

Abstract
1H Nuclear magnetic resonance (NMR) spectroscopy (400 MHz) was used in the context of food surveillance to develop a reliable analytical tool to differentiate brands of cola beverages and to quantify selected constituents of the soft drinks. The preparation of the samples required only degassing and addition of 0.1% of TSP in D2O for locking and referencing followed by adjustment of pH to 4.5. The NMR spectra obtained can be considered as “fingerprints” and were analyzed by principal component analysis (PCA). Clusters from colas of the same brand were observed, and significant differences between premium and discount brands were found. The quantification of caffeine, acesulfame-K, aspartame, cyclamate, benzoate, hydroxymethylfurfural (HMF), sulfite ammonia caramel (E 150D), and vanillin was simultaneously possible using external calibration curves and applying TSP as internal standard. Limits of detection for caffeine, aspartame, acesulfame-K, and benzoate were 1.7, 3.5, 0.8, and 1.0 mg/L, respectively. Hence, NMR spectroscopy combined with chemometrics is an efficient tool for simultaneous identification of soft drinks and quantification of selected constituents.
Keywords: NMR spectroscopy, cola, soft drinks, PCA, chemometrics
Introduction
High-resolution nuclear magnetic resonance spectroscopy (NMR) has become one of the most advantageous techniques for food quality control and authenticity assessment because of many benefits that other techniques do not provide.1 NMR spectroscopy is nondestructive, selective, and capable of simultaneous detection of a large number of compounds in complex mixtures without separation.2 This technique not only enables the quantitative determination of principal compounds but also chemometric classification: each spectrum can be treated as a fingerprint. Moreover, NMR spectroscopy is the best nontargeted method to use for food control without a priori knowledge about the composition, the origin, or the type of contamination of the samples.1
Previously, NMR and chemometrics were applied in beverage analysis to discriminate fruit juices,3−5 beer,6,7 or wine,8,9 but there is a sparsity of literature on the topic of NMR analysis applied to colas or soft drinks in general. Wilson et al.10 reported how NMR analysis was used in order to identify an unknown soft drink sample. Another NMR study was about the nontargeted detection of pesticide residues in soft drinks.11 NMR spectroscopy was introduced by Lachenmeier et al. for the quality control and authenticity assessment of beer in official food control.12
In the practice of quality control of alcohol-free beverages, we are often confronted with cola samples from questionable origin, which are labeled or sold from tap with premium brand names and are potentially counterfeited. Furthermore, we have to control the maximum limits of food additives according to European Union law. A large number of methods were published about the simultaneous separation and quantification of sweeteners, additives, and preservatives in soft drinks using reverse-phase high performance liquid chromatography (HPLC),13,14 UV spectrometry,14 mass spectrometry (MS),15 or capillary electrophoresis (CE),14,16 but to our knowledge, no study in the literature applied NMR spectroscopy to simultaneously quantify caffeine, artificial sweeteners, preservatives, and colors in the soft drinks.
This paper describes the investigation of a combined NMR and chemometric data analysis approach to distinguish different brands of cola drinks. We also explored the potential of high resolution 1H NMR to allow the identification and the quantification of selected components.
Materials and Methods
Samples and Chemicals
A total of 129 samples from different origins were analyzed using NMR. The samples were randomly selected from supermarkets in Karlsruhe, Germany, and in Strasbourg, France. Acesulfame-K, sodium benzoate, caffeine, vanillin, sodium cyclamate, aspartame, sodium azide, H3PO4, and HCl were purchased from Sigma Aldrich (Steinheim, Germany), hydroxymethylfurfural (HMF) from Acros Organics (Geel, Belgium), KOH and KH2PO4 from Merck (Darmstadt, Germany), and sulfite ammonia caramel (E 150D) from Couleur und Karamel GmbH (Offenbach, Germany).
The NMR buffer was prepared by dissolving 10.21 g of KH2PO4 and 9.75 mg of sodium azide in 50 mL of pure water and then by adjusting the pH to 4.0 with H3PO4 or KOH. Sodium azide was used for the preservation of the samples because this addition was proved to be advantageous, so that even longer storage of samples at room temperature in the autosampler did not lead to significant changes in the spectrum.
Sample and Standard Preparation
The soft drinks were degassed by ultrasonication for 10 min, and 800 μL of the degassed solution was combined with 100 μL of an internal standard (D2O containing 0.1% of TSP (sodium salt of 3-(trimethylsilyl)propionic acid-d4). The pH of the mixture was then adjusted to 4.50 by adding 101 μL of a buffer (pH 4.0) and 1–2 μL of HCl (1 M) or NaOH (1 M) using a titration unit BTpH (Bruker Biospin, Rheinstetten, Germany) which enabled the very precise adjustment of the pH of all the samples and standard solutions. A 600 μL amount of the final solution was poured into an NMR tube for direct measurement.
Stock solutions with concentrations of 1000 mg/L (caffeine, aspartame, acesulfame-K, cyclamate, and benzoate), of 5 g/L (E 150D), of 500 mg/L (vanillin), and of 10 g/L (HMF) were prepared in deionized water from the pure substances. By diluting the stock solutions in pure water, 15–20 calibration standards were prepared so that the calibration curves were accomplished across the concentration ranges encountered in the soft drinks. The calibration standards were then prepared by the same procedure as that used for the samples (i.e., internal standard and buffer additions as well as pH adjustment).
Optimization of Sample Preparation
The NMR spectra of cola samples show considerable shifts depending on pH. Therefore, the pH was buffered in all samples, so that the differences correspond to true differences in composition of the samples and not to pH effects. The automatic titration unit we have used allows the addition of very small quantities of acid or base (1 to 3 μL) in a short time (3–4 min). This ensures that the variations of composition caused by dilution are small for the multivariate analysis and that the titration is not time-consuming. The pH for the adjustment was set to 4.5 because this is near the pH of the samples after buffer addition (about 4.0), and therefore no large dilution has to occur. For quantitative analysis, the dilution effect was compensated by the use of TSP as internal standard.
1H NMR Measurements at 400 MHz
All NMR measurements were performed on a Bruker Avance 400 Ultrashield spectrometer (Bruker Biospin, Rheinstetten, Germany) equipped with a 5-mm SEI probe with Z-gradient coils, using a Bruker Automatic Sample Changer (B-ACS 120). 1H NMR spectra were acquired at 300.0 K without sample rotation, and 128 scans and 4 prior dummy scans of 65k points were acquired with a spectral width of 20.0234 ppm, a receiver gain of 28.5, an acquisition time of 10 s, and a recycle delay of 4 s. Water suppression was achieved using the NOESY-presaturation pulse sequence (Bruker 1D noesygppr1d pulse sequence) with irradiation at the water frequency during the recycle and mixing time delays.
The data were acquired automatically under the control of ICON-NMR (Bruker Biospin, Rheinstetten, Germany), requiring about 31 min per sample. All NMR spectra were phased, baseline-corrected, and calibrated by the TSP signal at 0.0 ppm.
Chemometrics
The spectral data in the region from 0 to 8 ppm were exported to Unscrambler X version 10.0.1 (Camo Software AS, Oslo, Norway). A bucket table was obtained from the complete set of spectra and analyzed by PCA. Details on the bucketing process of NMR spectra for multivariate data analysis were previously described.12
We tested several spectral regions for calculation: aliphatic (0–3 ppm), midfield (3–6 ppm), and aromatic (6–10 ppm) with 0.01 ppm bucket width. The technique of cross-validation was applied to determine the optimal number of principal components (PCs) needed to have robust models: this technique excludes some of the samples, models the remaining samples, and tests the models on the left-out samples, so the significant number of components and the expected prediction error can be estimated. Seven PCs were found sufficient for the discrimination of cola samples by PCA. Then the data were plotted in a coordinate system defined by two PCs in order to detect the key relationships in the data.
Quantification and Validation Studies
The spectra of cola samples were compared to the spectra of the standards. Separated peaks (i.e., peaks not overlapped or interfered with by matrix) corresponding to each substance were identified and integrated. It was found that in all cases a linear relationship exists between the concentration of the analyte and the ratio between the peak area of the analyte and the internal standard (TSP). Calibration plots were constructed for each substance. For the method validation, cola samples from Germany and France and standard solutions were analyzed several times daily (intraday, n = 5) and over several days (interday, n = 3). The limit of detection (LOD) and the limit of quantification (LOQ) were calculated according to the German Institute for Standardization standard DIN 3264517 using a separate calibration curve (Table 1). The recovery rate was ascertained by adding standard solution at two different concentrations to a real sample. For all calculations, statistical significance was assumed at below the 0.01 probability level.
Table 1. NMR Integration Regions and Concentration Ranges Used for Investigation of LOD, LOQ, and Linearity.
| compound | NMR range | working range (mg/L) | LOD/LOQ range (mg/L) |
|---|---|---|---|
| caffeine | 7.93–7.87 ppm (singlet) | 1–200 | 1–16 |
| aspartame | 3.11–3.03 ppm (multiplet): with citric acid | 3–350 | 3–10 |
| 2.76–2.73 ppm (doublet): without or few citric acid | 4–350 | 4–100 | |
| acesulfame-K | 5.70–5.65 ppm (quadruplet) | 1–300 | 1–10 |
| cyclamate | 2.01–1.92 ppm (multiplet) | 2–244 | 2–9 |
| benzoate | 7.62–7.56 ppm (multiplet) | 2–210 | 1–42 |
| E 150D | 2.23–2.21 ppm (singlet) | 41–2500 | 41–300 |
| HMF | 9.46–9.45 ppm (singlet) | 1–10 | 1–5 |
| vanillin | 9.73–9.72 ppm (singlet) | 1–20 | 1–10 |
Table 2. Results of Method Validation for Selected Compounds.
| aspartamea |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| caffeine | light sample | zero sample | acesulfame-K | cyclamate | benzoate | E150D | vanillin | HMF | |
| LOD (mg/L) | 1.7 | 4.6 | 2.9 | 0.75 | 1.6 | 0.97 | 41 | 0.6 | 0.7 |
| LOQ (mg/L) | 5.3 | 18.0 | 8.8 | 2.5 | 4.7 | 3.4 | 140 | 2.1 | 2.4 |
| precision intraday (%) | |||||||||
| standard solutionb | 4.2 (108) | 0.8 (240) | 1.0 (240) | 2.2 (160) | 1.2 (242) | 0.7 (210) | 2.7 (2.00) | 1.9 (20) | 18 (4.0) |
| authentic sampleb | 1.6 (100) | 5.4 (240) | 8.8 (390) | 2.8 (160) | 2.6 (253) | 2.2 (146) | 6.8 (2.05) | 1.9 (16) | 19 (4.7) |
| precision interday (%) | |||||||||
| standard solution | 4.0 | 1.2 | 2.3 | 2.9 | 0.8 | 0.9 | 2.6 | 5 | 18 |
| authentic sample | 1.5 | 6.3 | 6.7 | 3.1 | 3.7 | 2.8 | 6.5 | 3.1 | 17 |
| recovery range: at two concentrationsb | 100 (101) | 103 (100) | 107 (100) | 94 (100) | 102 (242) | 99 (84) | 99 (500) | 140 (20) | 67 (6.3) |
| 103 (51) | 103 (50) | 97 (50) | 96 (50) | 106 (133) | 96 (42) | 103 (250) | 164 (10) | 91 (3.1) | |
Two signals were used depending of the amount of citric acid: for the light sample, the doublet between 2.73 and 2.76 ppm was integrated, and for zero samples, the multiplet between 3.03 and 3.11 ppm was integrated.
Concentrations used for validation are given in parentheses (expressed in mg/L).
Results and Discussion
Nontargeted Multivariate Analysis
The NMR spectra of the soft drinks could be classified into two major groups: sugar-containing beverages and sugar-free drinks. Figure 1 shows the spectra of normal sugared samples as well as two sugar-free samples designated as “light” and “zero” cola from the same brand. The spectra of sugar-free and normal soft drinks with sugar could be easily differentiated even without PCA because of the large peaks of sucrose, glucose, and fructose between 6 and 3 ppm. Therefore, a separate PCA was made for cola drinks with sugar and for sugar-free samples (so-called light and zero colas). Cola samples with high quantities of caffeine (so-called “strong” colas), caffeine-free, or vanillin-containing drinks were removed from the models as outliers. The type of these special samples could be easily identified by the targeted analysis of the spectra in the aromatic region. Figure 2 presents the whole 1H NMR spectra of a premium-brand cola drink and of a discount-brand cola drink. Extensive spectral overlap was observed, especially in the midfield region. The differences between the spectra of sugared premium-brand cola and discount-brand cola drinks could not be differentiated by eye because of this high spectral complexity. The same problem occurred for the spectra of the sugar-free samples. Thus, a chemometric approach, such as PCA, was needed to interpret the NMR signals and to uncover hidden properties of the samples such as their type. In our case, the best PCA model with regard to classification ability was obtained in the 8–6 ppm region for the cola samples containing sugar and in the 3–0 ppm region for the sugar-free samples. Figures 3 and 4 show the results of the PCA: we could not only differentiate the premium cola brands from the discount brands but clusters from each brand were also clearly separated from each other. The PCA scores of the discount brand 6 and 7 were mixed in the same cluster because the beverages of these brands were probably produced in the same factory (some contract manufacturers conduct commissioned bottling for several discount brands according to our observations).
Figure 1.

1H NMR spectra of a normal sugared sample (A) compared to two sugar-free samples designated as light (B) and zero (C). All samples are from the same brand.
Figure 2.

1H NMR spectra of a premium brand cola (A) compared to a discount brand cola (B).
Figure 3.

Scatter plot of the PCA scores of sugar -containing colas in the aromatic region (8.0–6.0 ppm).
Figure 4.

Scatter plot of the PCA scores of sugar-free colas in the aliphatic region (3.0–0.25 ppm).
For the sugar-free samples, the PCA scores of brand 1 light and brand 1 light without caffeine were represented in the same group in the aliphatic region but the two types could be differentiated by analysis of the aromatic region or by the quantitative analysis. Therefore, 1H NMR combined with chemometrics can be used as a method to discriminate the premium-brand cola samples from the discount-brand cola samples. We also tried to separate the cola drinks by PCA according to their origin (France or Germany). While the French discount brand 3 was clearly distinguished from the German discount brands, as well as the premium brands, the separation was not as clear inside brand 1 (a brand that is filled at factories in both countries). For the normal, sugared type, no difference was detectable at all. However, the light and zero types showed a clear difference between countries. In France and Germany, different levels of aspartame and acesulfame-K are used, and especially sodium cyclamate is different, which is completely absent in the samples from France.
In an investigation by Wilson et al.,10 NMR spectroscopy was used for the identification of unknown samples of soft drink. 1H NMR spectra of samples of purified caffeine, purified sugar, purified citric acid, and soft drinks from popular brands were collected and compared to the spectrum of the unknown sample for identification. Our study confirms the suitability of the approach for a larger sample of commercial beverages. The discrimination and identification of 19 different soft drinks types is possible. Previously, chemometrics combined with other techniques such as a sensor device (so-called electronic tongue) were also used for discrimination of cola brands.18 Discrimination of soft drinks was also performed using a colorimetric sensor array and PCA.19 In comparison with these sensors, NMR spectroscopy provides highly reproducible spectra, allows the identification of marker compounds, and presents a high level of chemical discrimination, which enables differentiation between very similar types of samples. Moreover, this technique also permits the simultaneous acquisition of structural and quantitative information about a large number of targeted substances in the soft drinks.
Quantification Studies
The NMR range used for quantification and the linearity ranges are given in Table 1. The high correlation coefficients (R > 0.99) obtained for each calibration graph indicate a good linearity response within the concentration range studied for each compound. The results of the quantification are indicated in the Supporting Information. The Supporting Information also shows spectra examples for each targeted compound under investigation.
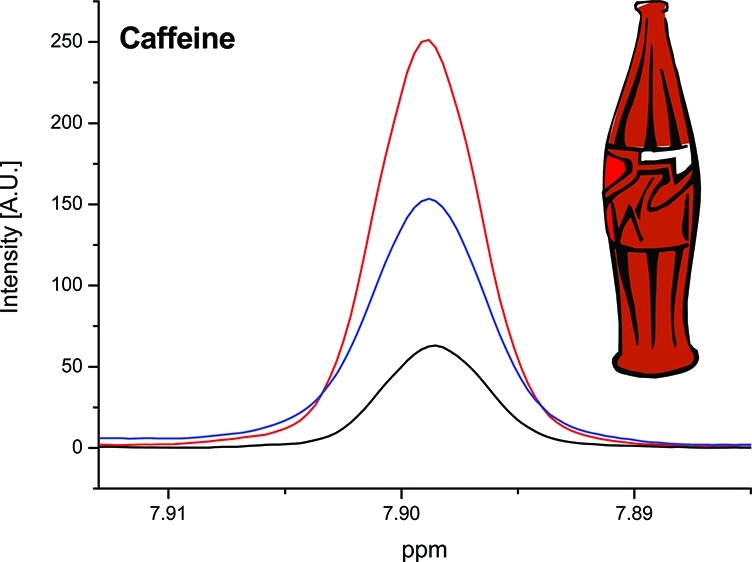
No Europe-wide legislation exists about maximum limits of caffeine, but the presence of caffeine has to be clearly labeled in drinks containing more than 150 mg/L, according to the European Directive 2002/67/EC. This rule applies to soft drinks and energy drinks containing caffeine. Figure 5 shows the signal of caffeine in standard solutions and a cola sample. LOD and LOQ for caffeine were 1.7 mg/L and 5.3 mg/L, respectively. Precisions with standards reached 4.2% (intraday) and 4.0% (interday) and with real samples 1.6% (intraday) and 1.5% (interday). Spiked recoveries ranged between 100% and 103%. We checked that the content of caffeine above 150 mg/L was labeled and that labeling was correct in all instances. In a previous study, a Fourier transform infrared (FTIR) spectroscopic method was developed for quantitative estimation using partial least-squares (PLS) regression.20 In contrast to this indirect chemometric FTIR approach, caffeine can be more straightforwardly quantified by NMR spectroscopy with a good precision using calibration curves with TSP as internal standard. Caffeine concentration was also determined using a microextraction of caffeine on the surface of a fused-silica fiber and gas chromatography with mass spectrometric detection (GC/MS).21 The practical LOD was found at about 2 mg/L, which is close to our value obtained with NMR. Another paper described the quantitative determination of caffeine in sugar-free soft drinks on reversed-phase C8 thin-layer chromatography plates using a surface sampling electrospray ionization system with tandem mass spectrometry detection.22 LODs for HPLC/UV and thin-layer chromatography/electrospray tandem mass spectrometry methods were 0.20 ng injected (0.50 μL) and 1.0 ng spotted on the plate, respectively. Such low limits of detection of caffeine are not possible with NMR spectroscopy but are also not required for the soft drinks, which contain in general around 100 mg/L of caffeine.
Figure 5.

400 MHz 1H NMR spectra of caffeine in standard solutions and the cola sample.
In cola drinks, the European Directive 94/35/EC sets the maximal concentration of aspartame at 600 mg/L, and we did not detect a concentration above this limit. For the quantification of aspartame, two different multiplets were integrated because of different amounts of citric acid among the sugar-free samples. In the range of 2.76 to 2.73 ppm, the peaks corresponding to aspartame and citric acid can overlap. Therefore, this signal was chosen for drinks containing only small quantities of citric acid (i.e., the so-called light cola drinks). For samples with high quantities of citric acid (the “zero” cola samples), the multiplet between 3.11 and 3.03 ppm was integrated (which is overlapped in “light” samples and in cyclamate-containing samples). The LODs of aspartame were 3.5 mg/L for light samples and 2.9 mg/L for zero samples, and the LOQs were 12 mg/L for light samples and 8.8 mg/L for zero samples. FTIR with attenuated total reflectance sampling accessory and PLS regression was previously used for the rapid determination of aspartame in soft drinks.23 Aspartame was found to average from 0.43 to 0.50 mg/L, similar to that found with our method, and with prediction errors ranging from 2.4% to 5.7%. In contrast with FTIR, the quantification of aspartame with NMR spectroscopy does not require the use of chemometrics.
Benzoate is limited to 150 mg/L (calculated as free acid) by the European Directive 95/2/EC, and the concentration of benzoate in the soft drinks analyzed did not exceed this limit. We found a LOD of 0.97 mg/L and a LOQ of 3.4 mg/L.
According to previous investigations, the analysis of carbonated beverages by capillary electrophoresis (CE) enabled the simultaneous determination of aspartame, benzoic acid, and caffeine in 2 min with minimal sample preparation.24 The LODs for caffeine, aspartame, and benzoic acid were 1.6, 18, and 4.0 mg/L, respectively. Another study compared UV spectrophotometry, liquid chromatography (LC), and CE for the quantification of caffeine, aspartame, and benzoic acid in soft drinks.14 The LODs found were 1.2, 0.57, 1.4 mg/L (LC) and 1.7, 0.29, 2.2 mg/L (CE) for caffeine, aspartame, and benzoic acid, respectively. The NMR analysis takes more time (31 min) but allows the simultaneous quantification of seven compounds in one single spectrum with limits of detection similar to that in LC or CE.
According to European Directive 94/35/EC, the concentration of acesulfame-K in soft drinks must not exceed 350 mg/L and the concentration in the cola drinks determined by NMR spectroscopy ranged between 40 and 165 mg/L. The LOD and LOQ obtained were 0.75 and 2.5 mg/L, respectively. Cyclamate is the sodium or calcium salt of cyclamic acid (cyclohexanesulfamic acid), and its limit was recently lowered from 400 to 250 mg/L (calculated as free acid). LOD and LOQ found were 1.6 and 4.7 mg/L. Cyclamate was not detected in cola drinks from France and was found around the maximum limit in the soft drinks from Germany. Cyclamate cannot be determined with HPLC or CE procedures using direct UV detection, as it does not absorb UV light at about 220 nm.25 However, the simultaneous determination of cyclamate, aspartame, and acesulfame-K was possible in 6 min by CE with capacitively coupled contactless conductivity detection.26 The relative standard deviation varied in the range of 1.5 −6.5%, and the recoveries were between 94% and 108%. In comparison, our NMR spectroscopy offers similar performance. Another study described the separation and simultaneous determination of aspartame, sodium cyclamate, and acesulfame-K by ion chromatography with a suppressed conductivity detector.27 The LODs found were at 0.87, 0.032, and 0.019 mg/L, respectively. Rates of recovery were between 98% and 105%, 99% and 104%, and 98% and 103%, respectively. NMR spectroscopy cannot give such low LODs, which are also not necessary to control the limits.
For the sulfite ammonia caramel (E150d), no maximum limit is established by EU law, although usage should be no higher than necessary to achieve the desired purpose. According to the Codex Alimentarius Standard for food additives,28 the maximum level of sulfite ammonia caramel in water-based flavored drinks is 50 000 mg/kg, and we found concentrations of caramel color of about 2000 mg/L which is below this limit. The LOD was 41 mg/L, and the LOQ was 140 mg/L. A simultaneous quantification of the class IV caramel, caffeine, aspartame, acesulfame-K, and sodium benzoate in soft drinks was performed by CE in 8 min.29 For all the food additives, a LOD of 5 mg/L was established and a LOQ of 10 mg/L was obtained. NMR spectroscopy shows similar limits (except for the caramel color) and enables the identification and quantification of other compounds such as cyclamate, vanillin, and HMF.
For vanillin and HMF, the precision and the recovery are not as satisfactory as for the others compounds because of the small quantities close to the limit of quantification, which are present in the samples. The LODs of HMF and vanillin were 0.74 and 0.63 mg/L, respectively, and the LOQs were 2.4 and 2.1 mg/L, respectively. No EU limit is established for the concentration of vanillin and of HMF in soft drinks. A method of quantification of HMF by HPLC in sugar-containing foods including soft drinks was developed, and the results of HMF varied around 0–3 mg/L.30 These results are close to the concentrations found by NMR spectroscopy (0–5 mg/L).
This investigation has shown that 1H NMR spectra of cola drinks can provide qualitative information about the compositional properties such as the brand and also quantitative information about a number of major components. Therefore, NMR spectroscopy combined with PCA is a suitable tool for soft drink authentication. Moreover, we can easily control the European maximum limits for additives. We observed that this technique can replace other traditional methods with lower discrimination ability and similar limits of detection and which often focus on the analysis of specific components of the sample. NMR is not yet a common analytical tool used in quality control; however, the application range has been increasing during the past decade. Advantages such as the high level of chemical discrimination, the minimal preparation of the sample, and the diversity of information make the technique interesting and in the future the usage of NMR spectroscopy will certainly increase in routine quality control.
Acknowledgments
The authors are grateful to Margit Boehm, Hannelore Heger, Bernd Siebler and Jürgen Geisser for excellent technical assistance.
Supporting Information Available
Results of the quantitative determination of substances by NMR. NMR resonances of selected compounds in standard solutions and cola samples. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Le Gall G.; Colquhoun I. J.. NMR Spectroscopy in food authentication. In Food authenticity and traceability;Lees M., Ed.; 2003; pp 131–155. [Google Scholar]
- Kidrič J. NMR study of beverages. Annu. Rep. NMR Spectrosc. 2008, 64, 161–171. [Google Scholar]
- Belton P. S.; Colquhoun I. J.; Kemsley E. K.; Delgadillo I.; Roma P.; Dennis M. J.; Sharman M.; Holmes E.; Nicholson J. K.; Spraul M. Application of chemometrics to 1H NMR spectra of apples juices: discrimination between apple varieties. Food Chem. 1998, 61 (1/2), 207–2213. [Google Scholar]
- Le Gall G.; Puaud M.; Colquhoun I. J. Discrimination between orange juice and pulp wash by 1H nuclear magnetic resonance spectroscopy: Identification of marker compounds. J. Agric. Food Chem. 2001, 49, 580–581. [DOI] [PubMed] [Google Scholar]
- Pelczer I.; D’Souza M.; Banik G.. NMR-based mixture analysis of juices and beverages using an integrated online cross-platform cheminformatics tool. Abstracts of Papers, 235th American Chemical Society National Meeting, New Orleans, LA, April 6–10, 2008; American Chemical Society: Washington, DC, 2008. [Google Scholar]
- Duarte I. F.; Barros A.; Almeida C.; Spraul M.; Gil A. M. Multivariate analysis of NMR and FTIR data as a potential tool for the quality control of beer. J. Agric. Food Chem. 2004, 52 (5), 1031–1038. [DOI] [PubMed] [Google Scholar]
- Almeida C.; Duarte I. F.; Barros A.; Rodrigues J.; Manfred S.; Gil A. M. Composition of beer by 1H NMR Spectroscopy: Effects of brewing site and date of production. J. Agric. Food Chem. 2006, 54 (3), 700–706. [DOI] [PubMed] [Google Scholar]
- Arvanitoyannis I. S.; Katsota M. N.; Psarra E. P.; Soufleros E. H.; Kallithraka S. Application of quality control methods for assessing wine authenticity: Use of multivariate analysis (chemometrics). Trends Food Sci. Technol. 1999, 10 (10), 321–336. [Google Scholar]
- Flemming H. L.; Van den Berg F.; Engelsen S. B. An exploratory chemometric study of 1H NMR spectra of table wines. J. Chemom. 2006, 20, 198–208. [Google Scholar]
- Wilson A.; Myers C.; Crull G.; Curtis M.; Patterson P. P. Analysis of soft drinks using Nuclear Magnetic Resonance Spectroscopy: A Mentorship. J. Chem. Educ. 1999, 76 (10), 1414–1416. [Google Scholar]
- Charlton A. J.; Robb P.; Donarski J. A.; Godward J. Non-targeted detection of chemical contamination in carbonated soft drinks using NMR Spectroscopy, variable selection and Chemometrics. Anal. Chim. Acta 2008, 618, 196–203. [DOI] [PubMed] [Google Scholar]
- Lachenmeier D. W.; Frank W.; Humpfer E.; Schäfer H.; Keller S.; Mörtter M.; Spraul M. Quality control of beer using High-resolution Nuclear Magnetic Resonance Spectroscopy and Multivariate Analysis. Eur. Food Res. Technol. 2005, 220, 215–221. [Google Scholar]
- Delaney M. F.; Pasko K. M.; Gsell D. S.; Korologos P. C.; Morawski J.; Krolikowski L. J.; Warren F. V. Determination of Aspartame, Caffeine, Saccharin, and Benzoic Acid in Beverages by High Performance Liquid Chromatography. J. Chem. Educ. 1985, 62 (7), 618. [Google Scholar]
- McDevitt V. L.; Rodriguez A.; Williams K. R. Analysis of Soft Drinks: UV Spectrophotometry, Liquid Chromatography, and Capillary Electrophoresis. J. Chem. Educ. 1998, 75 (5), 625. [Google Scholar]
- Bergen H. R.; Benson L. M.; Naylor S. Determination of Aspartame and Caffeine in Carbonated Beverages Utilizing Electrospray Ionization-Mass Spectroscopy. J. Chem. Educ. 2000, 77 (10), 1325. [Google Scholar]
- Ames J. M.; Royle L.; Nursten H. E. Capillary Electrophoresis of some Caffeinated Soft Drinks. ACS Symp. Series 2000, 754, 394–402. [Google Scholar]
- DIN 32645. Chemical analysis – Decision limit, detection limit and determination limit under repeatability conditions – Terms, methods, evaluation; Beuth Verlag: Berlin, Germany, 2008(in German). [Google Scholar]
- Lvova L.; Kim S. S.; Legin A.; Vlasov Y.; Yang J. S.; Cha G. S.; Nam H. All-solid-state electronic tongue and its application for beverages analysis. Anal. Chim. Acta 2002, 468, 303–314. [Google Scholar]
- Zhang C.; Suslick K. S. Colorimetric sensor array for soft drink analysis. J. Agric. Food Chem. 2007, 55 (2), 237–242. [DOI] [PubMed] [Google Scholar]
- Paradkar M. M.; Irudayaraj J. Rapid determination of caffeine content in soft drinks using FTIR-ATR spectroscopy. Food. Chem. 2002, 78, 261–266. [Google Scholar]
- Hawthorne S. B.; Miller D. J.; Pawliszyn J.; Arthur C. L. Solventless determination of caffeine in beverages using solid-phase microextraction with fused-silica fibers. J. Chromatogr. 1992, 603, 185–191. [DOI] [PubMed] [Google Scholar]
- Ford M. J.; Deibel M. A.; Tomkins B. A. Quantitative Thin-Layer Chromatography/ Mass Spectrometry Analysis of Caffeine Using a Surface Sampling Probe Electrospray Ionization Tandem Mass Spectrometry System. Anal. Chem. 2005, 77 (14), 4385–4389. [DOI] [PubMed] [Google Scholar]
- Khuranai H. K.; Cho I. K.; Shim J. Y.; Li Q. X.; Jun S. Application of Multibounce Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy and Chemomtrics for Determination of Aspartame in Soft Drinks. J. Agric. Food. Chem. 2008, 56 (3), 778–783. [DOI] [PubMed] [Google Scholar]
- Walker J. C.; Zaugg S. J.; Walker E. B. Analysis of beverages by capillar electrophoresis. J. Chromatogr., A 1997, 781, 481–485. [Google Scholar]
- Thomson C; Trennery O.; Kemmery V. C.; Miscellar B. Electrokinetic capillary chromatographic determination of articial sweeteners in low-Joule soft drinks and other foods. J. Chromatogr., A 1995, 694, 507–514. [DOI] [PubMed] [Google Scholar]
- Bergamo A. B.; Fracassi da Silva J. A.; Pereira de Jesus D. Simultaneous determination of aspartame, cyclamate, saccharin and acesulfame-K in soft drinks and tabletop sweeteners formulations by capillary electrophoresis with capacitevely coupled contactless conductivity detection. Food Chem. 2001, 124 (4), 1714–1717. [Google Scholar]
- Zhu Y.; Guo Y.; Ye M.; James F. S. Separation and simulateous determination of four artificial sweeteners in food and beverages by ion chromatography. J. Chromatogr., A 2005, 1085 (1), 143–146. [DOI] [PubMed] [Google Scholar]
- Codex alimentarius standard of food additives (CODEX STAN 192-1995), Codex Alimentarius Commission, Rome, Italy, 2011.
- Frazier R. A.; Inns E. L.; Dossi N.; Ames J. M.; Nursten H. E. Development of capillary simultaneous analysis of artificial sweeteners, preservatives and colours in soft drinks. J. Chromatogr., A 2000, 876, 213–220. [DOI] [PubMed] [Google Scholar]
- Makawi S. Z. A.; Taha M. I.; Zakaria B. A.; Siddig B.; Mahmod H.; Elhussein A. R. M. Gad kariem, E: A. Identification and Quantification of 5-Hydroxymethyl Furfural HMF in some Sugar-containing Food Products by HPLC. Pak. J. Nutr. 2009, 8 (9), 1391–1396. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.