as identified a number of new drug targets that revealed that SCC is a pathway activation-driven disease.
Our analysis revealed that there are indeed numerous overt pathway activation changes between AK, non-advanced SCC, and advanced SCC. We found systemic activation in advanced and non-advanced SCC compared to AK in many components of the RAS-RAF-ERK and AMPK-AKT-mTOR pathways. The observed increased phosphorylation of both subunits of AMPK in SCC compared to AK may initially appear to be contradictory to the known involvement of AMPK as a suppressor of tumor growth (24, 25). In the current study we observed increased phosphorylation of AMPK α at the Ser485 inhibitory site in SCC compared to normal epidermis. Inhibition at the Ser485 site can result in a downstream increase in mTOR signaling, which was indeed observed in SCCs. Interestingly, we also observed increased phosphorylation of AMPK at Ser108 on the β subunit in SCCs compared to AKs and UIA. Phosphorylation at the Ser108 site is thought to be required for AMPK activation (26) and would lead to downstream inactivation of mTOR. A potential explanation for these contrasting effects, described above, is the possibility that there are compensatory effects where one phosphorylation event activates downstream mTOR signaling and the other inhibits mTOR signaling. Despite these potential compensatory events we found mTOR signaling in SCCs to be highly activated. It must be pointed out that there are other pathways, such as the PI3K/AKT signaling pathway, that can lead directly to activation of mTOR downstream signaling, and these signaling events were indeed found to be elevated in SCC compared to AK.
Overall, our results are in keeping with the wealth of information generated from UV induced SCC carcinogenesis models, which have been primarily explored in mouse models. In totality, these UV induced SCC carcinogenesis models demonstrate the activation of abundant critical molecular targets and multiple signal transduction pathways that result in the induction of expression of specific genes that could lead to SCC (18–20, 27). UV is known to activate cell surface growth factor (i.e. EGFR) and cytokine (i.e. IL-1R) receptors which in turn lead to stimulation of mitogen-activated protein kinase (MAPK) signal transduction pathways and cellular responses like apoptosis, proliferation, inflammation, differentiation, and development (28, 29). Our group and others have demonstrated that the p38 MAPK pathway is activated after UV irradiation (17, 30–32). In addition, we have shown that activation of EGFR can lead to downstream activation of PI3K and AKT signaling pathways (40), which were also observed in this study.
The PI-3K-AKT signaling pathway, another important signaling pathway activated by UV (33–36), regulates the expression of cyclooxygenase 2 (COX-2) (37–39) which is transcriptionally regulated through altered phosphorylation of the cyclic AMP response element binding protein (CREB) (38, 39). In agreement with mouse studies, we found that UV-induced changes in human epidermis were seen in phospho-CREB, phospho-p38, phospho-GSK-3β, COX-2, and apoptosis (32). In another report, human skin exposed to low doses of UV activated the EGFR, the GTP-binding regulator protein p21RAS, and stimulates the MAPKs (ERKs, JNKs, and p38) all of which add to the strength of results from our study showing similar changes in cutaneous SCC (40). Another UV induced protein, mTOR, is regulated through activation of the PI-3K-AKT pathway and our findings reported herein reveal that this pathway and mTOR activation are significant components of human cutaneous SCC progression (41–43). These findings are in keeping with previous reports have shown that activation of this pathway is an important component in the development of human SCC (42–44).
There is limited information regarding expression of the majority of these signaling proteins in AK and SCC. Recently, Chen et al found that the percentage of phosphorylated AKT (Ser473) positive cells by immunohistochemistry was significantly higher in SCC than AK compared to normal skin and moreover was higher in SCCs with metastases. Results were similar for mTOR (Ser2448), 4EBP1 (Ser65), 70S6K1 (Thr421), p70S6K1 (Thr421 Ser424), and S6 (Ser6) (45).
In conclusion, while it is widely known that the majority of SCC proceed from an AK precursor lesion (2, 88), our pathway activation mapping study demonstrates dramatic alterations in the signaling architecture between AK and SCC in two independent (albeit small) sample sets. The results provide rationale to further study cutaneous SCC progression by functional pathway mapping techniques such as RPMA. Pathway activation portraits revealed distinct molecular derangements that are activated in a pathway-specific manner that transcend patient-patient heterogeneity and which can serve as an underpinning molecular rationale prospective clinical for selection and monitoring of efficacy of targeted inhibitors in prospective clinic. The signaling architecture of SCC (both advanced and non-advanced) was distinct from both AK and UIA. Signaling alterations that occur during the transition from AK to SCC were found and manifest in biochemically linked signaling networks.
Our finding that the signaling architecture of AK and normal upper inner arm skin are similar may be a factor of the reality that; 1) only a small proportion of AK progress to overt SCC (while the majority of SCCs arise from an existing AK), thus it is possible that few if any of AKs that we studied would actually have gone on to SCC, and 2) AKs appear to be a much more heterogeneous lesion than SCC; thus, we could postulate that the premalignant phenotype is a reflection of a natural selection “survival of the fittest” process where many different types of pathways are being turned on until the one that provides the necessary survival advantage is selected for and the SCC is the emergent, homogeneously selected clone.
These linked pathways, such as RAS-RAF-ERK and AMPK-AKT-mTOR, contain a number of important targets for molecularly targeted therapeutics. The ability to broadly measure and map the signaling architecture of SCC from tissue biopsy samples could provide a new opportunity to develop a personalized medicine approach whereby activated/phosphorylated signaling proteins are used as a companion diagnostic biomarker for upfront patient selection and stratification for targeted chemoprevention and treatment strategies. In the future, such an approach for NMSCs such as SCC would be philosophically similar to what is now occurring for melanoma. Based on our findings, we have shown in a preliminary fashion that cutaneous SCC development is driven by pathway activation. Genetic alterations caused by exposure to UV are likely the primary cause of this aberrant pathway activation leading to carcinogenesis. The next step in this work is to validate the activation of key signaling pathways in a larger sample set of skin biopsies, and confirm these results using other methods of phospho-protein expression analysis. If the RPMA technique proves to be a valid technique for determining activation (or de-activation) of key signaling pathways, we plan to use this as a prospective tool for selection of targeted inhibitors that can be used in combination for a true individualized approach to drug selection both in prevention of advanced disease in high-risk groups, and potentially in treatment of AK and SCC. This powerful technique can also be adapted to other epithelial tumor types in a similar manner in order to combat aberrant signaling network activation, as opposed to selection of therapy based only on over-expression (or under-expression) of single proteins and genes.
Supplementary Material
Acknowledgments
The authors appreciate the generous support of the College of Life Sciences at George Mason University as well as the human volunteers who participated in this study. The Cscape (Cancer Landscape) Program used was developed by Eli Lilly, Inc (K Gallagher, S McAhren, L Stancato) and utilized collaboratively for RPMA data analysis.
Grant/funding Support: This work was supported, in part, by PHS Grants CA027502 and CA023074 from the National Cancer Institute, National Institutes of Health. This publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
Footnotes
Conflict of Interest: Drs. Petricoin and Liotta have a financial interest in Theranostics Health, Inc.
References
- 1.Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58:S129–32. doi: 10.1016/j.jaad.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 2.Harvey I, Frankel S, Marks R, Shalom D, Nolan-Farrell M. Non-melanoma skin cancer and solar keratoses II analytical results of the South Wales Skin Cancer Study. Br J Cancer. 1996;74:1308–12. doi: 10.1038/bjc.1996.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ACS. Cancer Facts and Figures 2011. Atlanta: American Cancer Society; 2011. [Google Scholar]
- 4.Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. Ca: a Cancer Journal for Clinicians. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 5.Moller R, Reymann F, Hou-Jensen K. Metastases in dermatological patients with squamous cell carcinoma. Archives of Dermatology. 1979;115:703–5. doi: 10.1001/archderm.1979.04010060011017. [DOI] [PubMed] [Google Scholar]
- 6.Kwa RE, Campana K, Moy RL. Biology of cutaneous squamous cell carcinoma.[comment] Journal of the American Academy of Dermatology. 1992;26:1–26. doi: 10.1016/0190-9622(92)70001-v. [DOI] [PubMed] [Google Scholar]
- 7.Marks R, Rennie G, Selwood TS. Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet. 1988;1:795–7. doi: 10.1016/s0140-6736(88)91658-3. [DOI] [PubMed] [Google Scholar]
- 8.Graham J. Precancerous lesions of the skin. Prim Care. 1976;2:699–716. [Google Scholar]
- 9.Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. J Am Acad Dermatol. 2000;42:4–7. doi: 10.1067/mjd.2000.103342. [DOI] [PubMed] [Google Scholar]
- 10.Linden KG. Screening and early detection of skin cancer. Curr Oncol Rep. 2004;6:491–6. doi: 10.1007/s11912-004-0082-6. [DOI] [PubMed] [Google Scholar]
- 11.Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Archives of Dermatology. 2000;136:1524–30. doi: 10.1001/archderm.136.12.1524. [DOI] [PubMed] [Google Scholar]
- 12.Frankel DH, Hanusa BH, Zitelli JA. New primary nonmelanoma skin cancer in patients with a history of squamous cell carcinoma of the skin. Implications and recommendations for follow-up. J Am Acad Dermatol. 1992;26:720–6. doi: 10.1016/0190-9622(92)70100-t. [DOI] [PubMed] [Google Scholar]
- 13.Warino L, Tusa M, Camacho F, Teuschler H, Fleischer AB, Jr, Feldman SR. Frequency and cost of actinic keratosis treatment. Dermatol Surg. 2006;32:1045–9. doi: 10.1111/j.1524-4725.2006.32228.x. [DOI] [PubMed] [Google Scholar]
- 14.Brash DE. Roles of the transcription factor p53 in keratinocyte carcinomas. Br J Dermatol. 2006;154 (Suppl 1):8–10. doi: 10.1111/j.1365-2133.2006.07230.x. [DOI] [PubMed] [Google Scholar]
- 15.Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol. 2007;224:241–8. doi: 10.1016/j.taap.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jennings L, Schmults CD. Management of high-risk cutaneous squamous cell carcinoma. J Clin Aesthet Dermatol. 2010;3:39–48. [PMC free article] [PubMed] [Google Scholar]
- 17.Pfundt R, van Vlijmen-Willems I, Bergers M, Wingens M, Cloin W, Schalkwijk J. In situ demonstration of phosphorylated c-jun and p38 MAP kinase in epidermal keratinocytes following ultraviolet B irradiation of human skin. J Pathol. 2001;193:248–55. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH780>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 18.Bender K, Gottlicher M, Whiteside S, Rahmsdorf HJ, Herrlich P. Sequential DNA damage-independent and -dependent activation of NF-kappaB by UV. Embo J. 1998;17:5170–81. doi: 10.1093/emboj/17.17.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-Blight signalling. Nat Rev Cancer. 2004;4:23–35. doi: 10.1038/nrc1253. [DOI] [PubMed] [Google Scholar]
- 20.Bode AM, Dong Z. Molecular and cellular targets. Mol Carcinog. 2006;45:422–30. doi: 10.1002/mc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rapkiewicz A, Espina V, Zujewski JA, Lebowitz PF, Filie A, Wulfkuhle J, et al. The needle in the haystack: application of breast fine-needle aspirate samples to quantitative protein microarray technology. Cancer. 2007;111:173–84. doi: 10.1002/cncr.22686. [DOI] [PubMed] [Google Scholar]
- 22.Wulfkuhle JD, Speer R, Pierobon M, Laird J, Espina V, Deng J, et al. Multiplexed cell signaling analysis of human breast cancer applications for personalized therapy. J Proteome Res. 2008;7:1508–17. doi: 10.1021/pr7008127. [DOI] [PubMed] [Google Scholar]
- 23.Petricoin EF, 3rd, Espina V, Araujo RP, Midura B, Yeung C, Wan X, et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007;67:3431–40. doi: 10.1158/0008-5472.CAN-06-1344. [DOI] [PubMed] [Google Scholar]
- 24.Horman SR, Svoboda P, Luning Prak ET. The potential regulation of L1 mobility by RNA interference. J Biomed Biotechnol. 2006;2006:32713. doi: 10.1155/JBB/2006/32713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kayampilly PP, Menon KM. Follicle-stimulating hormone inhibits adenosine 5′-monophosphate-activated protein kinase activation and promotes cell proliferation of primary granulosa cells in culture through an Akt-dependent pathway. Endocrinology. 2009;150:929–35. doi: 10.1210/en.2008-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warden SM, Richardson C, O’Donnell J, Jr, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001;354:275–83. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyrrell RM. UV activation of mammalian stress proteins. Exs. 1996;77:255–71. [PubMed] [Google Scholar]
- 28.Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19:3485–95. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher GJ, Talwar HS, Lin J, Lin P, McPhillips F, Wang Z, et al. Retinoic acid inhibits induction of c-Jun protein by ultraviolet radiation that occurs subsequent to activation of mitogen-activated protein kinase pathways in human skin in vivo. J Clin Invest. 1998;101:1432–40. doi: 10.1172/JCI2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper S, MacGowan J, Young MR, Colburn NH, Bowden GT. A dominant negative c-jun inhibits UVB induced skin tumor size and burden in an SKH-1 hairless mouse model. Mol Can Res. 2003 [PubMed] [Google Scholar]
- 31.Mahns A, Wolber R, Stab F, Klotz LO, Sies H. Contribution of UVB and UVA to UV-dependent stimulation of cyclooxygenase-2 expression in artificial epidermis. Photochem Photobiol Sci. 2004;3:257–62. doi: 10.1039/b309067a. [DOI] [PubMed] [Google Scholar]
- 32.Einspahr JG, Bowden GT, Alberts DS, McKenzie N, Saboda K, Warneke J, et al. Cross-validation of murine UV signal transduction pathways in human skin. Photochem Photobiol. 2008;84:463–76. doi: 10.1111/j.1751-1097.2007.00287.x. [DOI] [PubMed] [Google Scholar]
- 33.Gonzales M, Bowden GT. Ultraviolet B (UVB) induction of the c-fos promoter is mediated by phospho-cAMP response element binding protein (CREB) binding to CRE and c-fos activator protein 1 site (FAP1) cis elements. Gene. 2002;293:169–79. doi: 10.1016/s0378-1119(02)00723-0. [DOI] [PubMed] [Google Scholar]
- 34.Gonzales M, Bowden GT. The role of PI 3-kinase in the UVB-induced expression of c-fos. Oncogene. 2002;21:2721–8. doi: 10.1038/sj.onc.1205366. [DOI] [PubMed] [Google Scholar]
- 35.Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001;18:461–6. doi: 10.3892/ijo.18.3.461. [DOI] [PubMed] [Google Scholar]
- 36.Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol Cancer Res. 2005;3:90–9. doi: 10.1158/1541-7786.MCR-04-0065. [DOI] [PubMed] [Google Scholar]
- 37.Chen W, Tang Q, Gonzales MS, Bowden GT. Role of p38 MAP kinases and ERK in mediating ultraviolet-B induced cyclooxygenase-2 gene expression in human keratinocytes. Oncogene. 2001;20:3921–6. doi: 10.1038/sj.onc.1204530. [DOI] [PubMed] [Google Scholar]
- 38.Tang Q, Chen W, Gonzales MS, Finch J, Inoue H, Bowden GT. Role of cyclic AMP responsive element in the UVB induction of cyclooxygenase-2 transcription in human keratinocytes. Oncogene. 2001;20:5164–72. doi: 10.1038/sj.onc.1204667. [DOI] [PubMed] [Google Scholar]
- 39.Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3beta in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer Res. 2001;61:4329–32. [PubMed] [Google Scholar]
- 40.Fisher GJ, Voorhees JJ. Molecular mechanisms of photoaging and its prevention by retinoic acid: ultraviolet irradiation induces MAP kinase signal transduction cascades that induce Ap-1-regulated matrix metalloproteinases that degrade human skin in vivo. J Investig Dermatol Symp Proc. 1998;3:61–8. [PubMed] [Google Scholar]
- 41.Affara NI, Trempus CS, Schanbacher BL, Pei P, Mallery SR, Bauer JA, et al. Activation of Akt and mTOR in CD34+/K15+ keratinocyte stem cells and skin tumors during multi-stage mouse skin carcinogenesis. Anticancer Res. 2006;26:2805–20. [PubMed] [Google Scholar]
- 42.Segrelles C, Lu J, Hammann B, Santos M, Moral M, Cascallana JL, et al. Deregulated activity of Akt in epithelial basal cells induces spontaneous tumors and heightened sensitivity to skin carcinogenesis. Cancer Res. 2007;67:10879–88. doi: 10.1158/0008-5472.CAN-07-2564. [DOI] [PubMed] [Google Scholar]
- 43.Segrelles C, Moral M, Lara MF, Ruiz S, Santos M, Leis H, et al. Molecular determinants of Akt-induced keratinocyte transformation. Oncogene. 2006;25:1174–85. doi: 10.1038/sj.onc.1209155. [DOI] [PubMed] [Google Scholar]
- 44.Checkley LA, Rho O, Moore T, Hursting S, Digiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-o-tetradecanoylphorbol-13-acetate. Cancer Prevention Research. 2011;4:1011–20. doi: 10.1158/1940-6207.CAPR-10-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen SJ, Nakahara T, Takahara M, Kido M, Dugu L, Uchi H, et al. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br J Dermatol. 2009;160:442–5. doi: 10.1111/j.1365-2133.2008.08903.x. [DOI] [PubMed] [Google Scholar]
- 46.www.cellsignal.com
- 47.http://code.google.com/apis/maps
- 48.http://www.cpan.org
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.