

Abstract
The PcF Toxin Family (Pfam 09461) includes the characterized phytotoxic protein PcF from Phytophthora cactorum, as well as several predicted protein effectors from other Phytophthora species recently identified by comparative genomics. Here we provide first evidence that such ‘putatives’, recombinantly expressed in bacteria and purified to homogeneity, similarly to PcF, can trigger defense-related responses on tomato, that is leaf withering and phenylalanine ammonia lyase induction, although with various degrees of effectiveness. In addition, structural prediction by computer-aided homology modeling and subsequent structural/functional comparison after rational engineering of the disulfide-structured protein fold by site-directed mutagenesis, highlighted the surface-exposed conserved amino acid stretch SK(E/C)C as a possible structural determinant responsible for the differential phytotoxicity within this family of cognate protein effectors.
Keywords: oomycete-plant pathogenesis, PcF toxin family, PAL induction, leaf withering, homology modeling, site-directed mutagenesis
Introduction
Oomycete pathogens belonging to Phytophthora spp. are a phylogenetically distinct group of eukaryotic fungus-like microorganisms1 including several species worldwide notorious for their phytopathogenicity, such as the potato and tomato late blight agent P. infestans,2 the soybean root and stem rot agent P. sojae,3 and the sudden oak death pathogen P. ramorum.4 Such pathogens are able to colonize their host plants by reprogramming the molecular network of target cells through an array of secreted signaling proteins, generally termed ‘effectors’, functioning either in the apoplast or inside the plant cell after translocation from the pathogen.5–8 Such effectors are thought to possess both an intrinsic function, supporting pathogen physiology at various levels, and a dual extrinsic function in plant pathogenesis, that is either promoting infection or triggering plant defense or both.9–11 At the sequence level, they share a limited degree of similarity, both to each other and to other proteins in databases, and only a few conserved motifs clearly signifying function, apart from the N-terminal secretion signals. These include the RXLR motif that is required for translocation of secreted oomycete effectors inside the host cell cytoplasm, and appears to identify those effectors directed to host intracellular targets.2,5,7,8,10,12 As a result, the oomycete effectors are currently grouped under different classes based on their modulatory activity toward plant defense and immunity.6 Furthermore, the oomycete effector secretome comprises several uncharacterized proteins resulting from large-scale comparative genomics efforts aimed at elucidating the molecular mechanisms of Phytophthora pathogenicity.2,13–15 This number of ‘putatives’ calls for functional characterization and mechanistic understanding of their action, widely considered pivotal for basic and applied science.16
The PcF Toxin Family (Pfam 09461) represents one group of small secreted proteins from Phytophthora spp. It has been originally named after the protein effector PcF (Phytophthora cactorum-Fragaria), discovered by us based on its toxic effect on both tomato and strawberry, and purified from the culture filtrates of a P. cactorum strain isolated from infected strawberry.17 The other family members have been related to PcF mainly on the basis of their conserved sequences, cysteine patterns, and N-terminal secretory signals.14,18 They are encoded by polymorphic genes and identified by the acronym small cysteine-rich (SCR) followed by their residues number.6 At NCBI, three SCR91 gene products18,19 are currently annotated into the PcF's family, as well as 17 SCR74 isoforms20 and one SCR70, all from the pathogen P. infestans. More recently, several orthologues from P. sojae and P. ramorum have also been reported and preliminarily annotated at NCBI trace archive.14 Notably, the P. infestans SCR74 genes appear upregulated during pathogen infection of both tomato and potato, and extensively polymorphic as a result of evolutionary forces of diversifying selection related to host-pathogen coevolution.20 Overall, these hints suggest conserved protein folds and functions. To date, however, PcF remains the only characterized member within its family.
Mature PcF is a 52-residue protein monomer tightly-bound by three disulfide bridges, that is processed before secretion by removal of a N-terminal signal peptide and by a peculiar modification of one of its three Pro residues to 4-hydroxyproline.17,21 Like most apoplastic proteins, PcF is acidic and resistant to heat denaturation and proteolysis, though it does not appear to inhibit most common proteases in vitro.17 Its 3D structure recently solved by NMR unraveled a peculiar disulfide-stabilized helix-loop-helix fold with a negative surface spot, highly suggestive of electrostatic interaction with yet unidentified target(s).22 The protein behaves as a necrosis-inducing factor, that is it triggers HR symptoms of localized cell-death when applied on leaves of both strawberry and tomato.17 In tomato, it also triggers intracellular induction of the key defense-related enzyme PAL21 and other PR genes, as well as extracellular leakage of electrolytes (Orsomando & Ruggieri, unpublished). Overall these defense-related responses highlight a direct role of PcF in plant recognition and pathogenesis, but its molecular action has not been identified yet. Typical of most protein effectors, it also has no clear database relative that can be of help for prediction of its biochemical function.
This report focuses on the bacterial expression of individual representative members belonging to the PcF Toxin Family, and on their preliminary characterization, structure prediction by homology modeling, and functional comparison after site-directed mutagenesis, aimed at investigating on the structural determinants of biological activity.
Results
Bacterial over-expression and mutagenesis strategy
Deposited proteins of the different PcF Toxin Family members are all illustrated in Figure 1. Their sequence-based alignment reveals a broad conservation with respect to the first characterized member PcF, and allows their subclassification in subgroups including P. infestans species identified as SCR91 and SCR74. These two sub-groups respectively share at least 33 and 14 identical residues with the 52-residue mature PcF sequence. Of these, 10 residues in particular appear positionally-conserved within the whole family (Fig. 1, top), interestingly including all six SS-bridged cysteines of PcF recognized as essential for structuring its helix-loop-helix core domain.22 Notably, only the SCR74 species possess two extra cysteines and thus one presumed additional SS bridge, not shared by other family members20 (Fig. 1, bottom).
Figure 1.

Clustal W2 multiple sequence alignment of the different PcF Toxin family members. Top panel, alignment of Phytophthora cactorum PcF and other Phytophthora infestans putative protein effectors, identified as SCR70, SCR91, and SCR74. Only the first amino acid sequence of each family sub-group is shown integrally. For the other members only differing residues are shown, while dots (·) represent identical residues. Dashes represent alignment gaps. UniProtKB/TrEMBL accession numbers are indicated in parentheses, except for the SCR74 sequence currently not deposited. The signal peptides, experimentally verified only for PcF17 or predicted by SignalP (http://www.cbs.dtu.dk/services/SignalP/) for all other members, encompass the first 21 residues. Therefore, mature proteins range from the twenty-second to the last residue shown. Conservation refers to the whole family: same residues are denoted; colon (:) indicates strongly conserved residue; full stop (.) indicates weakly conserved residue. Arrows point to protein species selected for characterization in this work. Bottom panel, alignment of predicted mature recombinant isoforms corresponding to the PcF Toxin family members obtained in this work as described in Materials and Methods. Asterisks (*) indicate residues mutated with respect to wild-type proteins, that is Q1E in the three SCR74 species obtained, and C25E in the not deposited SCR74 species resulting from site-directed mutagenesis. (a), CNBr cleavage sites, resulting in conversion of Met to homoserine (Hs). Gray boxes, the six conserved Cys residues were SS bridged according to the disulfide connectivities of PcF.22 White boxes highlight the two additional cysteines, unique to SCR74 species, predicted to form a fourth disulfide bridge. Underlined, the conserved SK(E/C)C motif identified in this study.
To obtain the proteins of interest in a soluble recombinant form, they were expressed in pET32b as thioredoxin-fused proteins and then released from the thioredoxin carrier by selective cleavage at the N-terminal Met residue by CNBr digestion. The additional carrier fragments because of CNBr cleavage at internal Met residues were resolved by C18-HPLC as described in “Materials and Methods” section. In view of this procedure, in order to avoid undesired protein fragmentation by CNBr, the sequences of interest were selected within the aforementioned SCR91 and SCR74 sub-groups based on number and position of internal Met residues. In particular, two Met-deficient SCR74 species, identified as B10 and D5,20 plus one SCR91 species showing a Met residue nearby the C-terminus and thus predicted to be shortened by three residues after CNBr cleavage (Fig. 1), were chosen. A third SCR74 species, containing one internal Met positioned seven residues before C-terminus, was selected because its shortening by CNBr digestion allows concomitant removal of one of the two not conserved cysteines. To this construct, site-directed mutagenesis was subsequently applied to replace the second additional cysteine (Cys25) with the glutamate conserved at this position in both PcF and SCR91 (Fig. 1, bottom). This approach was devised to obtain a mutant protein, herein named SCR74-C25E, entirely missing one additional SS bridge and thus resembling more closely the remainder of family members. Besides, C-terminal truncation by CNBr in such mutant protein, as well as in the aforementioned SCR91 species, were predicted to have negligible impact on a native-like protein fold, since the corresponding region in PcF is highly unstructured.22 The molecular characteristics predicted for each selected recombinant member of the PcF Toxin Family are summarized in Table I.
Table I.
Predicted molecular characteristics of recombinant PcF Toxin Family members
| Recombinant plasmid | Expressed protein | CNBr-digestion | N. of AA | pIa | ɛ280nma (M−1 cm−1) | Average massb |
|---|---|---|---|---|---|---|
| pET32b/PcF | Trx-PcF | No | 212 | 5.08 | 17460 | 22863.7 |
| PcF | Yes | 52 | 4.40 | 3355 | 5606.2 | |
| Mature wild-type PcF | – | 52 | 4.40 | 3355 | 5622.1c | |
| pET32b/SCR91 | Trx-SCR91 | No | 230 | 5.26 | 18950 | 24847.9 |
| SCR91 | Yes | 67 | 4.56 | 4845 | 7204.9d | |
| Predicted mature wild-type SCR91 | – | 70 | 4.78 | 4845 | 7590.3 | |
| pET32b/SCR74-B10 | Trx-SCR74-B10 | No | 213 | 5.49 | 17585 | 22897.9 |
| SCR74-B10 | Yes | 53 | 5.55 | 3480 | 5640.4 | |
| Predicted mature wild-type SCR74-B10 | – | 53 | 6.69 | 3480 | 5639.4 | |
| pET32b/SCR74-D5 | Trx-SCR74-D5 | No | 213 | 5.58 | 17585 | 22913.9 |
| SCR74-D5 | Yes | 53 | 6.03 | 3480 | 5656.4 | |
| Predicted mature wild-type SCR74-D5 | – | 53 | 6.85 | 3480 | 5655.4 | |
| pET32b/SCR74-C25E | Trx-SCR74-C25E | No | 213 | 5.59 | 17460 | 23101.1 |
| SCR74-C25E | Yes | 46 | 5.41 | 3355 | 5136.7d |
Bacterially-expressed recombinant proteins from the indicated plasmid constructs are presented either as thioredoxin (Trx) fusion proteins or as species resulting from CNBr cleavage (see corresponding sequences in Fig. 1, bottom). The characteristics of their wild-type counterparts are also shown for comparison. Molecular characteristics were predicted from sequences by using
ProtParam (http://expasy.org/cgi-bin/protparam) and
PeptideMass (http://expasy.org/tools/peptide-mass.html), assuming that all pairs of Cys residues form cystines. Wild-type PcF mass value
takes into account proline-49 posttranslational hydroxylation.16 Mass values of C-terminally CNBr-cleaved protein isoforms
take into account the conversion of methionine in homoserine lactone.
In accordance with the present strategy, the protein effectors were subsequently obtained from over-expressing bacteria as detailed in “Materials and Methods” section. First, in-frame fusion proteins with a thioredoxin carrier had to be generated by using a pET32b-based expression system. This was needed to yield high levels of soluble recombinant proteins, since expressing thioredoxin-free proteins alone mainly yielded inclusion bodies (not shown). Subsequently, quick purification by His-tag affinity chromatography of carrier-fused proteins, as well as by C18-HPLC separation of the carrier-free protein species was obtained. Finally, the five recombinant protein species depicted in Figure 1, bottom panel, have been obtained in adequate amounts for subsequent characterization. Typical yields of this protocol (referred to 1 L crude bacterial culture) were: ∼60 mg carrier-fused protein after His-tag affinity chromatography, and ∼1.5 mg carrier-free protein after CNBr digestion and C18 HPLC purification (i.e., ∼10% protein recovery on a molar basis).
Structural and functional characterization
Overall, five protein species were characterized, that is a recombinant PcF isoform, two full-length SCR74 species, one SCR91 species missing three C-terminal residues, and the above described SCR74-C25E mutant. Figure 2 summarizes the quality controls of each final preparation: all five predicted purified proteins were correctly identified by SDS-PAGE, N-terminal automated Edman sequencing, and MALDI-MS analyses (cf. Fig. 2 with predictions in Table I and Fig. 1, bottom). The same analyses also demonstrated the absence of contaminating protein species. In detail, the different proteins N-termini were as predicted by SignalP bioinformatic processing, with both PcF and SCR91 recombinant isoforms exactly matching their wild-type counterparts. Conversely, the three recombinant SCR74 species showed one conservative amino acid substitution at the first residue, arising from the cloning design (Fig. 1, bottom). The experimental protein mass values obtained by MALDI-MS (Fig. 2) were in excellent agreement with the predicted values calculated assuming full cysteine bridging (less than 1 Da difference in all cases, Table I), indicating the absence, in each recombinant species, of free thiol groups as well as other protein modifications, for example proline hydroxylation. Further IS-MS analyses also revealed no mass shifts after alkylating treatment carried out without prior protein reduction (not shown), thus confirming that six and eight SS-bridged cysteines, respectively, were originally present in SCR91 and SCR74 species, and involved in the formation of the three or four expected S–S bridges. Hence, due to cysteine conservation within the family and the obligatory integrity of disulfides for PcF function,21,22 the disulfide linkages of PcF were taken as template constraints for structural prediction of PcF's homologs (Fig. 1, bottom).
Figure 2.
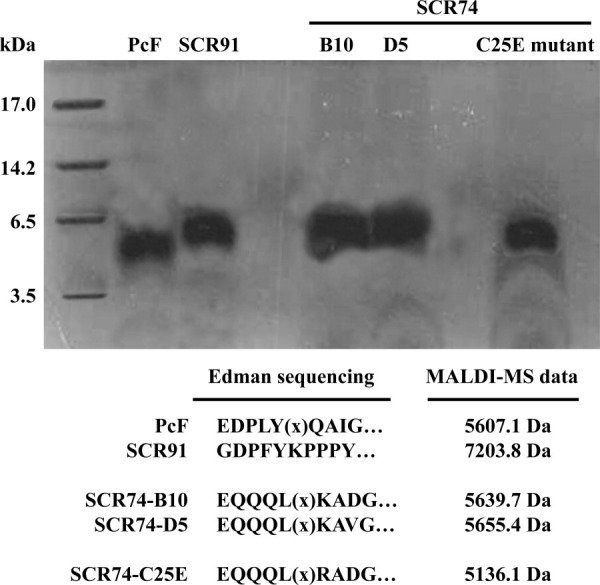
Tricine SDS-PAGE of final protein preparations (10 μg each) corresponding to wild-type and mutated PcF Toxin Family members. First lane, molecular size markers (Sigma M3546 ultra-low range): myoglobin (17 kDa), α-lactalbumin (14.2 kDa), aprotinin (6.5 kDa), and insulin, oxidized chain B (3.5 kDa). Bottom, summary of results from both automated Edman sequencing and MALDI-MS analyses. (x) indicate empty cycles corresponding to original Cys residues degraded by the Edman chemistry.
Subsequent bioassays were performed to test each recombinant protein effector for its ability to trigger both leaf withering and PAL induction in tomato seedlings. These functional tests were appropriately carried out using similar concentrations of each protein effector (10–18 μM range, Fig. 3 legend), that is 3- to 5-fold higher than previously reported PcF bioassays under similar conditions.21 As shown in Figure 3, all assayed members of the PcF Toxin Family induced on the plant similar morphological and biochemical symptoms with respect to their controls, and thus they appear to possess biological activity similar to PcF. In agreement with earlier observations,21 the appearance of apical withering on tomato leaflets was observed after ∼24 h treatment. Instead, the two authentic SCR74 species caused visible symptoms only after ∼48 h treatment (Fig. 3, top). Furthermore, PAL induction was observed in all specimens with comparable time-courses and similar peaking times at 2–3 h treatment, followed by a prolonged plateau of PAL activation. However, the extent of enzyme activity triggered by both authentic SCR74 species, with respect to the controls, was statistically significant (Student's t-test, P ≤ 0.068) and lower when compared to the other protein species (Fig. 3, bottom). The most striking feature was that such a functional behavior was lost when the disulfide-lacking SCR74-C25E mutant was used as the effector (Fig. 3). The gain-of-function observed after appropriate engineering of the protein to better imitate PcF's structural architecture, provided first hint that a minor structural change can dramatically affect the capability of perception/recognition by the target plant. This point was further addressed by homology modeling as detailed below.
Figure 3.
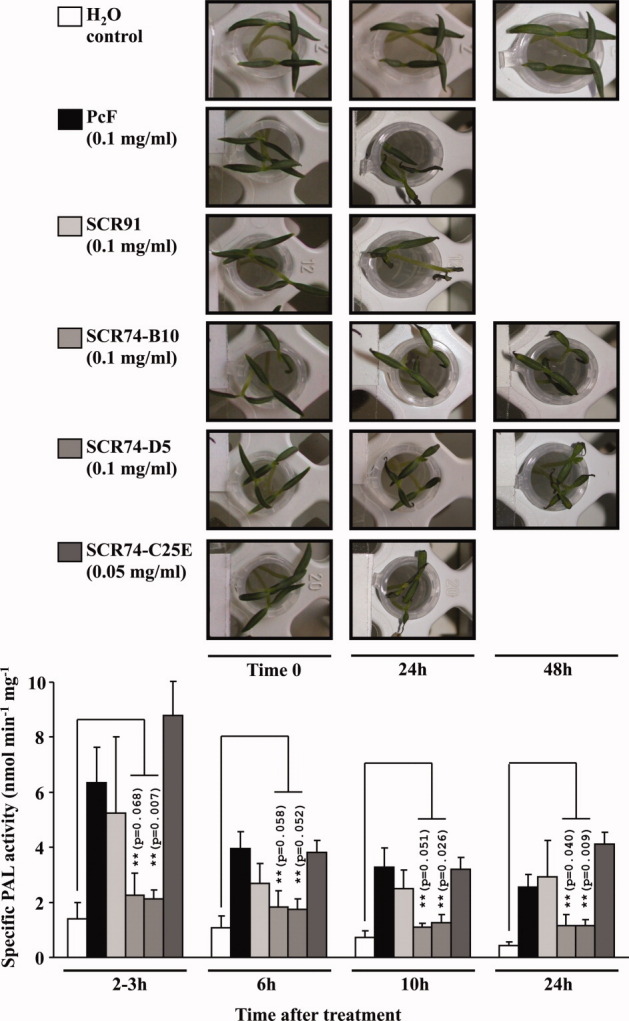
Morphological (top panel) and biochemical (bottom panel) stress symptoms triggered by the different selected members of the PcF Toxin Family on tomato seedlings (Lycopersicon esculentum var. Marmande). Bioassays were carried out in duplicate by dipping 10 days-old, root-excised tomato seedlings in eppendorf tubes containing either PcF (20 μg), or SCR91 (20 μg), or SCR74-B10 (20 μg), or SCR74-D5 (20 μg), or SCR74-C25E (10 μg) species dissolved in 200 μL distilled water. Controls were dipped in distilled water. At different times, the treated seedlings were observed for the appearance of leaf withering symptoms (top panel) and eventually collected and homogenized in liquid nitrogen to obtain crude protein extracts. The PAL specific activity was assayed in triplicate as described,21 and referred as mean value ± standard deviation for each sample (bottom panel histogram). (**), P values from Student's t-test (n = 3) comparing PAL induction by two authentic SCR74 species to the respective controls (P ≤ 0.02 in all other cases). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Homology modeling and structural comparison
Protein three-dimensional structures were predicted by automated web-based homology modeling and refined using molecular dynamics simulations. The resulting overall models of both authentic and mutated members selected among the PcF Toxin Family, that is species SCR91, SCR74-B10, and SCR74-C25E, are shown in Figure 4.
Figure 4.
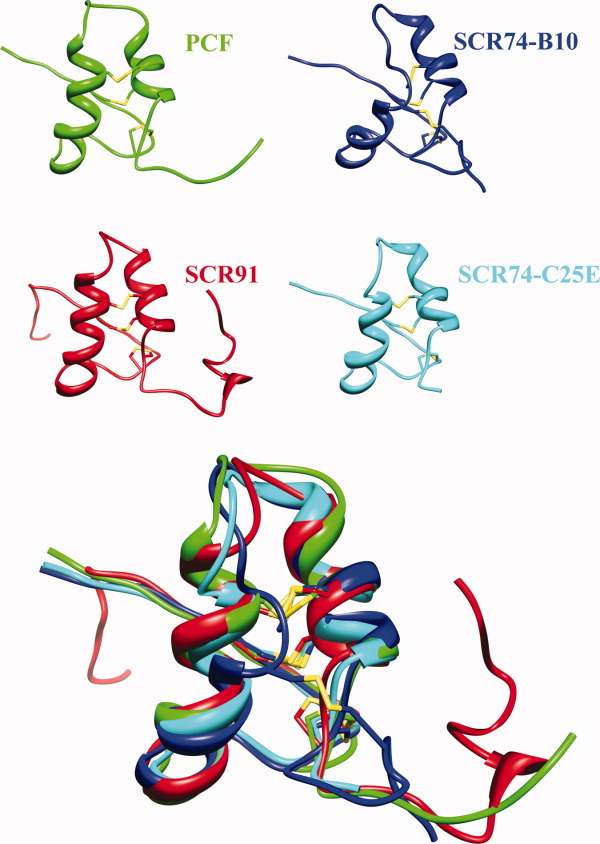
Ribbon representation of the NMR structure of PcF (PDB: 2BIC) together with the predicted models of SCR91, SCR74-B10, and SCR74-C25E species. All structures present N-terminus at the left. The disulfide bridges are depicted in yellow. Bottom panel, overlapped structures highlighting the local structural divergence of SCR74-B10 (blue) at the end of first helix. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
As expected, they share with PcF its all-alpha architecture with a helix-loop-helix core domain rigidly held in place by a peculiar disulfide bridging pattern, flanked by unstructured and flexible stretches at both N- and C-termini.22 The Ramachandran plots of refined model structures, as evaluated by PROCHECK, showed overall good quality. For SCR91 indeed 71.4% residues resulted in the most allowed regions, 25% in the additionally allowed regions, and 1.8% in the generously allowed and disallowed regions. SCR74-B10 showed 78.3% residues in the most allowed regions, 17.4% in the additionally allowed regions, and 2.2% in the generously allowed and disallowed regions. The mutant SCR74-C25E showed 71.7% residues in the most favored regions and 28.3% in the additionally allowed regions. Further analyses of RMSD (alpha carbon atoms), centers of mass, and number of hydrogen bonds formed as a function of time, showed great stability during the whole 2 ns simulation (not shown). Conversely, time-averaged RMSF analysis of the whole models, revealed some differential structural fluctuation. In fact, SCR91 appears fluctuating similarly to PcF,22 that is mainly at the level of its terminal ends, while the central structured protein core appears less flexible [Fig. 5(A)]. Instead, by comparing the RMSF profiles of SCR74-B10 and SCR74-C25E, a different fluctuation of residue 25 was observed, consistent with the fact that mutation appears to increase stability within the structured protein core [Fig. 5(B)]. Electrostatic potential surface areas are shown in Figure 6. The SCR91 species, again, resulted largely similar to PcF, showing the most extended patches of negative electrostatic potential clustered on one face of the helices plane [Fig. 6(A)]. In the same face, SCR74-B10 showed instead a predominant positive character, remarkably decreased in SCR74-C25E as a main consequence of the negatively charged point mutation introduced [Fig. 6(B,C)].
Figure 5.
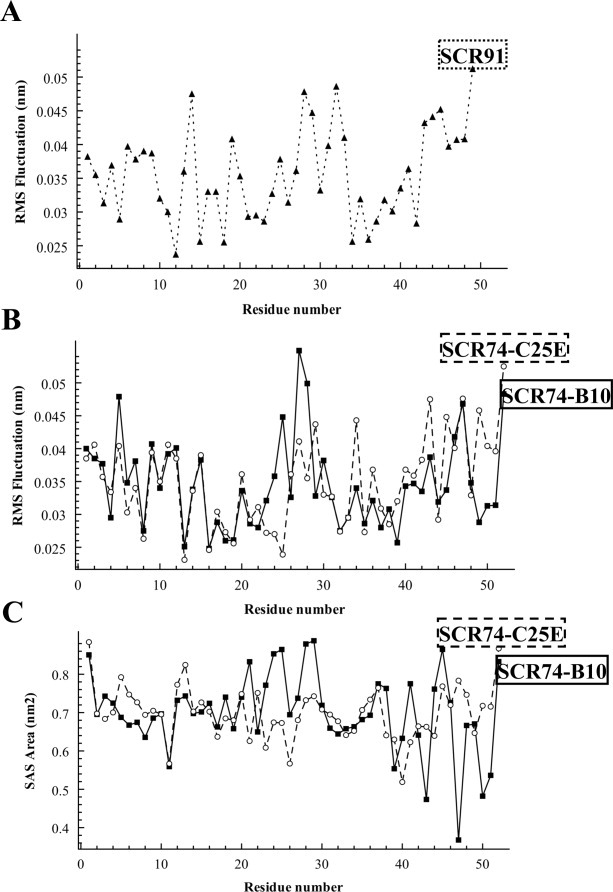
(A and B), Root Mean Square Fluctuation (RMSF) values, and (C) Solvent Accessible Surface (SAS) area of indicated protein models, all calculated per residue during the final 500 ps of molecular dynamics simulation.
Figure 6.
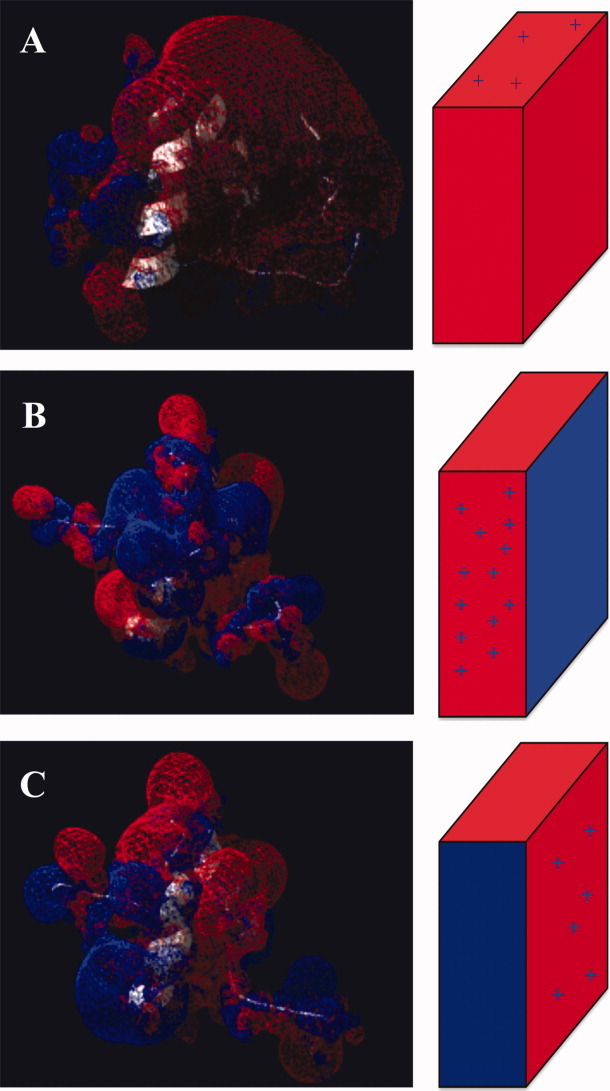
Electrostatic surface potential maps of (A) SCR91, (B) SCR74-B10, and (C) the SCR74-C25E mutant, calculated by solving the Poisson-Boltzmann equation through Swiss-PdbViewer. All protein molecules are shown in the same orientation. The sketch on the right side shows a simplified topological representation of charge distribution. Blue, indicates positively charged regions. Red, indicates negatively charged regions. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
By overviewing the structured elements in the three final models (Fig. 4, bottom), a relatively lower alignment of the helix-loop-helix domain of SCR74-B10 was observed with respect to both the PcF template and the other proteins. The STRIDE analysis23 confirmed such observation, highlighting a different secondary structure organization of the amino acid stretch forming the first alpha-helix. In particular, such a difference appears relevant by comparing authentic and modified SCR74 species: the mutation lengthens locally the alpha-helical arrangement, leading to a local fold clearly more similar to both PcF and SCR91 species [Fig. 7(A)]. Furthermore, DSSP analyses during the whole simulations revealed distinctive profiles for the three models, showing in particular fluctuation of residues forming the first helix of SCR74-B10 (Leu17-Cys26), unlike the corresponding residues of SCR74-C25E [Fig. 7(B)]. Focusing on the SCR74-B10 and SCR74-C25E models, analysis of the solvent accessible surface (SAS) also showed great variability in the region encompassing amino acid residues 20–30 [Fig. 5(C)], further highlighting the importance of residue 25 within this stretch.
Figure 7.
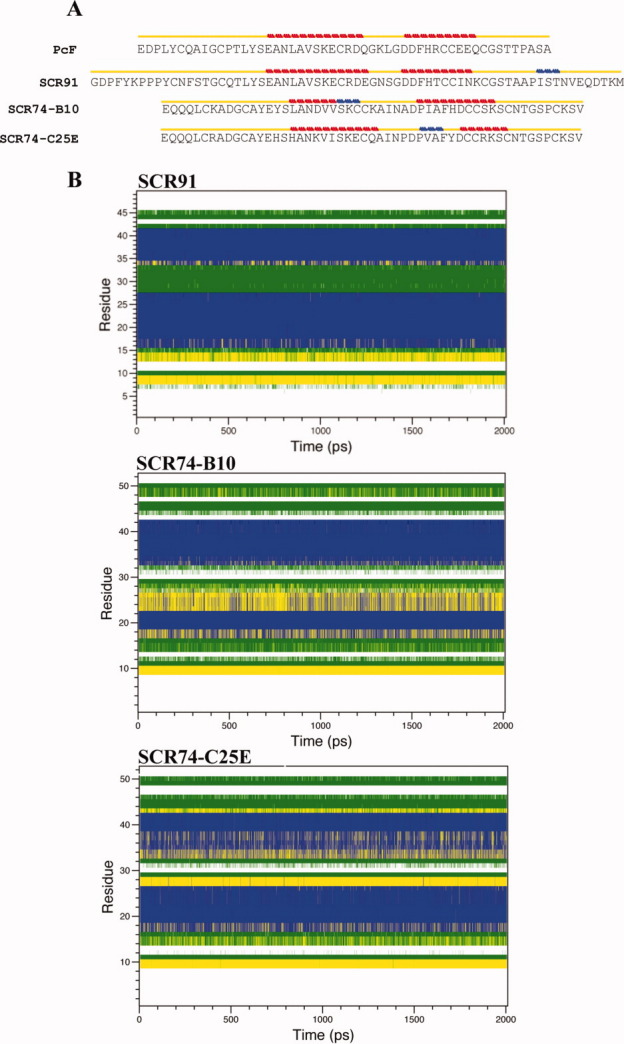
(A) Secondary structure-aided alignment of PcF, SCR91, SCR74-B10, and the SCR74-C25E mutant, performed by STRIDE program. Yellow line indicates a turn or a coil. Red line indicates an alpha-helix. Blue line indicates a 3–10 helix. (B) DSSP assignment of residues (mapped to the y-axis) during the evolution of a simulation (mapped to the x-axis) of SCR91, SCR74-B10, and SCR74-C25E. The different tertiary structure elements are color-coded as follows: coils in white, bends in green, turns in yellow, and alpha-helices in blue. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Discussion
The present report describes the structural and functional characterization of putative effectors belonging to the PcF Toxin Family (Pfam 09461), which includes members from relevant plant pathogenic oomycetes like P. infestans, P. sojae, and P. ramorum.2–4 Apart from PcF, all such proteins were uncharacterized until this study, and their bioactivity never experimentally verified before. We initially achieved their appropriate expression in bacteria, obtaining pure recombinant proteins with limited, if any, modifications with respect to their wild-type counterparts. In this regard, by improving the previously reported heterologs expression in yeast,21 the present bacterial PcF isoform shows no extra N-terminal residues. Like the yeast-overexpressed species, however, it lacks its naturally occurring proline-49 hydroxylation, apparently not relevant for bioactivity.21 Furthermore, we demonstrated the dual bioactivity towards tomato seedlings of all different members of the family, consisting on (i) the eliciting activity, that is the PAL enzyme activation, a typical defense reaction, and (ii) the toxic activity, that is the leaf withering. In this view, both authentic SCR91 and SCR74 species appear to function in a PcF-like manner, and thus their correct assignment into the PcF's family could be confirmed by the present study.
On the other hand, the observed effectiveness at inducing plant stress symptoms was not identical among the three different family sub-groups identified by primary structure alignments, whose representative members were PcF, one SCR91 and two polymorphic SCR74 species. Both last species in particular were shown to be less effective in either bioassay, as compared to the other members. This unpredicted finding drove subsequent structural studies in the attempt to find clues of this distinctive functional behavior. Indeed, average structural models of each PcF's homolog were extracted from molecular dynamics simulations and validated. Their overall predicted structures were found to be largely superimposable to PcF, comprising a similar disulfide-structured, tightly-bound helix-loop-helix domain, suitable for protecting the proteins in the harsh acidic and protease-rich apoplast environment where they are delivered during plant infection. As previously mentioned,22 such architectural fold is unusual across databases, thus likely representing a peculiar characteristic of this oomycete protein effectors family. Most remarkably, our computational study also allowed close inspection of slight structural differences among the PcF Toxin Family members, and in conjunction with functional comparison, revealed novel structural determinants of bioactivity. As a main result, a local structural divergence of SCR74s could be associated with their reduced triggering of plant stress symptoms that is the region of the structured protein core domain surrounding the Cys25 residue. This region, in fact, appears endowed with a relatively higher flexibility, a shorter helix structuring, and a positive character. Opposite structural features were found in the engineered mutant SCR74-C25E, consistent with its higher functional effectiveness, fully comparable to the remainder of family members. Such a gain-of-function mutation, therefore, substantiates the proposed structure/function relationship. Indeed, our mutagenesis experiments show that the relatively weaker functional effectiveness naturally exhibited by the SCR74 family members, can be significantly enhanced by a single amino acid replacement, yielding a locally enhanced alpha-helix structuring arising from the loss of one SS-bridge and concomitant rise of one negative charge. The differences at the level of predicted secondary and tertiary structures appear remarkable and consistent with the functional data: the stretches including the mutated residue 25 that is Cys25 in authentic SCR74 and Glu25 in mutated SCR74, show different arrangement at the end of the first helix, making the mutant protein structurally closer to PcF and SCR91 folds. The mismatching of this stretch, unaligned with respect to all other family members, is partly because of the constraint by the fourth disulfide bridge between Cys25 and Cys50, which is lost in the mutated form and naturally absent in all other family members. It seems like the fourth extra SS-bridge of SCR74 leads to relative local fold instability, thus negatively affecting its functional effectiveness towards the bioassayed plant.
On the other hand, the change of electrostatic potential consequent to the C25E mutation in SCR74 sequence can likewise be envisaged to be relevant for the function. Indeed, there is a dissimilar electrostatic potential distribution among the PcF Toxin Family members. Overall, they are acidic proteins with predicted Pi values ranging from 4.4 to 6.8, but the SCR74 species show a relatively higher positive potential character, clustered on the face that appears negative in the other proteins. Thus, also local increase of one negative charge in the engineered SCR74 mutant might explain its differential bioactivity as compared to the authentic SCR74 species, for example by favoring enhanced interaction capability with putative cationic targets, presently unknown. Nevertheless, on the basis of a “arms race” model, all PcF Toxin Family effectors have been tentatively classified as ‘candidate apoplastic effectors’ interacting with plant surface receptors.14,18,20 In keeping with this view, the same local spot of negative charges at the PcF surface has been previously proposed for interaction with a putative positively charged ligand, an early molecular event contributing to the observed effector selectivity.22
Altogether, these data converge to suggest that residue 25 and nearby amino acids form a surface-exposed structural determinant involved in the interaction with the plant target. Within this region, the included conserved motif SK(E/C)C (Fig. 1, bottom) might be proposed as a signature of bioactivity for the PcF Toxin Family.8,22 As confirmed by direct mutagenesis, the Glu25 residue included in the signature, present in both PcF and SCR91 sub-family members, supports stronger bioactivity in our bioassays as compared to SCR74, where it is replaced by a disulfide-forming Cys residue. PcF's orthologs from both P. sojae and P. ramorum appear to share most part of this signature (not shown) but do not exhibit the RXLR motif required for oomycete proteins translocation inside host plant cells.12 Even though, it appears not conserved in phylogenetically unrelated proteins, intriguingly, it is almost entirely conserved in the plant pollen allergen Ole e6 structure, where it is similarly localized at the end of the first α-helix and exposed at the protein surface, even in the absence of other sequence similarities (Fig. 8). Although, in both PcF and Ole e 6 the molecular details of their respective signaling function are still unknown, this localized structural similarity between a pathogen protein and a plant protein is indeed reminiscent of molecular mimicry.25,26
Figure 8.

Top panel, fold comparison between PcF (PDB: 2BIC) and the pollen allergen Ole e 6 from Olea europea (PDB: 1SS3). The structures present N-terminus at the left. The disulfide bridges are depicted in yellow. Bottom panel, structure-based sequence alignment performed by the SSM algorithm.24 Capital letters indicate alfa-helical regions. Bold letters indicate identical residues. Boxed, the structurally superimposed residues,22 as also depicted in the upper panel. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Further studies on the PcF Toxin Family members are needed to correlate their structural determinants to the biological activities expressed by either leaf toxicity or PAL elicitation. This knowledge might enable individual bioactivities modulation, for example by decreasing toxicity while enhancing defense elicitation, and better understanding of their mechanism of action and role in pathogenesis. Determination of 3D structures of the PcF Toxin Family members will hopefully enable a more refined analysis of structure/function relationships, and the efficient protocol for bacterial expression herein developed will help addressing this point.
Material and Methods
Materials
The P. infestans SCR91 clone MY-09-E-07 (GenBank AY961418; UniProtKB Q2M443) was provided by Dr. Francine Govers (Wageningen University, The Netherlands).19 The three SCR74 P. infestans clones corresponding to SCR74-B10 (GenBank AY723699-701; UniProtKB Q646W6), SCR74-D5 (GenBank AY723707-8; UniProtKB Q646V8), and to one not deposited polymorphic isoform herein referred as SCR74, were provided by Dr. Sophien Kamoun (The Sainsbury Laboratory, John Innes Centre, Norwich, UK).20 The E. coli strain BL21 and vector pET32b were from Novagen (San Diego, CA). The QuikChange™ Site-Directed Mutagenesis XL Kit was from Stratagene (La Jolla, CA). PD-10 columns were from GE Healthcare Life Sciences (Uppsala, Sweden). Other chemicals were from Sigma-Aldrich Corporation (St. Louis, MO).
Molecular cloning and bacterial expression
All protein effectors of interest in this study were cloned into plasmid pET32b, encoding the highly-soluble E. coli thioredoxin carrier27 fused in frame with the effector. Appropriate ORFs lacking predicted secretory signals were obtained by high-fidelity PCR amplification from each plasmid clone above, and directionally cloned in pET32b at NotI and EcoRI sites. The primer pairs used were (5′–3′ direction with restriction overhangs underlined) TATCCCATGGGGGACCCGTTCTAT and CGCAGAA TTCTTAGCTAAGATGCA for SCR91, and TATCCCA TGGAGCAGCAGCAGCTC and CGCAGAATTCTAG ACCGACTTGCATG for the three highly conserved SCR74 species. Likewise, mature PcF sequence was amplified from the vector pPIC9-PcF21 by the primers TATCCCATGGAGGACCCGCTGTAC and CG CAGAATTCTACGCGGAAGCTGG. The resulting recombinant plasmids pET32b/SCR91, pET32b/SCR74-B10, pET32b/SCR74-D5, and pET32b/PcF were sequence-verified28 and individually transformed into E. coli BL21 competent cells for protein expression. The recombinant plasmid pET32b/SCR74 was instead subjected to prior oligonucleotide-directed mutagenesis (see below). For bacterial expression, E. coli BL21 cells carrying each plasmid construct above were grown at 37°C into 1 L Luria-Bertani medium up to OD600 ∼0.6, then induced with 1 mM IPTG and harvested after 3 h incubation. Collected cells were resuspended in ∼20 mL ice-cold buffer A (50 mM Na-phosphate, pH 7.0, 0.5M NaCl, 1 mM PMSF), and homogenized twice using a French Press apparatus. His-tag affinity chromatography of each crude protein extract was carried out using a Qiagen Ni-NTA column (4 mL resin), equilibrated in buffer A, and then washed and eluted by imidazole as indicated by the manufacturer. Final protein preparations were desalted on PD-10 against 10 mM Tris/HCl, pH 7.2, and lyophilized. Yield and purification were evaluated by the Bio-Rad protein assay and SDS-PAGE,29 respectively.
CNBr cleavage and protein purification
Lyophilized, carrier-fused proteins, were resuspended at ∼7 mg/mL final concentration in 70% TFA in water containing 0.5M CNBr, and incubated in the dark for 3–5 h at room temperature. Digested mixtures were lyophilized, resuspended in distilled water, and injected in aliquots onto a C18 HPLC Supelco column (SupelcosilTM LC-18-T, 4.6 × 250 mm2, 5 μm, 300 Å), previously equilibrated at 1.3 mL/min in 0.1% TFA in water. Gradient elution was carried out by a linear increase of acetonitrile (0–90% in 50 mL) in the starting solvent. The chromatographic peak corresponding to each CNBr-released, carrier-free protein was identified by comparing its UV 230 nm elution profile with the profile of a control protein, obtained from the pET32b vector alone, digested in parallel. Collected fractions corresponding to not overlapping peaks were pooled, lyophilized, and stored at −20°C. The concentration of pure proteins was evaluated by UV280nm absorption using molar extinction coefficients calculated from their primary structures30 (Table I). Automated Edman sequencing was performed on a Procise Model 491 sequencer (Applied Biosystems), after protein immobilization on PVDF membranes (Millipore). MALDI-MS analyses (Omniflex, Bruker-Franzen Analytik, Bremen, Germany) were carried out on a sinapinic acid matrix, using a linear time-of-flight method.31 Cysteine alkylation and IS-MS analysis were carried out as described.17
Site-directed mutagenesis
Mutagenic PCR was performed by the Stratagene QuikChange Kit according to the manufacturer's instructions. The two mutagenic primers, annealing to the same target sequence on opposite strands of pET32b/SCR74 and appropriately designed to replace Cys-25 with glutamate, were (5′–3′ direction with mutated codon underlined) GCCAACAAAGT CATCAGCAAGGAGTGCCAAGCCATAAATCCGGAT and ATCCGGATTTATGGCTTGGCACTCCTTGCTGA TGACTTTGTTGGC. The resulting plasmid pET32b/SCR74-C25E was verified by DNA-sequencing28 and used to over-express the SCR74-C25E mutant as described earlier.
Bioassays
Both tests of leaf withering and PAL induction were carried out in duplicate on tomato seedlings (Lycopersicon esculentum var. Marmande) as previously described.17,21 The triplicate PAL activity values from treated and control samples were compared, for each induction time, by the Student's unpaired t-test for unequal variances using the Microsoft Excel version 2003 program.
Homology modeling and molecular dynamics
Raw model structures were built based on the PcF structural template (PDB ID: 2BIC)22 by using the automated protein-modeling server SWISS-MODEL.32 Prior to submission to the server, the primary structures of selected protein effectors were in silico processed by removal of their predicted N-terminal secretion signals and CNBr-cleaved fragments (Fig. 1, bottom). Structural stability of each model returned from the server was verified through molecular dynamics simulations using the GROMACS simulation package33–35 with the standard GROMOS96 force field,36 which allows to solve the Newton equations of motion for the desired system and to calculate variation of atomic coordinates as a function of time. Modeled structures were energy-minimized in vacuo using 1000 step of steepest descent in order to remove distorted geometries, and then embedded in a water box. A molecular dynamics simulation of 2 ns was applied to each model. During simulation, disulfide bond restraints were applied as follows: authentic SCR91 and mutant SCR74-C25E were restrained using the three known SS connectivities of PcF, that is Cys6-Cys40, Cys11-Cys44, and Cys26-Cys3922; authentic SCR74-B10 was restrained by an additional fourth disulfide linkage between the two unique cysteines not aligned (Fig. 1, bottom). For each protein, the last 500 ps of the 2 ns of molecular dynamics simulation, were used to compute an average structure that was finally minimized with 1000 steps of steepest descent. 3D rendering was carried out by the softwares Chimera37 and Swiss-PdbViewer (http://www.expasy.org/spdbv/).38 Other analyses were performed using scripts included in the GROMACS package. Model goodness and stability was evaluated by measuring RMSD of C-alphas as a function of time during the whole simulation. Structural fluctuation was examined on a residue-by-residue basis by computing the time-averaged RMSF of C-alphas. The SAS was also calculated for each residue. Finally, obtained average models were validated at the JCSG server (http://www.jcsg.org/prod/scripts/ validation/sv_final.cgi), using PROCHECK,39 ERRAT,40 and VERIFY3D41 tools. Electrostatic potential surfaces were computed by Swiss-PdbViewer. Secondary structures were analyzed by DSSP implemented in the GROMACS package42 and STRIDE (http://webclu.bio.wzw.tum.de/stride/).43
Acknowledgments
The authors wish to thank Dr. Francine Govers and Dr. Sophien Kamoun for providing the plasmid clones used in this study. They also thank Prof. Luigia Pazzagli and the CISM Mass Spectrometry Center (University of Florence, Italy) for their help performing MALDI-MS analyses.
Glossary
Abbreviations
- CNBr
cyanogen bromide
- HR
hypersensitive-response
- IPTG
isopropil β-1-tiogalattopiranoside
- IS-MS
ion spray-mass spectrometry
- MALDI-MS
matrix-assisted laser desorption/ionization-mass spectrometry
- ORF
open reading frame
- PAL
phenylalanine ammonia lyase
- PcF
Phytophthora cactorum-Fragaria
- PMSF
phenylmethylsulfonyl fluoride
- PR
pathogenesis-related
- PVDF
polyvinylidene difluoride
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- SCR
small cysteine-rich
- TFA
trifluoroacetic acid
References
- 1.Erwin DE, Ribeiro OK. Phytophthora disease worldwide. St. Paul, Minnesota: APS Press; 1996. [Google Scholar]
- 2.Haas BJ, Kamoun S, Zody MC, Jiang RH, Handsaker RE, Cano LM, Grabherr M, Kodira CD, Raffaele S, Torto-Alalibo T, Bozkurt TO, Ah-Fong AM, Alvarado L, Anderson VL, Armstrong MR, Avrova A, Baxter L, Beynon J, Boevink PC, Bollmann SR, Bos JI, Bulone V, Cai G, Cakir C, Carrington JC, Chawner M, Conti L, Costanzo S, Ewan R, Fahlgren N, Fischbach MA, Fugelstad J, Gilroy EM, Gnerre S, Green PJ, Grenville-Briggs LJ, Griffith J, Grünwald NJ, Horn K, Horner NR, Hu CH, Huitema E, Jeong DH, Jones AM, Jones JD, Jones RW, Karlsson EK, Kunjeti SG, Lamour K, Liu Z, Ma L, MacLean D, Chibucos MC, McDonald H, McWalters J, Meijer HJ, Morgan W, Morris PF, Munro CA, O'Neill K, Ospina-Giraldo M, Pinzón A, Pritchard L, Ramsahoye B, Ren Q, Restrepo S, Roy S, Sadanandom A, Savidor A, Schornack S, Schwartz DC, Schumann UD, Schwessinger B, Seyer L, Sharpe T, Silvar C, Song J, Studholme DJ, Sykes S, Thines M, Van de Vondervoort PJ, Phuntumart V, Wawra S, Weide R, Win J, Young C, Zhou S, Fry W, Meyers BC, Van West P, Ristaino J, Govers F, Birch PR, Whisson SC, Judelson HS, Nusbaum C. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–398. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 3.Tyler BM. Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol Plant Pathol. 2007;8:1–8. doi: 10.1111/j.1364-3703.2006.00373.x. [DOI] [PubMed] [Google Scholar]
- 4.Grunwald NJ, Goss EM, Press CM. Phytophthora ramorum: a pathogen with a remarkably wide host range causing sudden oak death on oaks and ramorum blight on woody ornamentals. Mol Plant Pathol. 2008;9:729–740. doi: 10.1111/j.1364-3703.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis JG, Rafiqi M, Gan P, Chakrabarti A, Dodds PN. Recent progress in discovery and functional analysis of effector proteins of fungal and oomycete plant pathogens. Curr Opin Plant Biol. 2009;12:399–405. doi: 10.1016/j.pbi.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol. 2006;44:41–60. doi: 10.1146/annurev.phyto.44.070505.143436. [DOI] [PubMed] [Google Scholar]
- 7.Schornack S, Huitema E, Cano LM, Bozkurt TO, Oliva R, Van Damme M, et al. Ten things to know about oomycete effectors. Mol Plant Pathol. 2009;10:795–803. doi: 10.1111/j.1364-3703.2009.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliva R, Win J, Raffaele S, Boutemy L, Bozkurt TO, Chaparro-Garcia A, et al. Recent developments in effector biology of filamentous plant pathogens. Cell Microbiol. 2010;12:705–715. doi: 10.1111/j.1462-5822.2010.01471.x. [DOI] [PubMed] [Google Scholar]
- 9.Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 10.Hogenhout SA, Van der Hoorn RA, Terauchi R, Kamoun S. Emerging concepts in effector biology of plant-associated organisms. Mol Plant Microbe Interact. 2009;22:115–122. doi: 10.1094/MPMI-22-2-0115. [DOI] [PubMed] [Google Scholar]
- 11.De Wit PJ, Mehrabi R, Van den Burg HA, Stergiopoulos I. Fungal effector proteins: past, present and future. Mol Plant Pathol. 2009;10:735–747. doi: 10.1111/j.1364-3703.2009.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Birch PR, Rehmany AP, Pritchard L, Kamoun S, Beynon JL. Trafficking arms: oomycete effectors enter host plant cells. Trends Microbiol. 2006;14:8–11. doi: 10.1016/j.tim.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Lamour KH, Win J, Kamoun S. Oomycete genomics: new insights and future directions. FEMS Microbiol Lett. 2007;274:1–8. doi: 10.1111/j.1574-6968.2007.00786.x. [DOI] [PubMed] [Google Scholar]
- 14.Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RH, Aerts A, et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006;313:1261–1266. doi: 10.1126/science.1128796. [DOI] [PubMed] [Google Scholar]
- 15.Raffaele S, Win J, Cano LM, Kamoun S. Analyses of genome architecture and gene expression reveal novel candidate virulence factors in the secretome of Phytophthora infestans. BMC Genomics. 2010;11:637. doi: 10.1186/1471-2164-11-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thines M, Kamoun S. Oomycete-plant coevolution: recent advances and future prospects. Curr Opin Plant Biol. 2010;13:427–433. doi: 10.1016/j.pbi.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Orsomando G, Lorenzi M, Raffaelli N, Dalla Rizza M, Mezzetti B, Ruggieri S. Phytotoxic protein PcF, purification, characterization, and cDNA sequencing of a novel hydroxyproline-containing factor secreted by the strawberry pathogen Phytophthora cactorum. J Biol Chem. 2001;276:21578–21584. doi: 10.1074/jbc.M101377200. [DOI] [PubMed] [Google Scholar]
- 18.Bos JIB, Armstrong M, Whisson SC, Torto TA, Ochwo M, Birch PRJ, et al. Intraspecific comparative genomics to identify avirulence genes from Phytophthora. New Phytol. 2003;159:63–72. doi: 10.1046/j.1469-8137.2003.00801.x. [DOI] [PubMed] [Google Scholar]
- 19.Kamoun S, Hraber P, Sobral B, Nuss D, Govers F. Initial assessment of gene diversity for the oomycete pathogen Phytophthora infestans based on expressed sequences. Fungal Genet Biol. 1999;28:94–106. doi: 10.1006/fgbi.1999.1166. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Bos JI, Armstrong M, Whisson SC, da Cunha L, Torto-Alalibo T, et al. Patterns of diversifying selection in the phytotoxin-like scr74 gene family of Phytophthora infestans. Mol Biol Evol. 2005;22:659–672. doi: 10.1093/molbev/msi049. [DOI] [PubMed] [Google Scholar]
- 21.Orsomando G, Lorenzi M, Ferrari E, de Chiara C, Spisni A, Ruggieri S. PcF protein from Phytophthora cactorum and its recombinant homologue elicit phenylalanine ammonia lyase activation in tomato. Cell Mol Life Sci. 2003;60:1470–1476. doi: 10.1007/s00018-003-3079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nicastro G, Orsomando G, Ferrari E, Manconi L, Desario F, Amici A, et al. Solution structure of the phytotoxic protein PcF: the first characterized member of the Phytophthora PcF toxin family. Protein Sci. 2009;18:1786–1791. doi: 10.1002/pro.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 24.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 25.Abramovitch RB, Janjusevic R, Stebbins CE, Martin GB. Type III effector AvrPtoB requires intrinsic E3 ubiquitin ligase activity to suppress plant cell death and immunity. Proc Natl Acad Sci U S A. 2006;103:2851–2856. doi: 10.1073/pnas.0507892103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stebbins CE, Galan JE. Structural mimicry in bacterial virulence. Nature. 2001;412:701–705. doi: 10.1038/35089000. [DOI] [PubMed] [Google Scholar]
- 27.McCoy J, Lavallie E. Expression and purification of thioredoxin fusion proteins. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb1608s28. Chapter 16: Unit16.8. [DOI] [PubMed] [Google Scholar]
- 28.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 30.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kussmann M, Roepstorff P. Sample preparation techniques for peptides and proteins analyzed by MALDI-MS. Methods Mol Biol. 2000;146:405–424. doi: 10.1385/1-59259-045-4:405. [DOI] [PubMed] [Google Scholar]
- 32.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 33.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 34.E Lindahl E, Hess B, van der Spoel D. A package for molecular simulation and trajectory analysis. J Mol Mod. 2001;7:306–317. [Google Scholar]
- 35.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;95:43–56. [Google Scholar]
- 36.Schuler LD, Daura X, van Gunsteren WF. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J Comput Chem. 2001;22:1205–1218. [Google Scholar]
- 37.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 38.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modelling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 39.Laskowski RA, MacArthur V, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 40.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 42.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 43.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]