

Abstract
During night (i.e., scotopic) vision in mammals, rod photoreceptor output is conveyed to ganglion cells (GCs), the output cells of the retina, by a specialized neural circuit comprising rod bipolar (RB) and AII amacrine cells. Here, we examined how intrinsic postsynaptic conductances in AIIs contribute to transmission of rod-derived signals. Using paired recordings from synaptically coupled RBs and AIIs, we found that a voltage-gated Na conductance in AII amacrines accelerated EPSPs arising from RB synaptic input. EPSPs also could be amplified by the Na conductance when AIIs were hyperpolarized below resting membrane potential, thereby increasing the availability of Na channels. AII amacrines are coupled electrically, and coupled AII amacrines likely receive common input from individual RBs. Na channel-mediated effects on EPSPs, however, appeared to occur at the single-cell rather than the AII network level. By recording light-evoked synaptic currents from GCs, we determined that the Na channel-dependent acceleration, but not amplification, of RB output by AII amacrines is reflected in the dynamics of AII synaptic output to retinal ganglion cells: synaptic inputs to both ON and OFF GCs are slowed equivalently, although not attenuated in amplitude, when Na channels in AIIs are blocked. Thus, during scotopic vision, Na conductances in AIIs serve to accelerate RB output.
Introduction
The rod bipolar (RB) cell pathway (Fig. 1) is well suited to transmit signals arising at visual threshold during rod-mediated scotopic vision in mammals (Bloomfield and Dacheux, 2001). Individual RBs receive input from many (∼20) rod photoreceptors. Similarly, individual AII amacrine cells receive input from many (∼20) RBs. AII amacrines are coupled electrically by gap junctions to each other and to multiple (∼15) ON cone bipolar cells (CBs) that provide excitatory input to retinal ganglion cells (GCs) (McGuire et al., 1984; Freed and Sterling, 1988; Tsukamoto et al., 2001). This anatomical convergence allows single GCs to pool the output of up to 10,000 rods; convergence and associated cellular and synaptic specializations enable signals elicited in a small fraction of the rods to modulate GC activity and to elicit a visual percept (Hecht et al., 1942; Barlow et al., 1971; Sakitt, 1972).
Figure 1.
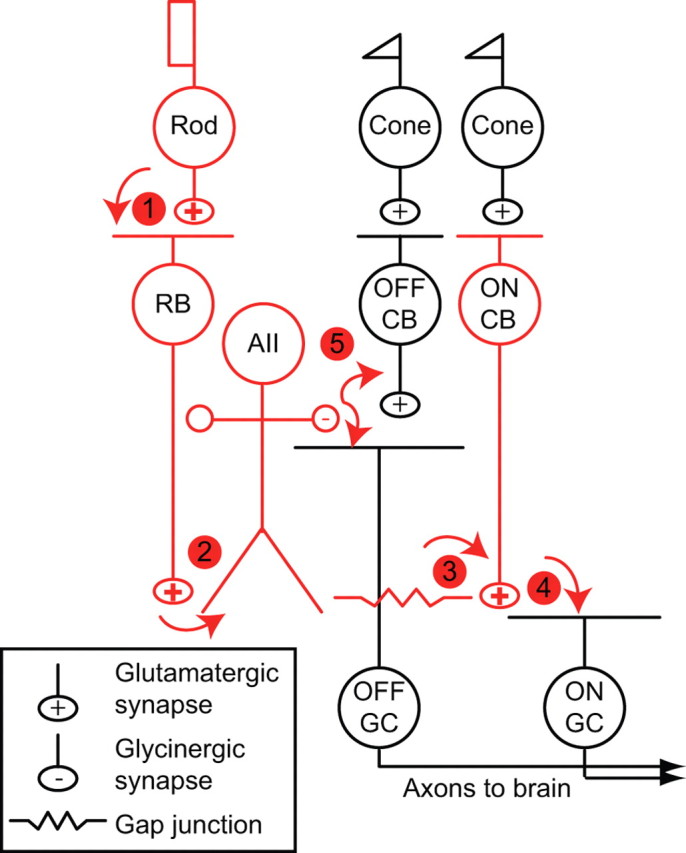
The rod bipolar pathway of the mammalian retina is illustrated schematically; its principal neurons are colored red, and its synapses are numbered. Cones make synapses with two classes of CB: ON and OFF CBs. ON and OFF CBs make excitatory synapses with GCs, the output cells of the retina. (1) Rods make synapses with an ON bipolar cell, the RB, and with a subset of OFF CBs (data not shown). (2) RBs contact AII amacrine cells. (3) AII amacrines are coupled by gap junctions to some types of ON CBs and to other AII amacrines. (4) ON CBs make synapses with ON GCs. (5) AII amacrines make inhibitory glycinergic synapses with some OFF CBs and OFF GCs. For a more complete discussion, see Bloomfield and Dacheux (2001).
Although the anatomy of the rod bipolar pathway has been characterized extensively, the mechanisms governing several interesting and potentially important characteristics of signal processing within it remain understood incompletely. For example, several studies indicate that light-evoked signals are high-pass filtered as they propagate through the rod bipolar pathway: light responses recorded in RBs are faster than those of rods (Field and Rieke, 2002), light responses recorded in AII amacrines are faster than those of RBs (Nelson, 1982; Trexler et al., 2005), and light responses recorded in GCs are faster still (Sampath et al., 2005). Both presynaptic and postsynaptic mechanisms likely underlie the acceleration of rod-derived signals at the RB–AII synapse. Presynaptically, exocytosis from RBs is transient, even during sustained presynaptic Ca2+ influx (Singer and Diamond, 2003; Trexler et al., 2005; Snellman et al., 2009). Postsynaptically, AII amacrines possess voltage-gated conductances (Boos et al., 1993) that may accelerate EPSPs.
Here, we investigated the role that intrinsic membrane conductances play in postsynaptic integration of RB input by AII amacrines. We show that a voltage-gated Na conductance accelerates the time course of signals measured in and downstream of AII amacrines.
Materials and Methods
Tissue preparation.
Experiments were performed on retinae isolated from C57BL/6 mice (4–8 weeks old). For slice experiments (RB and AII recordings), retinae from light-adapted mice were used. For recordings of GC light responses, retinae from dark-adapted (∼12 h) mice were isolated under infrared illumination (>900 nm). In all instances, retinae were isolated into bicarbonate buffered Ames' medium (Sigma) equilibrated with 95% O2/5% CO2 (carbogen). For retinal slice preparation, retinae then were embedded in low-melting temperature agarose (Sigma type VIIA, 3% in a HEPES-buffered saline), and slices (200 μm) were cut on a vibrating microtome (Microm). Slices were stored in carbogen-bubbled Ames' medium until use. For GC recordings, pieces of isolated retina were stored in carbogen-bubbled Ames' medium and transferred to a recording chamber after being placed GC-side up on a piece of filter paper (Anodisc 25; Whatman).
Data collection.
All experiments were performed at near-physiological temperature (∼32–34°C). Retinal slices were superfused with an artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 23 NaHCO3, 10 glucose, 1.25 NaH2PO4, 2.5 KCl, 1.1 CaCl2, 1.5 MgCl2, 2 Na-lactate, and 2 Na-pyruvate. Picrotoxin (100 μm), 1,2,5,6-tetrahydropyridin-4-yl) methylphosphinic acid (TPMPA) (50 μm), strychnine (0.5 μm), and niflumic acid (100 μm) were added to the ACSF to block GABAA receptor-activated, GABAC receptor-activated, glycine receptor-activated, and Ca2+-activated Cl channel-mediated currents, respectively. For recordings of K currents, 6,7-dinitroquinoxaline-2,3-dione (DNQX) (10 μm), tetrodotoxin (TTX) (500 nm), and CdCl2 (100 μm) also were added to the ACSF to block AMPA/kainate receptor-mediated, voltage-gated Na channel-mediated, and voltage-gated Ca channel-mediated currents, respectively. For recordings of Na currents, DNQX, CdCl2, tetraethylammonium chloride (TEA-Cl) (10 mm), and 4-aminopyridine (4-AP) (1 mm) were added to the ACSF to block AMPA/kainate receptor-mediated, voltage-gated Ca channel-mediated, and voltage-gated K channel-mediated currents, respectively. We noted some differences in Na channel-mediated events in ACSF and Ames' medium (qualitatively speaking, AIIs exhibited smaller spontaneous membrane potential fluctuations in Ames' medium), but these were not explored further. To simulate light responses chemically, l(+)-2-amino-4-phosphonobutyric acid (l-AP-4) (5 μm) was included in the ACSF, and (R,S)-α-cyclopropyl-4-phosphnophenylglycine (CPPG) (600 μm) was pressure ejected onto RB dendrites from a glass pipette pressurized by a Picospritzer III (Parker Hannifin). To measure the membrane time constant (τM), an exponential function was fit to the decay of a voltage waveform (average of at least 300 responses) elicited by current steps of −25 pA amplitude and 1 ms duration.
When recording light responses, flat-mounted retinae were superfused with Ames' medium or with ACSF (above) supplemented with 6.3 μm l-glutamate, 500 μm l-glutamine, and 0.9 μm l-aspartate. Neurons were visualized by transmitted light (>900 nm in the case of retinal ganglion cell recordings) video microscopy and targeted for whole-cell recordings. Pipettes were filled with the following solutions (in mm): 110 K-gluconate, 5 NaCl, 10 HEPES, 0.2 EGTA, 8 Tris-phosphocreatine, 4 MgATP, and 0.4 NaGTP for AII recording (when noted, QX-314-Cl [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium chloride] was added to the pipette solution to block Na currents within the recorded cell); 90 Cs-methanesulfonate, 20 TEA-Cl, 10 HEPES, 1 BAPTA, 8 Tris-phosphocreatine, 4 Mg-ATP, and 0.4 Na-GTP for RB recording; 105 Cs-methanesulfonate, 10 TEA-Cl, 20 HEPES, 10 EGTA, 2 QX-314-Cl, 5 Mg-ATP, and 0.5 Tris-GTP for retinal ganglion cell recording. When noted, TTX (500 nm) was added to the ACSF to block Na currents in all neurons. Access resistances were typically <40 MΩ for RBs and <25 MΩ for AII amacrines and GCs; compensation (∼75%) was used only for the GC recordings. Membrane potentials in voltage-clamp recordings are corrected for junction potentials of ∼10 mV.
Drugs were obtained from Sigma or Tocris Bioscience (except for TTX, which was from Alomone Labs). Recordings were made using a single MultiClamp 700B amplifier or two Axopatch 200B amplifiers (Molecular Devices). Synaptic transmission was elicited by stimulation of individual RBs at 20 s intervals. Recorded currents and potentials were low-pass filtered at 2–5 kHz and digitized at 10–20 kHz by ITC-16 or ITC-18 analog-to-digital boards (InstruTECH) controlled by software written in IGOR Pro (WaveMetrics). Recorded voltage-gated currents were leak subtracted (P/4 protocol). For light stimulation experiments, light from a 470 nm light-emitting diode (LED) was delivered to the recording chamber by a fiber optic cable. The spatially uniform stimulus illuminated a circular area 650 μm in diameter centered on the recorded cells. Calibrated photon fluxes at the preparation were converted to Rh*/rod using the spectral output of the LED, the absorption spectrum of rhodopsin, and an assumed collecting area of 0.5 μm2/rod. Most experiments used repeated presentations of a stimulus with a temporal trajectory drawn from a Gaussian distribution (0–60 Hz bandwidth). The contrast of this stimulus was defined as SD/mean. Synaptic currents were recorded at either ECl or ECation [to isolate excitatory (exc) and inhibitory (inh) inputs, respectively] and converted to conductances (Gexc and Ginh) on which cross-correlation analysis was performed.
Data analysis was performed using MATLAB (MathWorks), IGOR Pro, and Excel (Microsoft). We calculated the cross-correlation between postsynaptic conductances recorded in neighboring retinal ganglion cells and the autocorrelation for conductances in individual GCs according to the following equation: C(n) = NΣi=1 (xiyi−n)2/(xi2yi2)1/2, where x and y are measured postsynaptic conductances (comprising n points each), and xi and yi represent the difference between the response on a single trial and the mean response to repeated presentations of the same stimulus. To examine the cross-correlation function between light-evoked postsynaptic conductances in GCs on a trial-by-trial basis, we computed the cross-correlation function between responses on each trial and then averaged across the cross-correlation functions. Paired, two-tailed t tests were used to compare paired datasets; ANOVA was used for comparisons of two populations of data. In all cases, significance was accepted as p < 0.05. Unless indicated otherwise, data are presented as mean ± SEM, and illustrated traces are averages of 5–10 responses.
Results
AII amacrine cells possess voltage-gated Na and K conductances
First, we wanted to identify intrinsic membrane conductances that can interact with synaptic conductances. In accord with previous studies (Boos et al., 1993; Bloomfield and Xin, 2000; Veruki and Hartveit, 2002b), we recorded Na and K currents in AII amacrines. In voltage-clamp recordings with voltage-gated Ca and K channels blocked (see Materials and Methods), depolarization of the AII from −90 to −60 mV elicited an inward current that was blocked by TTX, indicating that it was mediated by voltage-gated Na channels (Fig. 2A). We could not characterize the voltage dependence of Na channel activation and inactivation using whole-cell recording because the voltage clamp appeared to be non-uniform over the surface of the cell (i.e., space clamp was inadequate): regenerative Na currents appeared at most potentials (Spruston et al., 1993; Williams and Mitchell, 2008). This is unsurprising given the elaborate, highly branched structure of the dendritic tree of the AII as well as electrical coupling between AII amacrines, the regenerative nature of Na channel-mediated depolarization, and the fast kinetics of Na channel activation (Bloomfield and Dacheux, 2001). We attempted to study the biophysical properties of the Na current in membrane patches excised from the soma, but we did not observe such currents in the somatic membrane, even when the majority of the soma was excised from the tissue slice as a nucleated outside-out patch (Fig. 2B). The Na conductance, then, appears to be localized to the AII dendrites, and, consequently, we turned to current-clamp recordings to examine GNa in more detail (below).
Figure 2.
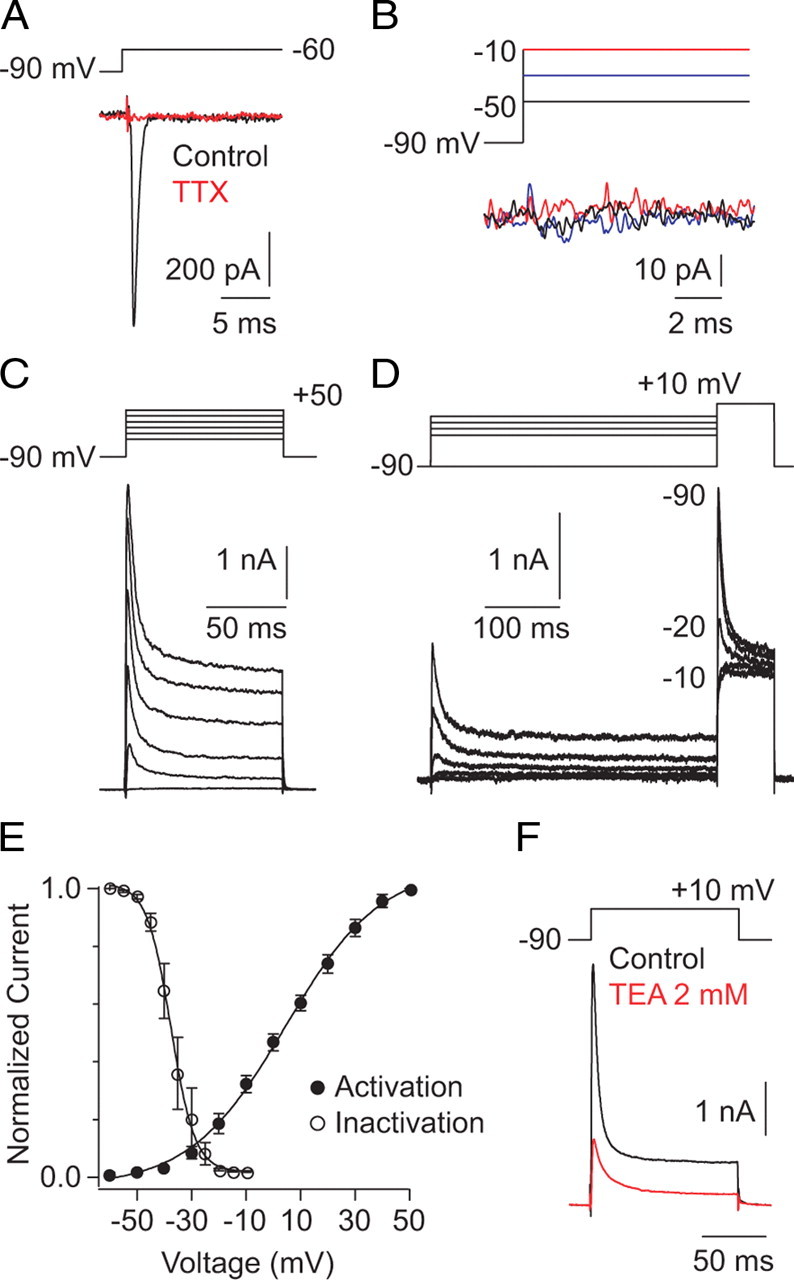
Evaluation of voltage-gated Na and K conductances in AII amacrines. A, A voltage step to −60 from −90 mV elicits an inactivating inward current that is blocked by TTX (n = 5). B, In a nucleated patch excised from the soma, no voltage-dependent currents are evident (n = 5). C, Whole-cell K currents recorded in AII amacrines after depolarizing 100 ms steps of 20 mV. Na and Ca currents channels were blocked with TTX and Cd2+, respectively. D, Steps to +10 mV were preceded by 500 ms prepulses to potentials between −65 and −10 mV (incremented by 5 mV; a subset of the responses are shown). E, Voltage dependence of K current activation and inactivation; a sigmoid function is fit to assist in visualization: note that little current is activated hyperpolarized to −40 mV, the range of VM elicited by EPSPs, and as a result of strong inactivation, very few channels are open at VREST. F, TEA (2 mm) dramatically suppressed the K current (n = 4).
To examine the voltage-gated K current, which was amenable to voltage-clamp characterization, depolarizing steps (100 ms) to potentials between −60 and +50 mV were delivered to AII amacrines from Vhold of −90 mV with voltage-gated Na and Ca channels blocked (see Materials and Methods). Partially inactivating currents were observed at potentials depolarized to −40 mV (Fig. 2C) (Boos et al., 1993). To examine the voltage dependence of current inactivation, the AII was stepped to +10 mV after prepulses to potentials between −90 and −10 mV (Fig. 2D). The transient component of the K current inactivated strongly and completely at potentials between −35 and −20 mV; the voltage dependence of current activation and inactivation is summarized in Figure 2E. Interestingly, because of their depolarized activation threshold, K channels will be activated only in conjunction with voltage-gated Na channels.
The K current recorded in AII amacrines after steps to +10 mV was sensitive to low concentrations of external TEA (Fig. 2F) (55 ± 6 or 72 ± 2% block at 1 or 2 mm, respectively; n = 4 for each condition). K currents also were blocked by 1 mm 4-AP but were unaffected by internal TEA at concentrations up to 10 mm (4-AP block 62 ± 6%; n = 5; data not shown). The transient and sustained portions of the current were affected approximately equivalently by these antagonists (percentage block for 2 mm TEA or 1 mm 4-AP, respectively: 72 ± 2 vs 77 ± 4 or 62 ± 6 vs 66 ± 3; n = 4 and 5), indicating that the two components arise from charge flux through the same type of channel. The voltage dependence and TEA sensitivity of the current are indicative of a Kv3 subunit-containing channel, likely a Kv3.4–Kv3.x heteromer (Rudy et al., 1999; Martina et al., 2003), but the biophysical properties of the current and the identity of the underlying channels were not explored any further.
AII amacrine cells exhibit Na channel-mediated spikelets
Next, we used current-clamp recordings to examine the effects of intrinsic membrane conductances on membrane voltage responses. The average resting membrane potential (VREST) of AIIs in our light-adapted slice preparation was −50 ± 1 mV; this value was the same (p = 0.92 by ANOVA; n = 27) in the presence and absence of DNQX (25 μm) to block excitatory inputs from RBs (picrotoxin, TPMPA, and strychnine also were present; see Materials and Methods). The fact that VM is unaffected by the presence of antagonists of fast synaptic transmission indicates that the resting potential is set by intrinsic membrane conductances rather than synaptic conductances. The observed VREST is slightly hyperpolarized to VREST of AII amacrines in the dark-adapted mouse retina (−46 ± 2 mV) (Dunn et al., 2006).
Injection of positive current at VREST elicited a membrane potential depolarization on which a series of “spikelets” rode (Fig. 3A). In some, but not all, recordings, we observed spontaneous spikelets at VREST (with no injected current) (Fig. 3A) (Veruki and Hartveit, 2002b). Both the evoked and spontaneous spikelets were blocked by TTX application, indicating that they arose from activation of voltage-gated Na channels.
Figure 3.
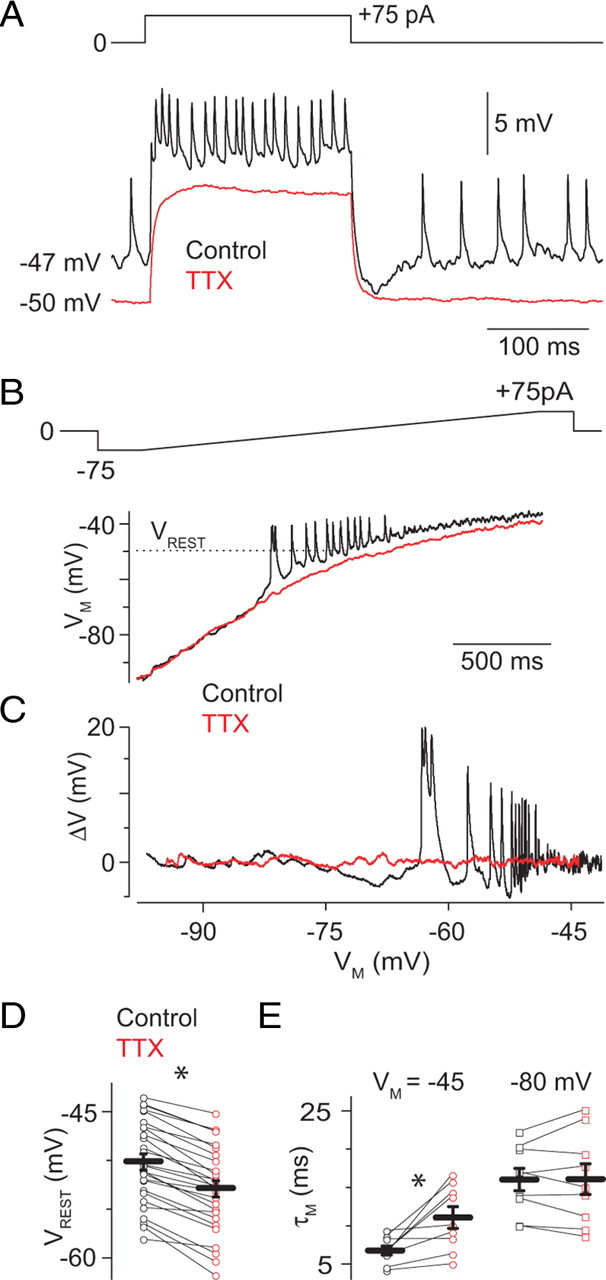
Current-clamp analysis of Na channel activity in AII amacrines. A, Positive current injection elicits membrane depolarization and small spikelets. Note that spontaneous spikelets are observed, at a lower frequency, at VREST (−47 mV) in the absence of injected current. Both evoked and spontaneous spikelets are blocked by TTX (500 nm). B, When a slowly varying current ramp is injected into the AII (−75 to 75 pA in 2 s), spikelets are evident at potentials near −60 mV (black). At potentials depolarized to VREST, spikelets are not seen, likely because of accumulated inactivation of Na channels, but membrane noise is increased. Spikelets and increased membrane noise are not observed in the presence of TTX. C, Deviations in VM around the baseline are plotted against membrane potential to illustrate the voltage range in which spikelets are generated, and membrane noise is increased by the TTX-sensitive Na conductance. D, TTX hyperpolarizes VM from −50 ± 1 to −53 ± 1 mV (n = 27), indicating that some GNa is open at rest. Gray and orange markers are observations from individual cells; black bars are means ± SEM. E, TTX increased τM at a potential depolarized to VREST (−45 mV) but not when VM was −80 mV, below the activation threshold of GNa (n = 9).
To determine the range of VM in which Na channels are active, we injected a slow (2 s) current ramp (from −75 to +75 pA at 0.075 pA/ms) (Fig. 3B) into AII amacrines; this changed VM from −87 ± 5 to −33 ± 1 mV (n = 6). Depolarization past a threshold VM of −57 ± 1 mV elicited spikelets (n = 6). In some cells (n = 3 of 6), spikelets could be observed at all potentials depolarized to threshold; in other cells (n = 3 of 6), spikelets were absent at potentials depolarized to VM of −41 ± 3 mV, presumably because of accumulated Na channel inactivation. For both sets of cells, the membrane was significantly noisier at VM more than −45 mV than at VM less than −60 mV, and data were pooled (RMS voltage at −40 vs −75 mV: 3.2 ± 0.7 vs 0.5 ± 0.1 mV; p = 0.008) (Fig. 3C). This increased membrane noise was suppressed by TTX, indicating that GNa does not inactivate completely at VM depolarized to rest (approximately −50 mV) (RMS voltage, 0.8 ± 0.1 vs 0.4 ± 0.1 mV; −40 vs −75 mV; n = 6; differences are significant at −40 but not −75 mV; p = 0.02 vs 0.4, respectively).
TTX also hyperpolarized the membrane by ∼3 mV (to −53 ± 1 mV; n = 27; p < 0.05) (Fig. 3A), demonstrating again that some Na channels are open at VREST. This assertion is supported by the observation that TTX lengthened τM at potentials near but not significantly hyperpolarized to VREST (Fig. 3E) (control vs TTX τM: 7 ± 1 vs 11 ± 1 ms at −45 mV; n = 9; p = 0.01; and 16 ± 2 vs 16 ± 2 ms at −80 mV; n = 9; p = 0.91; τM assayed as in Materials and Methods). Application of TEA (1.5 mm) in combination with TTX had at most a small additional effect on τM at depolarized but not hyperpolarized potentials (τM of 14 ± 1 ms at −45 mV and 16 ± 2 ms at −80 mV; n = 5 and 7 and p = 0.07 and 0.5, respectively), consistent with the observation that few K channels are active near VREST (Fig. 2E). That τM was longer at −80 mV than at −45 mV in the presence of TTX and TEA indicates that voltage-gated membrane conductances other than GNa and GK are active at depolarized potentials.
Our observations confirm that a voltage-gated Na conductance is found in the AII dendrites. This GNa is active near VREST and therefore may be functionally relevant in postsynaptic processing of synaptic inputs.
The dendritic Na conductance shapes EPSPs
To assay contributions of dendritic voltage-gated channels to postsynaptic integration over a range of AII membrane potentials, we recorded from pairs of synaptically coupled RBs and AII amacrines. The presynaptic RB was recorded in the voltage-clamp configuration and depolarized with voltage commands (depolarizing ramps of 100 ms beginning at −70 mV to potentials between −55 and −30 mV, followed by a 100 ms ramp repolarization to −70 mV) that had a time course and peak amplitude similar to light-evoked RB voltage responses (Euler and Masland, 2000; Trexler et al., 2005). These voltage commands elicited presynaptic Ca currents (Fig. 4Ai) and evoked EPSCs in AII amacrines (Vhold of −70 mV) (Fig. 4Aii,Aiii).
Figure 4.
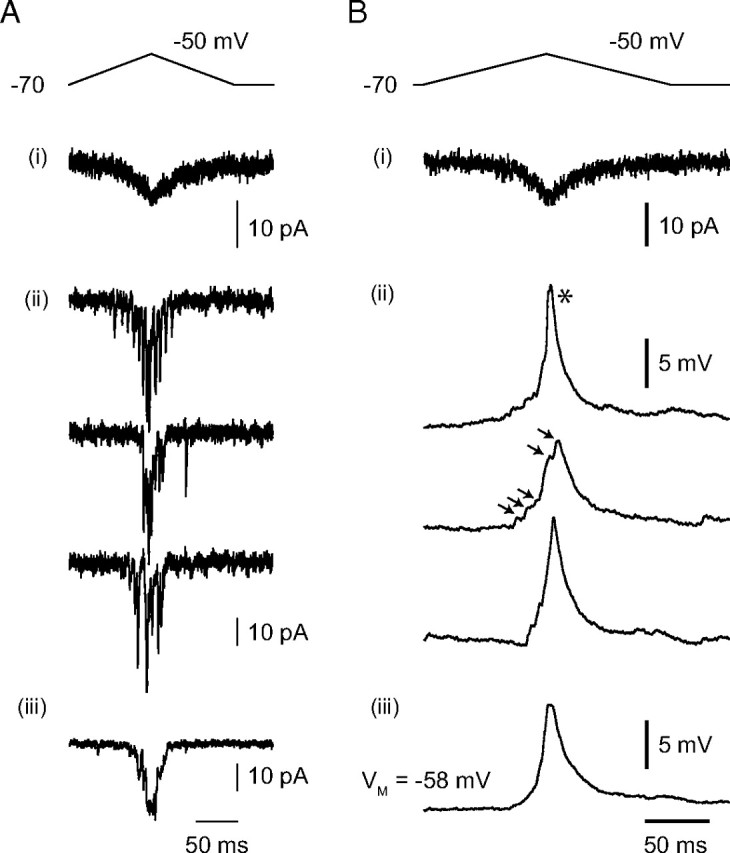
Representative AII responses to RB output. A, During a paired RB–AII recording, the voltage-clamped RB was depolarized with a waveform designed to mimic a light flash-evoked voltage response. This elicited a very small, slow presynaptic Ca current (i) and evoked exocytosis that was recorded as a postsynaptic current in a voltage-clamped AII (single responses in ii; average responses in iii). B, When the same AII was recorded in the current-clamp configuration, evoked EPSPs were observed (single responses in ii; average responses in iii). Note that spikelets are evident in some individual responses (asterisk) but not others. When spikelets are not evident, individual synaptic events can be resolved in the compound EPSP (arrowheads).
When the AII was recorded in the current-clamp configuration, exocytosis from RBs evoked EPSPs in the postsynaptic AII. The EPSPs illustrated in Figure 4B were recorded from a neuron that was hyperpolarized to approximately −60 mV by somatic current injection to reduce Na channel-mediated fluctuations in membrane potential (Fig. 3C). In some responses, the postsynaptic conductance triggered a spikelet that was observed as a depolarization with a uniform, rapid rising phase and an approximately monoexponential decay. In other responses, such spikelets were not evident, and it was possible to resolve individual synaptic events as inflections in the EPSP waveform (Fig. 4B, individual traces). These observations are consistent with a report of light-evoked spikelets in AII amacrine cells (Bloomfield and Xin, 2000).
The contribution of the postsynaptic GNa to postsynaptic integration was studied first by examining the effects of TTX on EPSPs evoked in AII amacrines by ramp depolarization of RBs. No current was injected into the AIIs; they were recorded at their VREST (−50 ± 1 mV; n = 10). Depolarization of the RB to −53 mV surpassed the presynaptic threshold necessary to evoke EPSPs on every trial; therefore, we call this presynaptic depolarization our “minimal” stimulus (Fig. 5A). This minimal-stimulus-evoked EPSP recorded at VREST was surprisingly large (10 ± 2 mV; n = 10), and presynaptic voltage ramps to more depolarized potentials evoked EPSPs of increasing amplitudes (up to a maximum of 13 ± 1 mV evoked by ramps to −30 mV). As observed previously, the appearance of spikelets in individual responses was variable (Fig. 5A). TTX applied to the slice attenuated dramatically the amplitude of the minimal-stimulus-evoked EPSPs (to 2 ± 1 mV) and had a much more modest effect on the amplitude of EPSPs evoked by more depolarizing presynaptic stimuli (to 12 ± 1 mV) (Fig. 5B1,B2). TTX also slowed the time course of all EPSPs. A simple way to represent this change in time course of the EPSPs, which exhibit a complex and variable waveform, was to measure the shift in the timing of the EPSP peak. In the presence of TTX, the time to EPSP peak was delayed by several milliseconds (6 ± 1 ms for ramps to −53 mV; 10 ± 2 ms for ramps to −30 mV) (Fig. 5B3).
Figure 5.
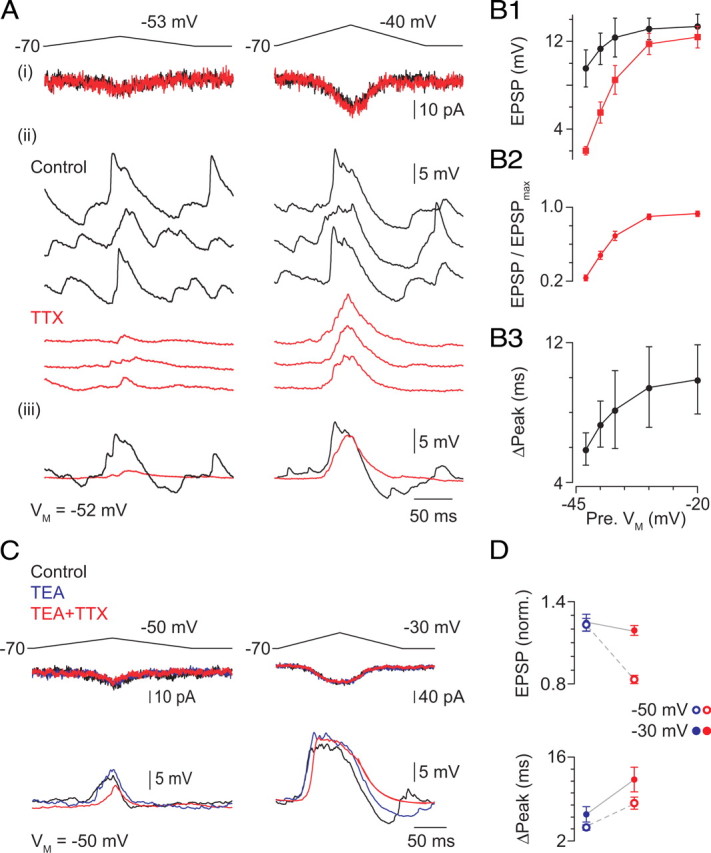
Voltage-dependent Na, but not K, channels make a significant contribution to the EPSP. A, Minimal (to −53 mV at left) and stronger (to −40 mV at right) presynaptic depolarizations elicit differently sized presynaptic Ca currents (i) and EPSPs (ii, individual responses; iii, averaged responses). EPSPs evoked by minimal stimulation are reduced in amplitude quite substantially by TTX; the effect of TTX on amplitude is weaker for larger EPSPs. TTX has no effect of the presynaptic Ca current. Note also that TTX eliminates fluctuations in VM. Summary data are presented in B. B1, The effect of TTX on EPSP amplitude varies with the magnitude of presynaptic depolarization, and TTX has no significant effect on the EPSPs evoked by the largest presynaptic depolarizations; this is illustrated clearly by the normalized data (TTX ÷ control) presented in B2: p < 0.001 for −53 and −50 mV; p < 0.05 for −47 and −40 mV; p > 0.05 for −30 mV. B3, TTX delays the peak of all EPSPs. Illustrated are the differences between the peak time in TTX and control conditions; the difference is significant in all instances: p < 0.01 for −53 and −50 mV; p < 0.05 for −47, −40, and −30 mV; n = 5–15 per point. C, TEA (2 mm) increased EPSP amplitude slightly. Application of TTX in the presence of TEA reduced the amplitude of the minimal stimulus evoked EPSP but not the EPSP evoked by a larger presynaptic depolarization. D, Summary of TEA and TEA + TTX effects on EPSP amplitude (top) and timing (bottom): EPSPs were evoked by presynaptic voltage ramps to either −50 or −30 mV.
We wanted to determine whether a voltage-gated K conductance was activated in the AII during the EPSP. To do this, we examined the effect of external TEA (2 mm) on EPSPs evoked by voltage ramps of different amplitudes delivered to the presynaptic RB (Fig. 5C). TEA had a small effect on both the amplitude and time course of the EPSP recorded at VM of −50 mV, close to the VREST of AII amacrines (Fig. 5C), but its effect was far smaller than that of TTX. The EPSP likely does not depolarize the AII sufficiently to activate many K channels: very little K current was observed at potentials hyperpolarized to −35 mV (Fig. 2E), the most depolarized potential reached by the largest EPSPs. In the presence of TEA, however, TTX reduced the amplitude of minimal-stimulation-evoked EPSPs, indicating that K conductance does not regulate the availability of Na channels during the EPSP. Additionally, in the presence of TEA, TTX slowed all EPSPs.
To assay the range of VM (recorded at the soma) over which the dendritic GNa can boost EPSPs, we varied VM by current injection and evoked EPSPs with a presynaptic voltage ramp to −50 mV (Fig. 6A). When the AII was hyperpolarized sufficiently to prevent the EPSP from activating Na channels (potentials less than −80 mV) or depolarized more than −45 mV to inactivate most Na channels, the effect of TTX on the EPSP was minimal (Fig. 6A–C). TTX, however, had a significant effect on EPSP amplitude and time course over a wide range of VM hyperpolarized to VREST (−55 to −75 mV) (Fig. 6B).
Figure 6.

The contribution of the Na conductance to the EPSP varies with VM. A, EPSPs were evoked by presynaptic depolarization (voltage command and presynaptic Ca currents at top) and recorded at different postsynaptic VM. Averaged traces are illustrated. EPSP amplitudes at different VM are plotted in B1. In the presence of TTX (500 nm), EPSP amplitude varied linearly with VM, as predicted by the change in driving force on the synaptic conductance. Under control conditions, however, the amplitude of the EPSPs was boosted significantly in the range of VM in which GNa is activated. Normalized data are illustrated in B2. p < 0.01 for −45 to −75 mV; p < 0.05 for −85 mV; p > 0.05 for −95 mV. B3, TTX delays the time to peak when VM permits activation of Na channels. p < 0.01 for −55 and −65 mV; p < 0.05 for −45 and −75 mV; p > 0.05 for −85 and −95 mV. Points represent the mean ± SEM of n = 4–13 paired recordings.
Network interactions are not required to boost EPSPs
Electrical coupling between AII amacrines and between AII amacrines and ON CBs is mediated by connexin36-containing gap junctions and is preserved in the light-adapted retinal slice preparation (Deans et al., 2002; Veruki and Hartveit, 2002a,b) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Because it has been postulated that Na channel-dependent boosting of EPSPs arising simultaneously in coupled AII amacrines can serve to eliminate synaptic noise in the AII network (Smith and Vardi, 1995), we took advantage of this arrangement to determine whether the observed effects of TTX were exerted on the level of individual AII amacrines or the AII network.
To do this, we made paired RB–AII recordings, as above, but we included the Na channel blocker QX-314 in the postsynaptic pipette to block GNa in the recorded AII only; there is no evidence that QX-314 crosses or blocks gap junctions (Fig. 7). Because a single RB contacts three to five AII amacrines (Tsukamoto et al., 2001), we presumed that the recorded RB made synapses onto AII amacrines that were coupled electrically to the recorded AII. When QX-314 was included in the postsynaptic recording pipette, EPSPs were smaller than control EPSPs recorded at approximately the same membrane potential (VM ≈ −55 mV; amplitude of 6 ± 1 vs 12 ± 2 mV) and were unaffected by TTX applied to the entire slice (Fig. 7A,B) (amplitude of 5 ± 1 mV; n = 5; p = 0.55). The amplitudes and time courses of EPSPs recorded from AII amacrines with internal QX-314 were indistinguishable statistically from those recorded from control AII amacrines in the presence of TTX: amplitude, 5 ± 1 vs 6 ± 1 mV; time of peak, 107 ± 2 vs 107 ± 1 ms; p = 0.85 and 0.79, respectively, by ANOVA), and spikelets were not evoked by synaptic input to these neurons. Thus, spikelets that may be evoked in a coupled AII by synaptic input are unable to propagate passively into the recorded AII because of the low-pass filter imposed by the gap junctions. Additionally, membrane potential changes elicited by spikelets in a coupled AII are not amplified by the recorded neuron. These observations are consistent with those made previously in a detailed study of electrical signaling between AIIs (Veruki and Hartveit, 2002b).
Figure 7.
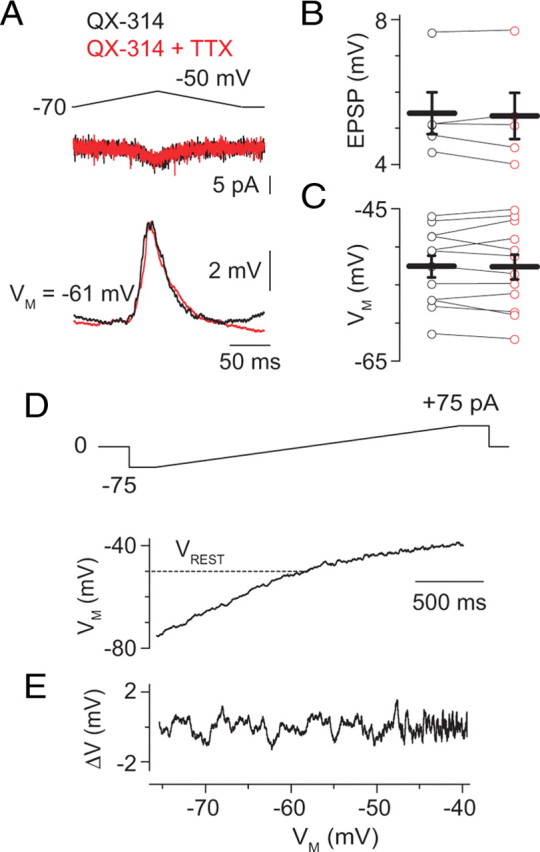
GNa-dependent effects on EPSPs are exerted at the single-neuron level. A, When QX-314 (5 mm) was included in the postsynaptic recording pipette, the EPSP was not affected by TTX (500 nm). B, C, Summary data illustrating that TTX did not affect EPSP amplitude (B; n = 5) or AII VM (C; n = 12) in the presence of internal QX-314. D, TTX effects on AII amacrines are abolished when QX-314 is present in the recording pipette: a slow current ramp from −75 to +75 pA in 2 s (top) induced voltage changes in an AII loaded with QX-314. For comparison, note Figure 1, D and E. E, As in Figure 3C, fluctuations in membrane potential, isolated by subtracting a smoothed curve from the trace illustrated, are plotted against VM (VREST of −50 mV).
Additionally, postsynaptic membrane potential was unaffected by TTX when internal QX-314 was present, and the voltage dependence of membrane noise was attenuated significantly in the presence of QX-314 (Fig. 7C–E). Together, these data indicate that boosting of excitatory synaptic inputs on AII dendrites occurs in an input-specific manner.
The Na conductance accelerates the output of the AII network
We wanted to determine how Na channel activation in AII amacrines affects their output. First, we examined the effect of TTX on light flash-evoked IPSCs in large OFF transient and sustained GCs, neurons that receive direct glycinergic synaptic input from AIIs (Manookin et al., 2008; Murphy and Rieke, 2008; Münch et al., 2009). We found that TTX slowed mildly the time course of IPSCs evoked at a range of flash strengths (from 0.01 to 0.5 R*/rod): the time to the half-maximal response, averaged over all flash strengths, was 97 ± 0.7% of control (n = 10 recorded GCs; p < 10−3; data not shown). We, however, were surprised to observe that TTX did not affect the amplitudes of IPSCs, even those evoked by the dimmest flashes (amplitude, 102% of control; p = 0.87).
Although the presynaptic stimulus delivered in our paired RB–AII recording experiments was designed to mimic the response of an RB to a light flash, the strength and timing of light-evoked synaptic input to AIIs certainly differs from the strength and timing of the EPSP evoked by the electrical stimulation of a single RB. Consequently, we wanted to examine the influence of the Na conductance of the AII to its synaptic output under conditions when it receives a more uniform and continuous input. To do this, we examined synaptic currents in both ON and OFF GCs evoked by a dim flickering light stimulus.
Because AII amacrines provide input to both ON and transient OFF GCs from distinct portions of their dendritic trees (Fig. 1: indirectly to ON GCs via the AII–ON CB gap junction and directly to transient OFF GCs via glycinergic synapses), signals in the RB pathway generate coincident changes in excitatory input to ON GCs and inhibitory input to transient OFF GCs (Murphy and F. Rieke, 2008). Here, we take advantage of this arrangement to examine the absolute and relative effects of TTX on light-evoked AII output to downstream targets.
We recorded simultaneously synaptic inputs elicited in ON and OFF GCs by a fluctuating light stimulus that produced on average 2–3 Rh*/rod/s; under these conditions, rod output is carried almost exclusively by the RB pathway (Murphy and Rieke, 2006). Fluctuations in light intensity generated strongly correlated excitatory and inhibitory conductances (Gexc and Ginh) in ON and transient OFF GCs, respectively (Fig. 8A).
Figure 8.

TTX slows the output of the AII network. A, A fluctuating light stimulus (top trace) elicits changes in the measured excitatory (Gexc; blue trace; Vhold of −70 mV) and inhibitory (Ginh; brown trace; Vhold of +10 mV) conductances recorded in neighboring ON and OFF GCs under control conditions (top) and in the presence of TTX (bottom). B, Cross-correlation measured between Gexc and Ginh in control (black) and TTX (red) conditions (mean ± SE; n = 4). The inset shows the peaks of the mean functions at higher temporal resolution. C, Cross-correlation measured in a given cell before and after TTX application; the −10 ms shift indicated that TTX slows the time-varying changes in both Gexc and Ginh (mean ± SE; n = 4). The inset shows the peaks of the mean functions at higher temporal resolution.
When TTX was applied to the preparation to block Na channels in AII amacrines, the amplitudes and relative timing of Gexc and Ginh were not altered (i.e., Gexc and Ginh remained correlated with a peak near 0 ms) (Fig. 8B). TTX, however, did slow the absolute timing of synaptic input to both ON and transient OFF GCs. This slowing was revealed as a shift of approximately −10 ms in the peaks of the cross-correlations of control and TTX Gexc for an ON cell and control and TTX Ginh for an OFF cell (Fig. 8C). As we observed for IPSCs evoked by single flashes, above, the amplitudes of Gexc and Ginh were not reduced by TTX. In fact, fluctuations in postsynaptic conductance tended to be larger in the presence of TTX: the average SD of the excitatory conductance was 176 ± 6% of control (p = 0.006), and the average SD of the inhibitory conductance was 139 ± 20% of control (p = 0.13).
To control for possible effects of TTX on RBs, we evoked synaptic currents in AII amacrines by pressure ejecting the mGluR6 antagonist CPPG onto RBs in the presence of the mGluR6 agonist l-AP-4; this evokes a chemical “light response” in RBs (Snellman and Nawy, 2002). TTX had no effect on either the amplitude or integral of these responses (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation indicates that TTX does not affect synaptic potentials in and transmitter release from RBs, consistent with the reported absence of voltage-gated Na channels in these neurons (Ma et al., 2005).
Although TTX does affect signal propagation in wide-field amacrine cells (Bloomfield, 1996), the fact that input to the ON and OFF pathways is affected equally by Na channel blockade makes it unlikely that the slowing of synaptic conductances reflects changes in the outputs of multiple neurons. We, however, cannot exclude the possibility that the effect of TTX on the amplitude of light-evoked conductances in GCs resulted from disinhibition of synaptic inputs onto GCs. Thus, our experiments indicate that, just as TTX slows the time course of individual EPSPs recorded in AII amacrines, TTX slows the output of the AII network. Because the correlation between Gexc and Ginh reported previously (Murphy and Rieke, 2008) was preserved in the presence of TTX (Fig. 8C), we conclude that the Na conductance of the AII affects its output to the ON and transient OFF channels similarly.
Discussion
We examined postsynaptic integration of RB output by the mammalian AII amacrine cell. Using paired RB–AII recordings, we determined that a dendritic GNa accelerates the time course and, under some conditions (i.e., after membrane hyperpolarization), amplifies postsynaptic responses to input from single RBs. Simultaneous recordings from ON and transient OFF GCs revealed that the speeding, but not the amplification, of rod-derived signals by GNa in the AII is reflected in the synaptic output of the AII network. Our experiments reveal a postsynaptic mechanism that may contribute to accelerating signals at the level of the AII network during rod-mediated vision.
Na current in an unconventional neuron
Because most neurons signal via action potentials, the process of synaptic integration usually is considered as the transformation of dendritic synaptic input into trains of action potentials originating at the axon initial segment. The interplay between dendritic and synaptic conductances during synaptic integration is complex and variable but generally is evaluated by assaying the efficacy with which synaptic inputs elicit action potentials (Häusser et al., 2000). The AII amacrine cell differs from most neurons in that it does not signal via action potentials (it exhibits only small, i.e., 5–10 mV, Na channel-dependent spikelets, and AII network output persists in the presence of TTX) (Fig. 8), and it is an axon-less neuron that both receives synaptic input and makes synaptic output on its dendrites. What, then, is the role of the voltage-gated GNa in these neurons?
Little is known about the Na channels found in AII amacrines. One in situ hybridization study indicates that they may be composed of Nav1.1 subunits (Kaneko and Watanabe, 2007), and one electrophysiological study involving localized TTX application indicates that most Na channels may be located in the soma and/or proximal dendrite (Tamalu and Watanabe, 2007). Nucleated patch recordings failed to demonstrate any somatic GNa, and our experimental observations demonstrate clearly that the activation of a dendritic GNa accelerates EPSPs arising at RB–AII synapses (Figs. 4, 5). The acceleration of the EPSP is propagated to neurons postsynaptic to the AII, the ON and transient OFF GCs, as demonstrated by the effect of TTX on the timing of synaptic conductances recorded in these neurons (Fig. 8B).
It is interesting to note that TTX did not have a differential effect on AII output to the ON or OFF pathways (Fig. 8C). Given the anatomical arrangement of input and output synapses on the AII dendritic tree (inputs from RBs are on distal dendrites, chemical output to OFF cells is from proximal dendrites, and gap junctions to ON CBs are in distal dendrites) (Tsukamoto et al., 2001), it might have been expected that GNa would affect preferentially output to the OFF pathway because output synapses are removed from the sites of synaptic input, whereas AII–ON CB gap junctions are near RB–AII synapses. That this is not the case suggests that Na channels may be distributed throughout the dendritic tree, as is the case in many conventional spiking neurons (Häusser et al., 2000).
Na channels and AII output
It is well known that AIIs express voltage-gated Na channels and are capable of generating TTX-sensitive spikelets in response to depolarization (Boos et al., 1993; Bloomfield and Xin, 2000; Veruki and Hartveit, 2002b; Tamalu and Watanabe, 2007). This has led to the hypothesis that Na channel-dependent spikelets may serve as a postsynaptic thresholding mechanism to amplify nonlinearly small synaptic inputs and thereby allow the AII to differentiate signals from synaptic noise (Smith and Vardi, 1995). Indeed, our recordings in the light-adapted retinal slice demonstrated that synaptic input evoked by depolarization of single RBs is capable of eliciting spikelets in AIIs (Fig. 4), thereby boosting the amplitude of EPSPs. The extent of this boosting depended on the postsynaptic membrane potential and that amplification of the EPSP was negligible at potentials at which Na channel availability was low (more than −45 mV) and at potentials at which the synaptic conductance cannot depolarize the membrane sufficiently to activate Na channels (less than −85 mV) (Fig. 6). Thus, the extent to which spikelets are evoked by photoreceptor activation may vary with ambient light intensity and/or the neuromodulatory state of the retina.
The generation of spikelets by the AII was not required for light-evoked synaptic output under scotopic conditions: TTX did not attenuate the amplitudes of light-evoked currents (or conductances) recorded in OFF GCs, reflecting glycinergic transmission from AIIs, or excitatory conductances recorded in ON GCs (Fig. 8). We can offer several explanations for the discrepancy between the effects of TTX on the amplitudes of EPSPs recorded in AIIs and the amplitudes of light-evoked synaptic conductances recorded in GCs: (1) Na channel availability may be modulated by ambient light intensity [although light-evoked spikelets have been recorded in dark-adapted AIIs (Bloomfield and Xin, 2000)]; (2) AIIs in the dark-adapted retina may be too depolarized to generate spikelets [VREST of −46 mV (Dunn et al., 2006)]; (3) the time course of light-evoked synaptic input to AIIs is not approximated well by our paired-recording experiments; and (4) depolarization arising from the synaptic conductance alone is sufficient to activate presynaptic Ca channels in the AII and saturate the rate of glycine release. Given that presynaptic Ca channels in AIIs appear to be activated maximally at quite hyperpolarized potentials [−40 mV (Habermann et al., 2003)], we favor some combination of the second and fourth possibilities.
Acceleration of RB output at the level of single AII amacrine cells
It has been recognized for some time that the RB–AII synapse acts as a high-pass filter to make rod-driven light responses faster in AII amacrines than in RBs (Nelson, 1982; Trexler et al., 2005). This acceleration has been attributed in part to the intrinsic dynamics of exocytosis from RB terminals (Singer and Diamond, 2003; Trexler et al., 2005; Snellman et al., 2009). Here, we provide the first experimental evidence for a significant postsynaptic modification of RB input by GNa in AII amacrines (Figs. 4, 5). Activation of a dendritic GNa by the synaptic conductance accelerates EPSPs and, under some conditions, boosts the amplitude of the EPSPs. The functional relevance of the speeding of the EPSP by GNa is illustrated by the slowing of AII network output to GCs in the presence of TTX under scotopic conditions (Fig. 8B).
The interaction between low-pass filtering (by gap junctions) and nonlinear acceleration and amplification (by Na channels) of synaptic inputs is not resolved completely by our experiments, but our study does provide some insight into the role that Na channels play in mediating responses to the output of individual RBs on both the levels of single AIIs and the AII network. In considering our paired recording experiments, we expect that the RBs from which we recorded provided synaptic input to AII amacrines that were coupled electrically to the postsynaptic AII. Given the strong electrical coupling between AII amacrines even in the light-adapted slice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (also a coupling coefficient ∼0.3 as measured by Veruki and Hartveit, 2002b) and the divergence of the output of a single RB to three to five AII amacrines (McGuire et al., 1984; Freed and Sterling, 1988; Tsukamoto et al., 2001), we believe it reasonable to expect that a portion of the recorded EPSP reflected a voltage change in a coupled AII. Importantly, our experimental results demonstrate that the responses of individual AII amacrines to input from single RBs is shaped by the activity of their Na channels, not that of Na channels in coupled neurons: TTX effects on AII membrane properties as well as EPSPs are abolished when QX-314 is included in the recording pipette (Fig. 6) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation is consistent with a previous report of passive signal propagation between electrically coupled AIIs (Veruki and Hartveit, 2002b).
The fact that local blockade of Na channels within a single AII abolished the effect on the EPSP of TTX application to the slice suggests to us that the majority of the boosting and acceleration of the EPSP in AII amacrines occurs in a cell-specific manner. This is consistent with observations that other types of signal processing in rod vision, e.g., nonlinear thresholding of rod responses by RBs and neural gain control driven by changes in ambient light intensity, occur at the level of individual synapses rather than in the retinal network as a whole (Field and Rieke, 2002; Sampath and Rieke, 2004; Dunn and Rieke, 2008). By processing RB output at the single-cell (or synapse) rather than the network level, the retina may ensure that the integrated signal produced by the network of AII amacrines is not dominated by the noisiest inputs; this may allow the RB–AII synapse to operate at a very high gain while noise in AII amacrine cell light responses remains essentially independent from background light intensity (Dunn et al., 2006; Dunn and Rieke, 2008). More generally, transformations applied before synaptic integration can serve very different functional roles than those applied after integration.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS-043365 and EY-017836 and the Alfred P. Sloan Foundation (J.H.S.), by NIH Grant EY-011850 and the Howard Hughes Medical Institute (F.R.), and by an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology at Northwestern University. We thank Dr. M. Martina for many helpful conversations and P. Chung and V. Dudley for technical assistance.
References
- Barlow HB, Levick WR, Yoon M. Responses to single quanta of light in retinal ganglion cells of the cat. Vision Res [Suppl] 1971;3:87–101. doi: 10.1016/0042-6989(71)90033-2. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA. Effect of spike blockade on the receptive-field size of amacrine and ganglion cells in the rabbit retina. J Neurophysiol. 1996;75:1878–1893. doi: 10.1152/jn.1996.75.5.1878. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA, Dacheux RF. Rod vision: pathways and processing in the mammalian retina. Prog Retin Eye Res. 2001;20:351–384. doi: 10.1016/s1350-9462(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Bloomfield SA, Xin D. Surround inhibition of mammalian AII amacrine cells is generated in the proximal retina. J Physiol. 2000;523:771–783. doi: 10.1111/j.1469-7793.2000.t01-1-00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos R, Schneider H, Wässle H. Voltage- and transmitter-gated currents of all-amacrine cells in a slice preparation of the rat retina. J Neurosci. 1993;13:2874–2888. doi: 10.1523/JNEUROSCI.13-07-02874.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–712. doi: 10.1016/s0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn FA, Rieke F. Single-photon absorptions evoke synaptic depression in the retina to extend the operational range of rod vision. Neuron. 2008;57:894–904. doi: 10.1016/j.neuron.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn FA, Doan T, Sampath AP, Rieke F. Controlling the gain of rod-mediated signals in the mammalian retina. J Neurosci. 2006;26:3959–3970. doi: 10.1523/JNEUROSCI.5148-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol. 2000;83:1817–1829. doi: 10.1152/jn.2000.83.4.1817. [DOI] [PubMed] [Google Scholar]
- Field GD, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002;34:773–785. doi: 10.1016/s0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- Freed MA, Sterling P. The ON-alpha ganglion cell of the cat retina and its presynaptic cell types. J Neurosci. 1988;8:2303–2320. doi: 10.1523/JNEUROSCI.08-07-02303.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann CJ, O'Brien BJ, Wässle H, Protti DA. AII amacrine cells express L-type calcium channels at their output synapses. J Neurosci. 2003;23:6904–6913. doi: 10.1523/JNEUROSCI.23-17-06904.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Spruston N, Stuart GJ. Diversity and dynamics of dendritic signaling. Science. 2000;290:739–744. doi: 10.1126/science.290.5492.739. [DOI] [PubMed] [Google Scholar]
- Hecht S, Shlaer S, Pirenne MH. Energy, quanta, and vision. J Gen Physiol. 1942;25:819–840. doi: 10.1085/jgp.25.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Watanabe S. Expression of Nav1.1 in rat retinal AII amacrine cells. Neurosci Lett. 2007;424:83–88. doi: 10.1016/j.neulet.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Ma YP, Cui J, Pan ZH. Heterogeneous expression of voltage-dependent Na+ and K+ channels in mammalian retinal bipolar cells. Vis Neurosci. 2005;22:119–133. doi: 10.1017/S0952523805222010. [DOI] [PubMed] [Google Scholar]
- Manookin MB, Beaudoin DL, Ernst ZR, Flagel LJ, Demb JB. Disinhibition combines with excitation to extend the operating range of the OFF visual pathway in daylight. J Neurosci. 2008;28:4136–4150. doi: 10.1523/JNEUROSCI.4274-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire BA, Stevens JK, Sterling P. Microcircuitry of bipolar cells in cat retina. J Neurosci. 1984;4:2920–2938. doi: 10.1523/JNEUROSCI.04-12-02920.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch TA, da Silveira RA, Siegert S, Viney TJ, Awatramani GB, Roska B. Approach sensitivity in the retina processed by a multifunctional neural circuit. Nat Neurosci. 2009;12:1308–1316. doi: 10.1038/nn.2389. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Rieke F. Network variability limits stimulus-evoked spike timing precision in retinal ganglion cells. Neuron. 2006;52:511–524. doi: 10.1016/j.neuron.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GJ, Rieke F. Signals and noise in an inhibitory interneuron diverge to control activity in nearby retinal ganglion cells. Nat Neurosci. 2008;11:318–326. doi: 10.1038/nn2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R. AII amacrine cells quicken time course of rod signals in the cat retina. J Neurophysiol. 1982;47:928–947. doi: 10.1152/jn.1982.47.5.928. [DOI] [PubMed] [Google Scholar]
- Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann N Y Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- Sakitt B. Counting every quantum. J Physiol. 1972;223:131–150. doi: 10.1113/jphysiol.1972.sp009838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath AP, Rieke F. Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron. 2004;41:431–443. doi: 10.1016/s0896-6273(04)00005-4. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Strissel KJ, Elias R, Arshavsky VY, McGinnis JF, Chen J, Kawamura S, Rieke F, Hurley JB. Recoverin improves rod-mediated vision by enhancing signal transmission in the mouse retina. Neuron. 2005;46:413–420. doi: 10.1016/j.neuron.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Singer JH, Diamond JS. Sustained Ca2+ entry elicits transient postsynaptic currents at a retinal ribbon synapse. J Neurosci. 2003;23:10923–10933. doi: 10.1523/JNEUROSCI.23-34-10923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RG, Vardi N. Simulation of the AII amacrine cell of mammalian retina: functional consequences of electrical coupling and regenerative membrane properties. Vis Neurosci. 1995;12:851–860. doi: 10.1017/s095252380000941x. [DOI] [PubMed] [Google Scholar]
- Snellman J, Nawy S. Regulation of the retinal bipolar cell mGluR6 pathway by calcineurin. J Neurophysiol. 2002;88:1088–1096. doi: 10.1152/jn.2002.88.3.1088. [DOI] [PubMed] [Google Scholar]
- Snellman J, Zenisek D, Nawy S. Switching between transient and sustained signalling at the rod bipolar-AII amacrine cell synapse of the mouse retina. J Physiol. 2009;587:2443–2455. doi: 10.1113/jphysiol.2008.165241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Williams SH, Johnston D. Voltage- and space-clamp errors associated with the measurement of electrotonically remote synaptic events. J Neurophysiol. 1993;70:781–802. doi: 10.1152/jn.1993.70.2.781. [DOI] [PubMed] [Google Scholar]
- Tamalu F, Watanabe S. Glutamatergic input is coded by spike frequency at the soma and proximal dendrite of AII amacrine cells in the mouse retina. Eur J Neurosci. 2007;25:3243–3252. doi: 10.1111/j.1460-9568.2007.05596.x. [DOI] [PubMed] [Google Scholar]
- Trexler EB, Li W, Massey SC. Simultaneous contribution of two rod pathways to AII amacrine and cone bipolar cell light responses. J Neurophysiol. 2005;93:1476–1485. doi: 10.1152/jn.00597.2004. [DOI] [PubMed] [Google Scholar]
- Tsukamoto Y, Morigiwa K, Ueda M, Sterling P. Microcircuits for night vision in mouse retina. J Neurosci. 2001;21:8616–8623. doi: 10.1523/JNEUROSCI.21-21-08616.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Hartveit E. Electrical synapses mediate signal transmission in the rod pathway of the mammalian retina. J Neurosci. 2002a;22:10558–10566. doi: 10.1523/JNEUROSCI.22-24-10558.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Hartveit E. AII (rod) amacrine cells form a network of electrically coupled interneurons in the mammalian retina. Neuron. 2002b;33:935–946. doi: 10.1016/s0896-6273(02)00609-8. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]