
Table I.
X-Ray Data Reduction and Crystallographic Refinement Statistics
| (A) X-ray data reduction statistics | |
|---|---|
| Space group | P422 |
| Unit cell parameters (a = b, c) | 105.45 Å, 330 Å |
| Wavelength | 0.99999 Å |
| Resolution (last shell) | 26.36–2.64 Å (2.78–2.64 Å) |
| R-mergea (last shell) | 0.063 (0.377) |
| Total no. of observations | 376,916 (12,653) |
| Total no. of unique reflections | 52,570 (6182) |
| Average I/σ(I) (last shell) | 19.9 (1.9) |
| Completeness (last shell) | 0.952 (0.797) |
| Redundancy (last shell) | 7.2 (2) |
| (B) Crystallographic refinement statistics | |
|---|---|
| Space group | P41212 |
| Unit cell parameters (a = b, c) | 105.45 Å, 330 Å |
| Low (high) resolution limit | 38.65–2.64 Å (2.71–2.64 Å) |
| No. of reflections, working set (last shell) | 49,850 (2626) |
| No. of reflections, test set (last shell) | 2666 (128) |
| R-factorb (last shell) | 0.2003 (0.2340) |
| R-freec (last shell) | 0.2294 (0.2703) |
| No. of residues | 1023 |
| No. of protein atoms | 8540 |
| No. of solvent atoms | 537 |
| Average B-factor (protein) | 65.8 Å2 |
| Average B-factor (solvent) | 54.8 Å2 |
| Overall anisotropy B11, B22, B33 | 0.61620 Å2, 0.61620 Å2, 1.23240 Å2 |
| R.m.s.d. from ideal values | |
| Bond length | 0.008 Å |
| Bond angle | 0.87° |
a
R-merge = 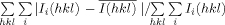 .
.
b
R-factor = 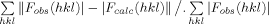 , where <|Fcalc|> denotes the expectation of |Fcalc(hkl)| used in defining the likelihood refinement target.
, where <|Fcalc|> denotes the expectation of |Fcalc(hkl)| used in defining the likelihood refinement target.
c
The free R-factor is a cross-validation residual calculated by using about 5% reflections, which were randomly chosen and excluded from the refinement.